
Abstract
Purpose
Human thyroid development is a complex and still unexplained process. Thyroid hemiagenesis is a congenital anomaly, where one of the thyroid lobes fails to develop. In the majority of patients with thyroid hemiagenesis, the genetic background remains unknown. The aim of the study was to search for novel genetic contributors to the etiology of thyroid hemiagenesis.
Methods
A cohort of 34 sporadic patients diagnosed with thyroid hemiagenesis and one three-generation family were subjected to comprehensive genomic examination. Initially, targeted screening of associated transcription factors, known to be linked to thyroid development, was performed. As a next step, genomic examinations were applied using high-resolution microarrays, whereas for the thyroid hemiagenesis family, additionally the whole exome sequencing was performed.
Results
Screening of transcription factors revealed no causative mutations in the studied cohort. Genomic examinations revealed the presence of four recurrent defects (three deletions and one duplication) affecting highly conservative proteasome genes PSMA1, PSMA3, and PSMD3. In a thyroid hemiagenesis family a splice site mutation in a proteasome gene PSMD2 (c.612T > C cDNA.1170T > C, g.3271T > C) was found in both affected mother and daughter.
Conclusions
Our results shed a new light on etiology of thyroid hemiagenesis, so far suspected to be linked only to mutations in the genes directly involved in the thyroid development. We demonstrated, for the first time, that genomic alterations in proteasome-associated genes co-occur in patients presenting this developmental anomaly.
Similar content being viewed by others
Introduction
Thyroid hemiagenesis (THA) is a rare congenital anomaly occurring when one of the thyroid lobes fails to develop. The incidence of the disorder is estimated at 0.05–0.5% of the general population. THA occurs usually as an isolated feature, more frequently in women than in men [1]. THA belongs to the broad and heterogeneous spectrum of thyroid dysgenesis, but in contrast to the other more severe forms, clinical consequences of abnormality rarely include congenital hypothyroidism [2]. The accurate etiology of THA remains unknown, even though the abnormality was found to be an inherited condition in a few families, signifying its genetic origin [3]. Additionally, THA family members commonly present other thyroid developmental anomalies (i.e., thyroid agenesis, ectopy or thyroglossal duct cyst) [4]. This, in turn, suggests common genetic background of different thyroid developmental anomalies, but also contribution of other factors modulating expressivity and severity of the definitive phenotype. The genes known to regulate thyroid embryogenesis (i.e., NKX2-1, FOXE1, PAX8) are rarely altered in THA patients, and only one THA familial case was reported to be caused by a heterozygous mutation in PAX8 [5]. Valuable insights into the etiology of the condition provide genetic syndromes, where THA appears as a part of complex phenotype. THA is occasionally concomitant with Williams and DiGeorge syndromes [6]. In both disorders the responsible altered genetic regions are bearing SHH and TBX genes. Their mutated mouse orthologues were evidenced to cause THA in an animal model [7]. TBX1 knockout mice presented normal development of follicular cells, whereas due to disturbed formation of ultimobranchial bodies the gland lacked C cells [7]. The animal THA model however, does not reflect accurately the human species, because in surgical specimens obtained from the THA patients the presence of normal C cells was demonstrated [8]. Human SHH gene is responsible for severe developmental conditions like holoprosencephaly, and there are no evidences of SHH allelism, that may lead to milder phenotype [9]. Other clues coming from THA mouse knockouts encouraged researchers to pay particular attention on human homeobox gene family. The Hoxa3 null mice particularly show diminished number of follicular cells, hypoplasia or absence of one thyroid lobe which corresponds to human THA [10]. In the recent study by Kizys et al. HOXA3, HOXB3, HOXD3, and PITX2 genes were analyzed in THA, however no causative mutations were found [11].
Lack of success in identification of common genetic mechanism in THA indicate the need to search for other genetic targets. In this study, we aimed to look for novel genetic factors that contribute to the development of THA in a uniquely large cohort of patients.
Patients and methods
The studied group consisted of 34 unrelated patients and two familial cases, with THA as an inherited condition transmitted from mother to daughter. The diagnosis of THA was confirmed by ultrasound examination and scintiscan. Recruited patients did not present any concomitant congenital developmental disorders. DNA of all affected individuals and first-degree relatives in THA family was obtained from blood samples. A population cohort of 100 individuals originating from the same region of Poland was used as a control group.
Genetic examinations
Firstly, we analyzed the genes which role in the thyroid development was documented in previous studies. Sequencing of PAX8 (NM_003466), TTF1 (NKX2-1, NM_001079668), TTF2 (FOXE1, NM_004473), HHEX (NM_002729), SHH (NM_000193), and TBX1 (NM_080647) genes encompassed coding sequences, with neighboring intronic regions. Microarray analyses was performed using Illumina Infinium Human OmniExpress-12v1.0 beadchip (San Diego, USA) and then for selected samples on Affymetrix CytoScanHD arrays (2,7M, Santa Clara, USA). For assays, 200–250 ng of genomic DNA was used. Array scans were managed using Illumina GenomeStudio and Affymetrix Chromosome Analysis Suite (ChAS). Log2 R ratios and B-allele frequencies were used for copy number variations (CNVs) identification in all samples. Population specific CNVs were filtered out (control cohort of 100 healthy individuals were analyzed previously using CytoScan 750 K arrays and included in Poznan University of Medical Sciences database) and then identified abnormalities were compared with CNVs annotated in the database of genomic variants. Findings were validated using Real Time qPCR (ΔΔCT method, Life Technologies). Complete genomic coordinates regarding THA patients were deposited in ClinVar database (Table 1). The whole exome analysis was conducted in a three-generation family, presenting transmission of THA as an isolated feature (pedigree presented on Supplementary Fig. 2). Four family members were examined (proband and their first-degree relatives). A total of 1 μg of genomic DNA from subjects was used for the construction of a library with the TruSeq DNA Sample Preparation Kit (Illumina). Whole exome enrichment was performed with the use of DNA library and TruSeq Exome Enrichment Kit (Illumina). For pathogenicity evaluation, the following features were applied: (a) gene/transcript annotations (downloaded from UCSC GenomeBrowser, hg18), (b) known sequence variants from dbSNP (version 132), 1000 Genomes project (The 1000 Genomes Project Consortium). NGS findings were validated using Sanger sequencing. Strategy presented on Supplementary Fig. 1.
Prediction of mutations impact and protein network analysis
The identified mutations were analyzed for their impact on protein conformation and functionality. The amino acid changes were examined using Sift [12], and PolyPhen2 [13] as well as Splice Site mutations using Human Splicing Finder (HSF3.0) [14], and MutationTaster [15]. CNVs encompassing genes, were subjected to a search for phenotype relevancy using GENECODIS, whereas functional annotation was conducted with the database for annotation, visualization, and integrated discovery [16]. P-values and numerical scores were calculated to rank networks according to their degree of relevance in regards to the different gene lists.
Results
Sequencing of known thyroid transcription factor genes (PAX8, NKX2-1, FOXE1, and HHEX) and selected targets, demonstrated in previous studies to contribute to the thyroid development (TBX1, SHH) did not reveal mutations in THA patients. This prompted us to look for genomic abnormalities across sporadic patients, as well as one THA family. The examination using microarrays revealed CNVs (ranging in size from 5 Kb up to 3.2 Mb). We focused particularly on cytoregions bearing genes emerging 63 unique deleterious CNVs. Three deletions turned out to be found in more than one sporadic patient. Analogous investigation was conducted for duplications and selection of 18 gains in copy number were established. Within these loci, a total number of 24 genes were identified and one duplication was found in two unrelated patients. In total four recurrent CNVs (three deletions, one duplication) were detected. We explored DGV to find overlaps with CNVs reported in large control populations, and we found an ultra-rare variability for three out of four regions (not present for PSMD3 gene, where point mutation in THA family was identified). All four regions were statistically significant and not coincidently enriched comparing to large control populations. Microarray data are summarized in Table 1. Recurrent deletions were harboring highly conservative proteasome genes PSMA1 (NM_002786, two patients), PSMA3 (NM_002788, two patients) and additionally deletion encompassing PSMD3 gene (NM_002809, found in one sporadic patient). In the studied group of patients, deletions of two other proteasome-associated genes VPS13C (NM_020821, two patients) and RAD23B (NM_002874, one patient) were detected.
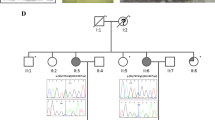
Exome-wide sequencing in a THA family resulted in identification of 93 alterations. One change, was a splice site mutation in a proteasome gene PSMD2 (c.612 T > C cDNA.1170 T > C, g.3271 T > C, NM_002808) found in affected mother and daughter, and regarding microarray findings it was particularly interesting. The mutation introduced an exonic splice sequence (silencer motif), knocking out one PSMD2 allele. Both microarray and whole exome sequencing (WES) data were concordant and subsequently indicating haploinsufficiency of core proteasome genes (Fig. 1).

Proteasome and protein units shown to be altered in THA sporadic patients (microarray studies, encircled in red) and familial case (WES, an arrow). The corresponding gene names as follow: Rpn1—PSMD2, Rpn3— PSMD3, α6—PSMA1, α7—PSMA3
Recurrent abnormalities were found in 7 out of 34 patients (20%), and therefore an attempt was made to emerge THA pathways, based on functional interactions between other detected genes/regions. We used all identified CNVs (63 deleterious, 18 gains), bearing in total 175 known genes. This examination resulted in identification of four significant clusters: (a) protein degradation via proteasome, (b) transmembrane transport (solute carriers), (c) cytoskeleton maintenance (actin related) and (d) transcriptional regulation, respectively (Supplementary Table 1). Except proteasome-related genes, none of the above genes altered in more than one patient.
Discussion
The development of thyroid gland in vertebrates is a multistep complex process. Recently, a comprehensive review summarizing the contribution of transcription factors in thyroid embryogenesis and their role in the differentiation of thyroid follicular cells was published [17]. Mutations of these genes are associated with broad spectrum of clinical features i.e., thyroid hypoplasia, ectopy, agenesis or even syndromal phenotypes characterized by multiple organ development failure. Although the knowledge of the mechanisms involved in the thyroid development has been expanded, in a substantial portion of patients with thyroid dysgenesis it was not possible to identify causative mutations. This suggests that other unknown genes may contribute to the disorder. The aim of the present study was to search for genetic abnormalities explaining the origin of thyroid bilobation failure in humans presenting as an isolated condition.
The transcription factors (TTFs) (PAX8, FOXE1, HHEX or, NKX2-1) represent different classes of conservative transcription factors. They show expression across various tissues, but appear together exclusively in the thyroid. Their interaction is crucial for proper organogenesis, and single TTF perturbation is leading to severe dysgenesis [18]. In order to identify THA genetic background, a cohort THA patients and a THA family were screened for mutations in TTFs. This examination failed to find any abnormalities in known thyroid genes in THA patients under study. This supports previous findings, that patients with isolated THA in majority do not present mutations in TTFs [4]. Therefore, we searched for defects in other genes that would affect thyroid bud migration, or stabilization of the bilobar thyroid structure. We identified recurrent abnormalities affecting proteasome genes comprising recently an attractive target for the studies on developmental mechanisms. The major function of proteasomes in the cell is degradation of ubiquitinated proteins. Their functional insufficiency leads to accumulation of undegraded protein aggregates, which may exert a toxic effect on the cell. Mutations in proteasome related genes are linked to a broad spectrum of phenotypes encompassing neurological, autoimmune, and developmental disorders [19–21]. We found recurrent deletions of proteasome genes, accounting for portion of patients, that nevertheless is unlikely coincidental. All of the genes—PSMA1, PSMA3 and PSMD3 as well as PSMD2 gene in familial case present haploinsufficiency. None of those genes has been linked to the human disease so far. The detected c.612T > C mutation in a PSMD2 gene, which is transmitted together with a THA in a familial case, introduces an exonic splicing silencer sequence. Remarkably, all the identified genes are coding proteins that orchestrates proteasome, although they represent different functional subunits (PSMA1, PSMA3—core particle and PSMD3 as well as PSMD2—regulatory particles), depicted on Fig. 1. The proteasome-related pathomechanism was additionally supported by deletions of other proteasome associated genes VPS13C (two patients) and RAD23B (one patient).
The development of the thyroid is indispensably linked to the cardiovascular system due to juxtaposition of thyroid bud and cardiogenic mesoderm. This fact is supported by high occurrence of heart defects in children with congenital hypothyroidism [22]. On the other hand, the dysfunction of proteasomal-ubiquitin system is recently thoroughly explored in the context of cardiac diseases [23, 24].
Heritability of thyroid dysgenesis accounts for only about 2% of patients and for those cases, the contribution of monogenic background is evidenced (TTFs). So far none of the hypotheses proposed by the Abramowicz et al. (epigenetic regulation, early somatic mutation, stochastic events) referring to missing causes of thyroid developmental anomalies including THA were evidenced [25]. Taking into account high variability and a high frequency of de novo occurring genomic events (particularly CNVs), the model of autosomal dominant transmission proposed by the Macchia et al. is still valid and feasible [5].
In conclusion, our results shed a new light on etiology of THA, so far suspected to be linked only to mutations in the genes directly involved in thyroid development, like TTFs. We demonstrated, for the first time, that genomic alterations in proteasome-associated genes co-occur in patients presenting this developmental anomaly.
References
M. Ruchala, E. Szczepanek, W. Szaflarski, J. Moczko, A. Czarnywojtek, L. Pietz, M. Nowicki, M. Niedziela, M. Zabel, J. Kohrle, J. Sowinski, Increased risk of thyroid pathology in patients with thyroid hemiagenesis: results of a large cohort case–control study. Eur. J. Endocrinol. 162(1), 153–160 (2010). doi:10.1530/EJE-09-0590
E. Al Taji, H. Biebermann, Z. Limanova, O. Hnikova, J. Zikmund, C. Dame, A. Gruters, J. Lebl, H. Krude, Screening for mutations in transcription factors in a Czech cohort of 170 patients with congenital and early-onset hypothyroidism: identification of a novel PAX8 mutation in dominantly inherited early-onset non-autoimmune hypothyroidism. Eur. J. Endocrinol. 156(5), 521–529 (2007). doi:10.1530/EJE-06-0709
H.O. Rajmil, J. Rodriguez-Espinosa, J. Soldevila, J. Ordonez-Llanos, Thyroid hemiagenesis in two sisters. J. Endocrinol. Invest. 7(4), 393–394 (1984). doi:10.1007/BF03351023
M. Castanet, L. Leenhardt, J. Leger, A. Simon-Carre, S. Lyonnet, A. Pelet, P. Czernichow, M. Polak, Thyroid hemiagenesis is a rare variant of thyroid dysgenesis with a familial component but without Pax8 mutations in a cohort of 22 cases. Pediatr. Res. 57(6), 908–913 (2005). doi:10.1203/01.PDR.0000161409.04177.36
P.E. Macchia, P. Lapi, H. Krude, M.T. Pirro, C. Missero, L. Chiovato, A. Souabni, M. Baserga, V. Tassi, A. Pinchera, G. Fenzi, A. Gruters, M. Busslinger, R. Di Lauro, PAX8 mutations associated with congenital hypothyroidism caused by thyroid dysgenesis. Nat. Genet. 19(1), 83–86 (1998). doi:10.1038/ng0598-83
V. Cammareri, G. Vignati, G. Nocera, P. Beck-Peccoz, L. Persani, Thyroid hemiagenesis and elevated thyrotropin levels in a child with Williams syndrome. Am. J. Med. Genet. 85(5), 491–494 (1999). doi:10.1002/(SICI)1096-8628(19990827)85:5<491::AID-AJMG11>3.0.CO;2-Z
H. Fagman, J. Liao, J. Westerlund, L. Andersson, B.E. Morrow, M. Nilsson, The 22q11 deletion syndrome candidate gene Tbx1 determines thyroid size and positioning. Hum. Mol. Genet. 16(3), 276–285 (2007). doi:10.1093/hmg/ddl455
M. Ruchala, E. Szczepanek, P. Sujka-Kordowska, M. Zabel, M. Biczysko, J. Sowinski, The immunohistochemical demonstration of parafollicular cells and evaluation of calcium-phosphate balance in patients with thyroid hemiagenesis. Folia Histochem. Cytobiol. 49(2), 299–305 (2011)
H. Fagman, M. Grande, A. Gritli-Linde, M. Nilsson, Genetic deletion of sonic hedgehog causes hemiagenesis and ectopic development of the thyroid in mouse. Am. J. Pathol. 164(5), 1865–1872 (2004). doi:10.1016/S0002-9440(10)63745-5
N.R. Manley, M.R. Capecchi, The role of Hoxa-3 in mouse thymus and thyroid development. Development 121(7), 1989–2003 (1995)
M.M. Kizys, S. Nesi-Franca, M.G. Cardoso, M.Y. Harada, M.C. Melo, M.I. Chiamolera, M.R. Dias-da-Silva, R.M. Maciel, The absence of mutations in homeobox candidate genes HOXA3, HOXB3, HOXD3 and PITX2 in familial and sporadic thyroid hemiagenesis. J. Pediatr. Endocrinol. Metab. 27(3–4), 317–322 (2014). doi:10.1515/jpem-2013-0289
P. Kumar, S. Henikoff, P.C. Ng, Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4(7), 1073–1082 (2009)
I.A. Adzhubei, S. Schmidt, L. Peshkin, V.E. Ramensky, A. Gerasimova, P. Bork, A.S. Kondrashov, S.R. Sunyaev, A method and server for predicting damaging missense mutations. Nat. Methods 7(4), 248–249 (2010)
F.O. Desmet, D. Hamroun, M. Lalande, G. Collod-Beroud, M. Claustres, C. Beroud, Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 37(9), e67 (2009)
J.M. Schwarz, D.N. Cooper, M. Schuelke, D. Seelow, MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11(4), 361–362 (2014)
W. Huang da, B.T. Sherman, R.A. Lempicki, Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4(1), 44–57 (2009). doi:10.1038/nprot.2008.211
L.P. Fernandez, A. Lopez-Marquez, P. Santisteban, Thyroid transcription factors in development, differentiation and disease. Nat. Rev. Endocrinol. 11(1), 29–42 (2015). doi:10.1038/nrendo.2014.186
H. Fagman, L. Andersson, M. Nilsson, The developing mouse thyroid: embryonic vessel contacts and parenchymal growth pattern during specification, budding, migration, and lobulation. Dev. Dyn. 235(2), 444–455 (2006). doi:10.1002/dvdy.20653
J.M. Deger, J.E. Gerson, R. Kayed, The interrelationship of proteasome impairment and oligomeric intermediates in neurodegeneration. Aging Cell 14(5), 715–724 (2015). doi:10.1111/acel.12359
A.V. Gomes, Genetics of proteasome diseases. Scientifica 2013, 637629 (2013). doi:10.1155/2013/637629
E.S. Lobanova, S. Finkelstein, N.P. Skiba, V.Y. Arshavsky, Proteasome overload is a common stress factor in multiple forms of inherited retinal degeneration. Proc. Natl Acad. Sci. USA 110(24), 9986–9991 (2013). doi:10.1073/pnas.1305521110
A. Olivieri, M.A. Stazi, P. Mastroiacovo, C. Fazzini, E. Medda, A. Spagnolo, S. De Angelis, M.E. Grandolfo, D. Taruscio, V. Cordeddu, M. Sorcini; Study Group for Congenital Hypothyroidism, A population-based study on the frequency of additional congenital malformations in infants with congenital hypothyroidism: data from the Italian Registry for Congenital Hypothyroidism (1991–1998). J. Clin. Endocrinol. Metab. 87(2), 557–562 (2002). doi:10.1210/jcem.87.2.8235
J.M. Predmore, P. Wang, F. Davis, S. Bartolone, M.V. Westfall, D.B. Dyke, F. Pagani, S.R. Powell, S.M. Day, Ubiquitin proteasome dysfunction in human hypertrophic and dilated cardiomyopathies. Circulation 121(8), 997–1004 (2010). doi:10.1161/CIRCULATIONAHA.109.904557
S. Schlossarek, N. Frey, L. Carrier, Ubiquitin-proteasome system and hereditary cardiomyopathies. J. Mol. Cell. Cardiol. 71, 25–31 (2014). doi:10.1016/j.yjmcc.2013.12.016
M.J. Abramowicz, G. Vassart, S. Refetoff, Probing the cause of thyroid dysgenesis. Thyroid 7(3), 325–326 (1997)
D. Pinto, C. Marshall, L. Feuk, S.W. Scherer, Copy-number variation in control population cohorts. Hum. Mol. Genet. 16 Spec No. 2, R168–R173 (2007). doi:10.1093/hmg/ddm241
G.M. Cooper, B.P. Coe, S. Girirajan, J.A. Rosenfeld, T.H. Vu, C. Baker, C. Williams, H. Stalker, R. Hamid, V. Hannig, H. Abdel-Hamid, P. Bader, E. McCracken, D. Niyazov, K. Leppig, H. Thiese, M. Hummel, N. Alexander, J. Gorski, J. Kussmann, V. Shashi, K. Johnson, C. Rehder, B.C. Ballif, L.G. Shaffer, E.E. Eichler, A copy number variation morbidity map of developmental delay. Nat. Genet. 43(9), 838–846 (2011). doi:10.1038/ng.909
T.H. Shaikh, X. Gai, J.C. Perin, J.T. Glessner, H. Xie, K. Murphy, R. O’Hara, T. Casalunovo, L.K. Conlin, M. D’Arcy, E.C. Frackelton, E.A. Geiger, C. Haldeman-Englert, M. Imielinski, C.E. Kim, L. Medne, K. Annaiah, J.P. Bradfield, E. Dabaghyan, A. Eckert, C.C. Onyiah, S. Ostapenko, F.G. Otieno, E. Santa, J.L. Shaner, R. Skraban, R.M. Smith, J. Elia, E. Goldmuntz, N.B. Spinner, E.H. Zackai, R.M. Chiavacci, R. Grundmeier, E.F. Rappaport, S.F. Grant, P.S. White, H. Hakonarson, High-resolution mapping and analysis of copy number variations in the human genome: a data resource for clinical and research applications. Genome Res. 19(9), 1682–1690 (2009). doi:10.1101/gr.083501.108
R.E. Mills, K. Walter, C. Stewart, R.E. Handsaker, K. Chen, C. Alkan, A. Abyzov, S.C. Yoon, K. Ye, R.K. Cheetham, A. Chinwalla, D.F. Conrad, Y. Fu, F. Grubert, I. Hajirasouliha, F. Hormozdiari, L.M. Iakoucheva, Z. Iqbal, S. Kang, J.M. Kidd, M.K. Konkel, J. Korn, E. Khurana, D. Kural, H.Y. Lam, J. Leng, R. Li, Y. Li, C.Y. Lin, R. Luo, X.J. Mu, J. Nemesh, H.E. Peckham, T. Rausch, A. Scally, X. Shi, M.P. Stromberg, A.M. Stutz, A.E. Urban, J.A. Walker, J. Wu, Y. Zhang, Z.D. Zhang, M.A. Batzer, L. Ding, G.T. Marth, G. McVean, J. Sebat, M. Snyder, J. Wang, K. Ye, E.E. Eichler, M.B. Gerstein, M.E. Hurles, C. Lee, S.A. McCarroll, J.O. Korbel, P. Genomes, Mapping copy number variation by population-scale genome sequencing. Nature 470(7332), 59–65 (2011). doi:10.1038/nature09708
A. Itsara, G.M. Cooper, C. Baker, S. Girirajan, J. Li, D. Absher, R.M. Krauss, R.M. Myers, P.M. Ridker, D.I. Chasman, H. Mefford, P. Ying, D.A. Nickerson, E.E. Eichler, Population analysis of large copy number variants and hotspots of human genetic disease. Am. J. Hum. Genet. 84(2), 148–161 (2009). doi:10.1016/j.ajhg.2008.12.014
Acknowledgements
The study was funded by Polish National Science Center (grant No DEC-2011/03/D/NZ5/06142) and by National Multidisciplinary Laboratory of Functional Nanomaterials NanoFun nr POIG.02.02.00-00-025/09 (Innovative Economy Operational Program, Priority Axis 2: R&D Infrastructure, Action 2.2: Support of Formation of Common Research Infrastructure of Scientific Units)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no competing interests.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Budny, B., Szczepanek-Parulska, E., Zemojtel, T. et al. Mutations in proteasome-related genes are associated with thyroid hemiagenesis. Endocrine 56, 279–285 (2017). https://doi.org/10.1007/s12020-017-1287-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12020-017-1287-4