B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments

Abstract
:1. Introduction
2. Normal Development and Homeostasis of B Lymphocytes
3. Abnormalities in B Cell Tolerance and Regulation—Role in SLE and LN Pathogenesis
4. Perturbations in Circulating and Infiltrating B Cell Subsets—Role in SLE and LN Pathogenesis
5. Effect of Immunosuppressive Treatments on B Cells and Implications on the Choice of Therapies in Lupus nephritis
5.1. Conventional Immunosuppressive Treatments for SLE and LN
5.2. Biologics and Emerging Therapies or SLE and LN
6. Future Directions and Concluding Remarks
Funding
Conflicts of Interest
References
- Yap, D.Y.; Tang, C.S.; Ma, M.K.; Lam, M.F.; Chan, T.M. Survival analysis and causes of mortality in patients with lupus nephritis. Nephrol. Dial. Transplant. 2012, 27, 3248–3254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernatsky, S.; Boivin, J.F.; Joseph, L.; Manzi, S.; Ginzler, E.; Gladman, D.D.; Urowitz, M.; Fortin, P.R.; Petri, M.; Barr, S.; et al. Mortality in systemic lupus erythematosus. Arthritis Rheum. 2006, 54, 2550–2557. [Google Scholar] [CrossRef] [PubMed]
- Yap, D.Y.; Chan, T.M. Lupus Nephritis in Asia: Clinical Features and Management. Kidney Dis. 2015, 1, 100–109. [Google Scholar] [CrossRef] [PubMed]
- Isenberg, D.; Appel, G.B.; Contreras, G.; Dooley, M.A.; Ginzler, E.M.; Jayne, D.; Sanchez-Guerrero, J.; Wofsy, D.; Yu, X.; Solomons, N. Influence of race/ethnicity on response to lupus nephritis treatment: The ALMS study. Rheumatology 2010, 49, 128–140. [Google Scholar] [CrossRef] [Green Version]
- Yap, D.Y.; Lai, K.N. Pathogenesis of renal disease in systemic lupus erythematosus-the role of autoantibodies and lymphocytes subset abnormalities. Int. J. Mol. Sci. 2015, 16, 7917–7931. [Google Scholar] [CrossRef]
- Yung, S.; Chan, T.M. Anti-dsDNA antibodies and resident renal cells-heir putative roles in pathogenesis of renal lesions in lupus nephritis. Clin. Immunol. 2017, 185, 40–50. [Google Scholar] [CrossRef]
- Bagavant, H.; Fu, S.M. Pathogenesis of kidney disease in systemic lupus erythematosus. Curr. Opin. Rheumatol. 2009, 21, 489–494. [Google Scholar] [CrossRef] [Green Version]
- Esdaile, J.M.; Abrhamowicz, M.; Joseph, L.; MacKenzie, T.; Li, Y.; Danoff, D. Laboratory tests as predictors of disease exacerbations in systemic lupus erythematosus. Why some tests fail. Arthritis Rheum. 1996, 39, 370–378. [Google Scholar] [CrossRef]
- Esdaile, J.M.; Joseph, L.; Abrahamowicz, M.; Li, Y.; Danoff, D.; Clarke, A.E. Routine immunologic tests in systemic lupus erythematosus: Is there a need for more studies? J. Rheumatol. 1996, 23, 1891–1896. [Google Scholar]
- Moroni, G.; Radice, A.; Giammarresi, G.; Qualini, S.; Gallelli, B.; Leoni, A.; Li Vecchi, M.; Messa, P.; Sinico, R.A. Are laboraotry tests useful for monitoring the activity of lupus nephritis? A 6-year prospective study in a cohort of 228 patients with lupus nephritis. Ann. Rheum. Dis. 2009, 68, 234–237. [Google Scholar] [CrossRef]
- Pieper, K.; Grimbacher, B.; Eibel, H. B-cell biology and development. J. Allergy Clin. Immunol. 2013, 131, 959–971. [Google Scholar] [CrossRef]
- Dorner, T.; Radbruch, A.; Burmester, G.R. B-cell-directed therapies for autoimmune disease. Nat. Rev. Rheumatol. 2009, 5, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Malkiel, S.; Barlev, A.N.; Atisha-Fregoso, Y.; Suurmond, J.; Diamond, B. Plasma Cell Differentiation Pathways in Systemic Lupus Erythematosus. Front. Immunol. 2018, 9, 427. [Google Scholar] [CrossRef] [PubMed]
- Karrar, S.; Cunninghame Graham, D.S. Abnormal B Cell Development in Systemic Lupus Erythematosus: What the Genetics Tell Us. Arthritis Rheumatol. 2018, 70, 496–507. [Google Scholar] [CrossRef] [PubMed]
- Medvedovic, J.; Ebert, A.; Tagoh, H.; Busslinger, M. Pax5: A master regulator of B cell development and leukemogenesis. Adv. Immunol. 2011, 111, 179–206. [Google Scholar] [CrossRef]
- Itoh-Nakadai, A.; Hikota, R.; Muto, A.; Kometani, K.; Watanabe-Matsui, M.; Sato, Y.; Kobayashi, M.; Nakamura, A.; Miura, Y.; Yano, Y.; et al. The transcription repressors Bach2 and Bach1 promote B cell development by repressing the myeloid program. Nat. Immunol. 2014, 15, 1171–1180. [Google Scholar] [CrossRef]
- Huang, C.; Geng, H.; Boss, I.; Wang, L.; Melnick, A. Cooperative transcriptional repression by BCL6 and BACH2 in germinal center B-cell differentiation. Blood 2014, 123, 1012–1020. [Google Scholar] [CrossRef]
- Muto, A.; Ochiai, K.; Kimura, Y.; Itoh-Nakadai, A.; Calame, K.L.; Ikebe, D.; Tashiro, S.; Igarashi, K. Bach2 represses plasma cell gene regulatory network in B cells to promote antibody class switch. EMBO J. 2010, 29, 4048–4061. [Google Scholar] [CrossRef] [Green Version]
- Yap, D.Y.; Lai, K.N. The role of cytokines in the pathogenesis of systemic lupus erythematosus-from bench to bedside. Nephrology 2013, 18, 243–255. [Google Scholar] [CrossRef]
- Mackay, F.; Schneider, P. Cracking the BAFF code. Nat. Rev. Immunol. 2009, 9, 491–502. [Google Scholar] [CrossRef] [Green Version]
- Almaani, S.; Rovin, B.H. B-cell therapy in lupus nephritis: An overview. Nephrol. Dial. Transplant. 2019, 34, 22–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gross, J.A.; Johnston, J.; Mudri, S.; Enselman, R.; Dillon, S.R.; Madden, K.; Xu, W.; Parrish-Novak, J.; Foster, D.; Lofton-Day, C.; et al. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature 2000, 404, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Tackey, E.; Lipsky, P.E.; Illei, G.G. Rationale for interleukin-6 blockade in systemic lupus erythematosus. Lupus 2004, 13, 339–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, B.B.; Bi, E.; Chen, H.; Yu, J.J.; Ye, B.H. IL-21 and CD40L synergistically promote plasma cell differentiation through upregulation of Blimp-1 in human B cells. J. Immunol. 2013, 190, 1827–1836. [Google Scholar] [CrossRef] [PubMed]
- Meffre, E. The establishment of early B cell tolerance in humans: Lessons from primary immunodeficiency diseases. Ann. N. Y. Acad. Sci. 2011, 1246, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Girschick, H.J.; Grammer, A.C.; Nanki, T.; Vazquez, E.; Lipsky, P.E. Expression of recombination activating genes 1 and 2 in peripheral B cells of patients with systemic lupus erythematosus. Arthritis Rheum. 2002, 46, 1255–1263. [Google Scholar] [CrossRef]
- Sinai, P.; Dozmorov, I.M.; Song, R.; Schwartzberg, P.L.; Wakeland, E.K.; Wulfing, C. T/B-cell interactions are more transient in response to weak stimuli in SLE-prone mice. Eur. J. Immunol. 2014, 44, 3522–3531. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.M. Mechanisms and functions for the duration of intercellular contacts made by lymphocytes. Nat. Rev. Immunol. 2009, 9, 543–555. [Google Scholar] [CrossRef]
- Nashi, E.; Wang, Y.; Diamond, B. The role of B cells in lupus pathogenesis. Int. J. Biochem. Cell Biol. 2010, 42, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Tipton, C.M.; Fucile, C.F.; Darce, J.; Chida, A.; Ichikawa, T.; Gregoretti, I.; Schieferl, S.; Hom, J.; Jenks, S.; Feldman, R.J.; et al. Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat. Immunol. 2015, 16, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Boes, M.; Schmidt, T.; Linkemann, K.; Beaudette, B.C.; Marshak-Rothstein, A.; Chen, J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc. Natl. Acad. Sci. USA 2000, 97, 1184–1189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- William, J.; Euler, C.; Christensen, S.; Shlomchik, M.J. Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 2002, 297, 2066–2070. [Google Scholar] [CrossRef] [PubMed]
- Odegard, J.M.; Marks, B.R.; DiPlacido, L.D.; Poholek, A.C.; Kono, D.H.; Dong, C.; Flavell, R.A.; Craft, J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J. Exp. Med. 2008, 205, 2873–2886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herlands, R.A.; Christensen, S.R.; Sweet, R.A.; Hershberg, U.; Shlomchik, M.J. T cell-independent and toll-like receptor-dependent antigen-driven activation of autoreactive B cells. Immunity 2008, 29, 249–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Niu, X.; Liu, H.; Zhang, M.; Chen, M.; Deng, S. Up-regulation of transcription factor Blimp1 in systemic lupus erythematosus. Mol. Immunol. 2013, 56, 574–582. [Google Scholar] [CrossRef]
- Guimaraes, P.M.; Scavuzzi, B.M.; Stadtlober, N.P.; Franchi Santos, L.; Lozovoy, M.A.B.; Iriyoda, T.M.V.; Costa, N.T.; Reiche, E.M.V.; Maes, M.; Dichi, I.; et al. Cytokines in systemic lupus erythematosus: Far beyond Th1/Th2 dualism lupus: Cytokine profiles. Immunol. Cell Biol. 2017, 95, 824–831. [Google Scholar] [CrossRef]
- Salazar-Camarena, D.C.; Ortiz-Lazareno, P.C.; Cruz, A.; Oregon-Romero, E.; Machado-Contreras, J.R.; Munoz-Valle, J.F.; Orozco-Lopez, M.; Marin-Rosales, M.; Palafox-Sanchez, C.A. Association of BAFF, APRIL serum levels, BAFF-R, TACI and BCMA expression on peripheral B-cell subsets with clinical manifestations in systemic lupus erythematosus. Lupus 2016, 25, 582–592. [Google Scholar] [CrossRef]
- Hoyer, B.F.; Moser, K.; Hauser, A.E.; Peddinghaus, A.; Voigt, C.; Eilat, D.; Radbruch, A.; Hiepe, F.; Manz, R.A. Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J. Exp. Med. 2004, 199, 1577–1584. [Google Scholar] [CrossRef] [Green Version]
- Nickerson, K.M.; Christensen, S.R.; Shupe, J.; Kashgarian, M.; Kim, D.; Elkon, K.; Shlomchik, M.J. TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J. Immunol. 2010, 184, 1840–1848. [Google Scholar] [CrossRef] [Green Version]
- Christensen, S.R.; Shupe, J.; Nickerson, K.; Kashgarian, M.; Flavell, R.A.; Shlomchik, M.J. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 2006, 25, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Mackay, F.; Woodcock, S.A.; Lawton, P.; Ambrose, C.; Baetscher, M.; Schneider, P.; Tschopp, J.; Browning, J.L. Mice transgenic for BAFF develop lymphocytic disorders along with autoimmune manifestations. J. Exp. Med. 1999, 190, 1697–1710. [Google Scholar] [CrossRef] [PubMed]
- Arkatkar, T.; Jacobs, H.M.; Du, S.W.; Li, Q.Z.; Hudkins, K.L.; Alpers, C.E.; Rawlings, D.J.; Jackson, S.W. TACI deletion protects against progressive murine lupus nephritis induced by BAFF overexpression. Kidney Int. 2018, 94, 728–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petri, M.; Stohl, W.; Chatham, W.; McCune, W.J.; Chevrier, M.; Ryel, J.; Recta, V.; Zhong, J.; Freimuth, W. Association of plasma B lymphocyte stimulator levels and disease activity in systemic lupus erythematosus. Arthritis Rheum. 2008, 58, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Kitani, A.; Hara, M.; Hirose, T.; Harigai, M.; Suzuki, K.; Kawakami, M.; Kawaguchi, Y.; Hidaka, T.; Kawagoe, M.; Nakamura, H. Autostimulatory effects of IL-6 on excessive B cell differentiation in patients with systemic lupus erythematosus: Analysis of IL-6 production and IL-6R expression. Clin. Exp. Immunol. 1992, 88, 75–83. [Google Scholar] [CrossRef]
- Arkatkar, T.; Du, S.W.; Jacobs, H.M.; Dam, E.M.; Hou, B.; Buckner, J.H.; Rawlings, D.J.; Jackson, S.W. B cell-derived IL-6 initiates spontaneous germinal center formation during systemic autoimmunity. J. Exp. Med. 2017, 214, 3207–3217. [Google Scholar] [CrossRef]
- Ryffel, B.; Car, B.D.; Gunn, H.; Roman, D.; Hiestand, P.; Mihatsch, M.J. Interleukin-6 exacerbates glomerulonephritis in (NZB x NZW) F1 mice. Am. J. Pathol. 1994, 144, 927–937. [Google Scholar]
- Finck, B.K.; Chan, B.; Wofsy, D. Interleukin 6 promotes murine lupus in NZB/NZW F1 mice. J. Clin. Investig. 1994, 94, 585–591. [Google Scholar] [CrossRef] [Green Version]
- Liang, B.; Gardner, D.B.; Griswold, D.E.; Bugelski, P.J.; Song, X.Y. Anti-interleukin-6 monoclonal antibody inhibits autoimmune responses in a murine model of systemic lupus erythematosus. Immunology 2006, 119, 296–305. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Wu, T.H.; Yu, C.L.; Lu, J.Y.; Tsai, Y.Y. Increased excretions of beta2-microglobulin, IL-6, and IL-8 and decreased excretion of Tamm-Horsfall glycoprotein in urine of patients with active lupus nephritis. Nephron 2000, 85, 207–214. [Google Scholar] [CrossRef]
- Herrera-Esparza, R.; Barbosa-Cisneros, O.; Villalobos-Hurtado, R.; Avalos-Diaz, E. Renal expression of IL-6 and TNFalpha genes in lupus nephritis. Lupus 1998, 7, 154–158. [Google Scholar] [CrossRef]
- Nakou, M.; Papadimitraki, E.D.; Fanouriakis, A.; Bertsias, G.K.; Choulaki, C.; Goulidaki, N.; Sidiropoulos, P.; Boumpas, D.T. Interleukin-21 is increased in active systemic lupus erythematosus patients and contributes to the generation of plasma B cells. Clin. Exp. Rheumatol. 2013, 31, 172–179. [Google Scholar] [PubMed]
- Ozaki, K.; Spolski, R.; Ettinger, R.; Kim, H.P.; Wang, G.; Qi, C.F.; Hwu, P.; Shaffer, D.J.; Akilesh, S.; Roopenian, D.C.; et al. Regulation of B cell differentiation and plasma cell generation by IL-21, a novel inducer of Blimp-1 and Bcl-6. J. Immunol. 2004, 173, 5361–5371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bubier, J.A.; Sproule, T.J.; Foreman, O.; Spolski, R.; Shaffer, D.J.; Morse, H.C., 3rd; Leonard, W.J.; Roopenian, D.C. A critical role for IL-21 receptor signaling in the pathogenesis of systemic lupus erythematosus in BXSB-Yaa mice. Proc. Natl. Acad. Sci. USA 2009, 106, 1518–1523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herber, D.; Brown, T.P.; Liang, S.; Young, D.A.; Collins, M.; Dunussi-Joannopoulos, K. IL-21 has a pathogenic role in a lupus-prone mouse model and its blockade with IL-21R.Fc reduces disease progression. J. Immunol. 2007, 178, 3822–3830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawalha, A.H.; Kaufman, K.M.; Kelly, J.A.; Adler, A.J.; Aberle, T.; Kilpatrick, J.; Wakeland, E.K.; Li, Q.Z.; Wandstrat, A.E.; Karp, D.R.; et al. Genetic association of interleukin-21 polymorphisms with systemic lupus erythematosus. Ann. Rheum. Dis. 2008, 67, 458–461. [Google Scholar] [CrossRef] [Green Version]
- Webb, R.; Merrill, J.T.; Kelly, J.A.; Sestak, A.; Kaufman, K.M.; Langefeld, C.D.; Ziegler, J.; Kimberly, R.P.; Edberg, J.C.; Ramsey-Goldman, R.; et al. A polymorphism within IL21R confers risk for systemic lupus erythematosus. Arthritis Rheum. 2009, 60, 2402–2407. [Google Scholar] [CrossRef] [Green Version]
- Devarapu, S.K.; Anders, H.J. Toll-like receptors in lupus nephritis. J. Biomed. Sci. 2018, 25, 35. [Google Scholar] [CrossRef] [Green Version]
- Celhar, T.; Hopkins, R.; Thornhill, S.I.; De Magalhaes, R.; Hwang, S.H.; Lee, H.Y.; Yasuga, H.; Jones, L.A.; Casco, J.; Lee, B.; et al. RNA sensing by conventional dendritic cells is central to the development of lupus nephritis. Proc. Natl. Acad. Sci. USA 2015, 112, E6195–E6204. [Google Scholar] [CrossRef] [Green Version]
- Weindel, C.G.; Richley, L.J.; Bolland, S.; Mehta, A.J.; Kearney, J.F.; Huber, B.T. B cell autophagy mediates TLR7-dependent autoimmunity and inflammation. Autophagy 2015, 11, 1010–1024. [Google Scholar] [CrossRef] [Green Version]
- Applequist, S.E.; Wallin, R.P.; Ljunggren, H.G. Variable expression of toll-like receptor in murine innate and adaptive immune cell lines. Int. Immunol. 2002, 14, 1065–1074. [Google Scholar] [CrossRef] [Green Version]
- Lau, C.M.; Broughton, C.; Tabor, A.S.; Akira, S.; Falvell, R.A.; Mamula, M.J.; Christensen, S.R.; Shlomchik, M.J.; Viglianti, G.A.; Rifkin, I.R.; et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/toll-like receptor 7 engagement. J. Exp. Med. 2005, 202, 1171–1177. [Google Scholar] [CrossRef]
- Leadbetter, E.A.; Rifkin, I.R.; Hohlbaum, A.M.; Beadudette, B.C.; Schlomchik, M.J.; Marshak-Rothstein, A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and toll-like receptors. Nature 2002, 416, 603–607. [Google Scholar] [CrossRef]
- Boule, M.W.; Broughton, C.; Mackay, F.; Akira, S.; Marshak-Rothstein, A.; Rifkin, I.R. Toll-like receptor 9-dependent and independent dendritic cell activation by chromatin-immunoglobulin G complexes. J. Exp. Med. 2004, 199, 1631–1640. [Google Scholar] [CrossRef] [Green Version]
- Jackson, S.W.; Scharping, N.E.; Kolhatkar, N.S.; Khim, S.; Schwartz, M.A.; Li, Q.Z.; Hudkins, K.L.; Alpers, C.E.; Liggitt, D.; Rawlings, D.J. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J. Immunol. 2014, 192, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Capolunghi, F.; Roasado, M.M.; Cascioli, S.; Girolami, E.; Bordasco, S.; Vivarelli, M.; Ruggiero, B.; Cortis, E.; Insalaco, A.; Fanto, N.; et al. Pharmacological inhibition of TLR9 activation blocks autoantibody production in human B cells from SLE patients. Rheumatol. Oxf. 2010, 49, 2281–2289. [Google Scholar] [CrossRef] [Green Version]
- Chauhan, S.K.; Singh, W.; Rai, R.; Rai, M.; Rai, G. Distinct autoantibody profiles in systemic lupus erythematosus patients are selectively associated with TLR7 and TLR9 upregulation. J. Clin. Immunol. 2013, 33, 954–964. [Google Scholar] [CrossRef]
- Sekine, H.; Watanabe, H.; Gilkeson, G.S. Enrichment of anti-glomerular antigen antibody-producing cells in the kidneys of MRL/MpJ-Fas(lpr) mice. J. Immunol. 2004, 172, 3913–3921. [Google Scholar] [CrossRef] [Green Version]
- Espeli, M.; Bokers, S.; Giannico, G.; Dickinson, H.A.; Bardsley, V.; Fogo, A.B.; Smith, K.G. Local renal autoantibody production in lupus nephritis. J. Am. Soc. Nephrol. 2011, 22, 296–305. [Google Scholar] [CrossRef] [Green Version]
- Chan, O.T.; Hannum, L.G.; Haberman, A.M.; Madaio, M.P.; Shlomchik, M.J. A novel mouse with B cells but lacking serum antibody reveals an antibody-independent role for B cells in murine lupus. J. Exp. Med. 1999, 189, 1639–1648. [Google Scholar] [CrossRef]
- Chan, O.T.; Madaio, M.P.; Shlomchik, M.J. B cells are required for lupus nephritis in the polygenic, Fas-intact MRL model of systemic autoimmunity. J. Immunol. 1999, 163, 3592–3596. [Google Scholar]
- Odendahl, M.; Jacobi, A.; Hansen, A.; Feist, E.; Hiepe, F.; Burmester, G.R.; Lipsky, P.E.; Radbruch, A.; Dorner, T. Disturbed peripheral B lymphocyte homeostasis in systemic lupus erythematosus. J. Immunol. 2000, 165, 5970–5979. [Google Scholar] [CrossRef] [Green Version]
- Dorner, T.; Jacobi, A.M.; Lee, J.; Lipsky, P.E. Abnormalities of B cell subsets in patients with systemic lupus erythematosus. J. Immunol. Methods 2011, 363, 187–197. [Google Scholar] [CrossRef]
- Tiller, T.; Tsuiji, M.; Yurasov, S.; Velinzon, K.; Nussenzweig, M.C.; Wardemann, H. Autoreactivity in human IgG+ memory B cells. Immunity 2007, 26, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Jacobi, A.M.; Odendahl, M.; Reiter, K.; Bruns, A.; Burmester, G.R.; Radbruch, A.; Valet, G.; Lipsky, P.E.; Dorner, T. Correlation between circulating CD27high plasma cells and disease activity in patients with systemic lupus erythematosus. Arthritis Rheum. 2003, 48, 1332–1342. [Google Scholar] [CrossRef]
- Hahn, B.H.; McMahon, M.A.; Wilkinson, A.; Wallace, W.D.; Daikh, D.I.; Fitzgerald, J.D.; Karpouzas, G.A.; Merrill, J.T.; Wallace, D.J.; Yazdany, J.; et al. American College of Rheumatology guidelines for screening, treatment, and management of lupus nephritis. Arthritis Care Res. 2012, 64, 797–808. [Google Scholar] [CrossRef] [Green Version]
- Bertsias, G.K.; Tektonidou, M.; Amoura, Z.; Aringer, M.; Bajema, I.; Berden, J.H.; Boletis, J.; Cervera, R.; Dorner, T.; Doria, A.; et al. Joint European League Against Rheumatism and European Renal Association-European Dialysis and Transplant Association (EULAR/ERA-EDTA) recommendations for the management of adult and paediatric lupus nephritis. Ann. Rheum. Dis. 2012, 71, 1771–1782. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) Glomerulonephritis Work Group. KDIGO Clinical Practice Guidelines for Glomerulonephritis. Kidney Int. Suppl. 2012, 2, 139–274. [Google Scholar]
- Mok, C.C.; Yap, D.Y.; Navarra, S.V.; Liu, Z.H.; Zhao, M.H.; Lu, L.; Takeuchi, T.; Avihingsanon, Y.; Yu, X.Q.; Lapid, E.A.; et al. Overview of lupus nephritis management guidelines and perspective from Asia. Nephrology 2014, 19, 11–20. [Google Scholar] [CrossRef]
- Chan, T.M.; Li, F.K.; Tang, C.S.; Wong, R.W.; Fang, G.X.; Ji, Y.L.; Lau, C.S.; Wong, A.K.; Tong, M.K.; Chan, K.W.; et al. Efficacy of mycophenolate mofetil in patients with diffuse proliferative lupus nephritis. N. Engl. J. Med. 2000, 343, 1156–1162. [Google Scholar] [CrossRef] [Green Version]
- Ginzler, E.M.; Dooley, M.A.; Aranow, C.; Kim, M.Y.; Buyon, J.; Merrill, J.T.; Petri, M.; Gilkeson, G.S.; Wallace, D.J.; Weisman, M.H.; et al. Mycophenolate mofetil or intravenous cyclophosphamide for lupus nephritis. N. Engl. J. Med. 2005, 353, 2219–2228. [Google Scholar] [CrossRef] [Green Version]
- Contreras, G.; Pardo, V.; Leclercq, B.; Lenz, O.; Tozman, E.; O’Nan, P.; Roth, D. Sequential therapies for proliferative lupus nephritis. N. Engl. J. Med. 2004, 350, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Appel, G.B.; Contreras, G.; Dooley, M.A.; Ginzler, E.M.; Isenberg, D.; Jayne, D.; Li, L.S.; Mysler, E.; Sanchez-Guerrero, J.; Solomons, N.; et al. Mycophenolate mofetil versus cyclophosphamide for induction treatment of lupus nephritis. J. Am. Soc. Nephrol. 2009, 20, 1103–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fassbinder, T.; Saunders, U.; Mickholz, E.; Jung, E.; Becker, H.; Schluter, B.; Jacobi, A.M. Differential effects of cyclophosphamide and mycophenolate mofetil on cellular and serological parameters in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2015, 17, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Jiang, Z.; Jiang, Y.; Ma, N.; Wang, K.; Zhang, Y. Changes in immune cell frequencies after cyclophosphamide or mycophenolate mofetil treatments in patients with systemic lupus erythematosus. Clin. Rheumatol. 2012, 31, 951–959. [Google Scholar] [CrossRef]
- Dooley, M.A.; Jayne, D.; Ginzler, E.M.; Isenberg, D.; Olsen, N.J.; Wofsy, D.; Eitner, F.; Appel, G.B.; Contreras, G.; Lisk, L.; et al. Mycophenolate versus azathioprine as maintenance therapy for lupus nephritis. N. Engl. J. Med. 2011, 365, 1886–1895. [Google Scholar] [CrossRef] [Green Version]
- Yap, D.Y.; Ma, M.K.; Mok, M.M.; Tang, C.S.; Chan, T.M. Long-term data on corticosteroids and mycophenolate mofetil treatment in lupus nephritis. Rheumatology 2013, 52, 480–486. [Google Scholar] [CrossRef] [Green Version]
- Yap, D.Y.H.; Tang, C.; Ma, M.K.M.; Mok, M.M.Y.; Chan, G.C.W.; Kwan, L.P.Y.; Chan, T.M. Longterm Data on Disease Flares in Patients with Proliferative Lupus Nephritis in Recent Years. J. Rheumatol. 2017, 44, 1375–1383. [Google Scholar] [CrossRef]
- Rollinghoff, M.; Schrader, J.; Wagner, H. Effect of azathioprine and cytosine arabinoside on humoral and cellular immunity in vitro. Clin. Exp. Immunol. 1973, 15, 261–269. [Google Scholar]
- Gorski, A.; Korczak-Kowalska, G.; Nowaczyk, M.; Paczek, L.; Gaciong, Z. The effect of azathioprine on terminal differentiation of human B lymphocytes. Immunopharmacology 1983, 6, 259–266. [Google Scholar] [CrossRef]
- Eickenberg, S.; Mickholz, E.; Jung, E.; Nofer, J.R.; Pavenstadt, H.J.; Jacobi, A.M. Mycophenolic acid counteracts B cell proliferation and plasmablast formation in patients with systemic lupus erythematosus. Arthritis Res. Ther. 2012, 14, R110. [Google Scholar] [CrossRef] [Green Version]
- Yap, D.Y.; Lee, P.; Tam, C.; Yam, I.; Yung, S. B cell subsets and signatures in lupus nephritis patients receiving mycophenolate or azathioprine maintenance. J. Am. Soc. Nephrol. 2018, 29, 336. [Google Scholar]
- Yap, D.Y.; Lee, P.; Tam, C.; Yam, I.; Yung, S.; Chan, T.M. Proliferation and changes in cellular signatures in memory B cells from lupus nephritis patients receiving mycophenolate or azathioprine maintenance. J. Am. Soc. Nephrol. 2019, 30, 63. [Google Scholar]
- Salles, G.; Barrett, M.; Foa, R.; Maurer, J.; O’Brien, S.; Valente, N.; Wenger, M.; Maloney, D.G. Rituximab in B-Cell Hematologic Malignancies: A Review of 20 Years of Clinical Experience. Adv. Ther. 2017, 34, 2232–2273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gopal, A.K.; Press, O.W. Clinical applications of anti-CD20 antibodies. J. Lab. Clin. Med. 1999, 134, 445–450. [Google Scholar] [CrossRef]
- Chen, D.R.; Cohen, P.L. Living life without B cells: Is repeated B-cell depletion a safe and effective long-term treatment plan for rheumatoid arthritis? Int. J. Clin. Rheumtol. 2012, 7, 159–166. [Google Scholar] [CrossRef]
- Boross, P.; Leusen, J.H. Mechanisms of action of CD20 antibodies. Am. J. Cancer Res. 2012, 2, 676–690. [Google Scholar]
- Ahuja, A.; Shupe, J.; Dunn, R.; Kashgarian, M.; Kehry, M.R.; Shlomchik, M.J. Depletion of B cells in murine lupus: Efficacy and resistance. J. Immunol. 2007, 179, 3351–3361. [Google Scholar] [CrossRef]
- Bekar, K.W.; Owen, T.; Dunn, R.; Ichikawa, T.; Wang, W.; Wang, R.; Barnard, J.; Brady, S.; Nevarez, S.; Goldman, B.I.; et al. Prolonged effects of short-term anti-CD20 B cell depletion therapy in murine systemic lupus erythematosus. Arthritis Rheum. 2010, 62, 2443–2457. [Google Scholar] [CrossRef] [Green Version]
- Albert, D.; Dunham, J.; Khan, S.; Stansberry, J.; Kolasinski, S.; Tsai, D.; Pullman-Mooar, S.; Bamack, F.; Striebich, C.; Looney, R.J.; et al. Variability in the biological response to anti-CD20 B cell depletion in systemic lupus erythematosus. Ann. Rheum. Dis. 2008, 67, 1724–1731. [Google Scholar] [CrossRef]
- Gomez Mendez, L.M.; Cascino, M.D.; Garg, J.; Katsumoto, T.R.; Brakeman, P.; Dall’Eram, M.; Looney, R.J.; Rovin, B.; Deagone, L.; Brunetta, P. Peripheral Blood B Cell Depletion after Rituximab and Complete Response in Lupus Nephritis. Clin. J. Am. Soc. Nephrol. 2018, 13, 1502–1509. [Google Scholar] [CrossRef] [Green Version]
- Vital, E.M.; Dass, S.; Buch, M.H.; Henshaw, K.; Pease, C.T.; Martin, M.F.; Ponchel, F.; Rawstron, A.C.; Emery, P. B cell biomarkers of rituximab responses in systemic erythematosus. Arthritis Rheum. 2011, 63, 3038–3047. [Google Scholar] [CrossRef] [PubMed]
- Looney, R.J.; Anolik, J.H.; Campbell, D.; Felgar, R.E.; Young, F.; Arend, L.J.; Sloand, J.A.; Rosenblatt, J.; Sanz, I. B cell depletion as a novel treatment for systemic lupus erythematosus: A phase I/II dose-escalation trial of rituximab. Arthritis Rheum. 2004, 50, 2580–2589. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; Neuwelt, C.M.; Wallace, D.J.; Shanahan, J.C.; Latinis, K.M.; Oates, J.C.; Utset, T.O.; Gordon, C.; Isenberg, D.A.; Hsieh, H.J.; et al. Efficacy and safety of rituximab in moderately-to-severely active systemic lupus erythematosus: The randomized, double-blind, phase II/III systemic lupus erythematosus evaluation of rituximab trial. Arthritis Rheum. 2010, 62, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Furie, R.; Latinis, K.; Looney, R.J.; Fervenza, F.C.; Sanchez-Guerrero, J.; Maciuca, R.; Zhang, D.; Garg, J.P.; Brunetta, P.; et al. Efficacy and safety of rituximab in patients with active proliferative lupus nephritis: The Lupus Nephritis Assessment with Rituximab study. Arthritis Rheum. 2012, 64, 1215–1226. [Google Scholar] [CrossRef] [PubMed]
- Md Yusof, M.Y.; Shaw, D.; El-Sherbiny, Y.M.; Dunn, E.; Rawstron, A.C.; Emery, P.; Vital, E.M. Predicting and managing primary and secondary non-response to rituximab using B-cell biomarkers in systemic lupus erythematosus. Ann. Rheum. Dis. 2017, 76, 1829–1836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazarus, M.N.; Turner-Stokes, T.; Chavele, K.M.; Isenberg, D.A.; Ehrenstein, M.R. B-cell numbers and phenotype at clinical relapse following rituximab therapy differ in SLE patients according to anti-dsDNA antibody levels. Rheumatology 2012, 51, 1208–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weidenbusch, M.; Rommele, C.; Schrottle, A.; Anders, H.J. Beyond the LUNAR trial. Efficacy of rituximab in refractory lupus nephritis. Nephrol. Dial. Transplant. 2013, 28, 106–111. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Lagares, C.; Croca, S.; Sangle, S.; Vital, E.M.; Catapano, F.; Martinez-Berriotxoa, A.; Garcia-Hernandez, F.; Callejas-Rubio, J.L.; Rascon, J.; D’Cruz, D.; et al. Efficacy of rituximab in 164 patients with biopsy-proven lupus nephritis: Pooled data from European cohorts. Autoimmun. Rev. 2012, 11, 357–364. [Google Scholar] [CrossRef]
- Condon, M.B.; Ashby, D.; Pepper, R.J.; Cook, H.T.; Levy, J.B.; Griffith, M.; Cairns, T.D.; Lightstone, L. Prospective observational single-centre cohort study to evaluate the effectiveness of treating lupus nephritis with rituximab and mycophenolate mofetil but no oral steroids. Ann. Rheum. Dis. 2013, 72, 1280–1286. [Google Scholar] [CrossRef]
- Pepper, R.; Griffith, M.; Kirwan, C.; Levy, J.; Taube, D.; Pusey, C.; Lightstone, L.; Cairns, T. Rituximab is an effective treatment for lupus nephritis and allows a reduction in maintenance steroids. Nephrol. Dial. Transplant. 2009, 24, 3717–3723. [Google Scholar] [CrossRef] [Green Version]
- Fanouriakis, A.; Kostopoulou, M.; Alunno, A.; Aringer, M.; Bajema, I.; Boletis, J.N.; Cervera, R.; Doria, A.; Gordon, C.; Govoni, M.; et al. 2019 update of the EULAR recommendations for the management of systemic lupus erythematosus. Ann. Rheum. Dis. 2019, 78, 736–745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rovin, B.; Aroca Martinez, G.; Alvarez, A.; Fragoso Ioyo, H.E.; Zuta, A.E.; Furie, R.; Brunetta, P.; Schindler, T.; Hassan, I.; Cascino, M.; et al. A Phase 2 Randomized, Controlled Study of Obinutuzumab with Mycophenolate and Corticosteroids in Proliferative Lupus Nephritis. In Proceedings of the American Society of Nephrology Kidney Week 2019, FR-OR136, Washington, DC, USA, 5–10 November 2019. [Google Scholar]
- Merrill, J.T.; June, J.; Koumpouras, F.; Machua, W.; Khan, M.F.; Askanase, A.D.; Sheikh, S.Z.; Khosroshahi, A.; Foster, P.; Zack, D.J. Results of a phase 2, double-blind, randomized, placebo-controlled study of a reversible B cell inhibitor, XmAb5871, in systemic lupus erythematosus. Lupus Sci. Med. 2019, 6, A1–A227. [Google Scholar]
- Wallace, D.J.; Gordon, C.; Strand, V.; Hobbs, K.; Petri, M.; Kalunian, K.; Houssiau, F.; Tak, P.P.; Isenberg, D.A.; Kelley, L.; et al. Efficacy and safety of epratuzumab in patients with moderate/severe flaring systemic lupus erythematosus: Results from two randomized, double-blind, placebo-controlled, multicentre studies (ALLEVIATE) and follow-up. Rheumatology 2013, 52, 1313–1322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clowse, M.E.; Wallace, D.J.; Furie, R.A.; Petri, M.A.; Pike, M.C.; Leszczyński, P.; Neuwelt, C.M.; Hobbs, K.; Keiserman, M.; Duca, L.; et al. Efficacy and Safety of Epratuzumab in Moderately to Severely Active Systemic Lupus Erythematosus: Results from Two Phase III Randomized, Double-Blind, Placebo-Controlled Trials. Arthritis Rheumatol. 2017, 69, 362–375. [Google Scholar]
- Alexander, T.; Sarfert, R.; Klotsche, J.; Kuhl, A.A.; Rubbert-Roth, A.; Lorenz, H.M.; Rech, J.; Hoyer, B.F.; Cheng, Q.; Waka, A.; et al. The proteasome inhibitor bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann. Rheum. Dis. 2015, 74, 1474–1478. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, Z.; Huang, L.; Hou, J.; Zhou, M.; Huang, X.; Hu, W.; Liu, Z. The short-term efficacy of bortezomib combined with glucocorticoids for the treatment of refractory lupus nephritis. Lupus 2017, 26, 952–958. [Google Scholar] [CrossRef] [PubMed]
- Kansal, R.; Richardson, N.; Neeli, I.; Khawaja, S.; Chamberlain, D.; Ghani, M.; Ghani, Q.U.; Balazs, L.; Beranova-Giorgianni, S.; Giorgianni, F.; et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yan, M.; Seshasayee, D.; Wang, H.; Lee, W.; French, D.M.; Grewal, I.S.; Cochran, A.G.; Gordon, N.C.; Yin, J.; et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity 2002, 17, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Ramskold, D.; Parodis, I.; Lakshmikanth, T.; Sippl, N.; Khademi, M.; Chen, Y.; Zickert, A.; Mikes, J.; Achour, A.; Amara, K.; et al. B cell alterations during BAFF inhibition with belimumab in SLE. EBioMedicine 2019, 40, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Navarra, S.V.; Guzman, R.M.; Gallacher, A.E.; Hall, S.; Levy, R.A.; Jimenez, R.E.; Li, E.K.; Thomas, M.; Kim, H.Y.; Leon, M.G.; et al. Efficacy and safety of belimumab in patients with active systemic lupus erythematosus: A randomised, placebo-controlled, phase 3 trial. Lancet 2011, 377, 721–731. [Google Scholar] [CrossRef]
- Furie, R.; Petri, M.; Zamani, O.; Cervera, R.; Wallace, D.J.; Tegzova, D.; Sanchez-Guerrero, J.; Schwarting, A.; Merrill, J.T.; Chatham, W.W.; et al. A phase III, randomized, placebo-controlled study of belimumab, a monoclonal antibody that inhibits B lymphocyte stimulator, in patients with systemic lupus erythematosus. Arthritis Rheum. 2011, 63, 3918–3930. [Google Scholar] [CrossRef] [PubMed]
- Dooley, M.A.; Houssiau, F.; Aranow, C.; D’Cruz, D.P.; Askanase, A.; Roth, D.A.; Zhong, Z.J.; Cooper, S.; Freimuth, W.W.; Ginzler, E.M.; et al. Effect of belimumab treatment on renal outcomes: Results from the phase 3 belimumab clinical trials in patients with SLE. Lupus 2013, 22, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Stohl, W.; Hiepe, F.; Latinis, K.M.; Thomas, M.; Scheinberg, M.A.; Clarke, A.; Aranow, C.; Wellborne, F.R.; Abud-Mendoza, C.; Hough, D.R.; et al. Belimumab reduces autoantibodies, normalizes low complement levels, and reduces select B cell populations in patients with systemic lupus erythematosus. Arthritis Rheum. 2012, 64, 2328–2337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isenberg, D.A.; Petri, M.; Kalunian, K.; Tanaka, Y.; Urowitz, M.B.; Hoffman, R.W.; Morgan-Cox, M.; Iikuni, N.; Silk, M.; Wallace, D.J. Efficacy and safety of subcutaneous tabalumab in patients with systemic lupus erythematosus: Results from ILLUMINATE-1, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.T.; van Vollenhoven, R.F.; Buyon, J.P.; Furie, R.A.; Stohl, W.; Morgan-Cox, M.; Dickson, C.; Anderson, P.W.; Lee, C.; Berclaz, P.Y.; et al. Efficacy and safety of subcutaneous tabalumab, a monoclonal antibody to B-cell activating factor, in patients with systemic lupus erythematosus: Results from ILLUMINATE-2, a 52-week, phase III, multicentre, randomised, double-blind, placebo-controlled study. Ann. Rheum. Dis. 2016, 75, 332–340. [Google Scholar] [CrossRef] [Green Version]
- Rovin, B.H.; Dooley, M.A.; Radhakrishnan, J.; Ginzler, E.M.; Forrester, T.D.; Anderson, P.W. The impact of tabalumab on the kidney in systemic lupus erythematosus: Results from two phase 3 randomized, clinical trials. Lupus 2016, 25, 1597–1601. [Google Scholar] [CrossRef]
- Ginzler, E.M.; Wax, S.; Rajeswaran, A.; Copt, S.; Hillson, J.; Ramos, E.; Singer, N.G. Atacicept in combination with MMF and corticosteroids in lupus nephritis: Results of a prematurely terminated trial. Arthritis Res. Ther. 2012, 14, R33. [Google Scholar] [CrossRef] [Green Version]
- Merrill, J.T.; Shanahan, W.R.; Scheinberg, M.; Kalunian, K.C.; Wofsy, D.; Martin, R.S. Phase III trial results with blisibimod, a selective inhibitor of B-cell activating factor, in subjects with systemic lupus erythematosus (SLE): Results from a randomised, double-blind, placebo-controlled trial. Ann. Rheum. Dis. 2018, 77, 883–889. [Google Scholar] [CrossRef]
- Lorenzetti, R.; Janowska, I.; Smulski, C.R.; Frede, N.; Henneberger, N.; Walter, L.; Schleyer, M.T.; Huppe, J.M.; Staniek, J.; Salzer, U.; et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J. Autoimmun. 2019, 101, 145–152. [Google Scholar] [CrossRef]
- Daikh, D.I.; Finck, B.K.; Linsley, P.S.; Hollenbaugh, D.; Wofsy, D. Long-term inhibition of murine lupus by brief simultaneous blockade of the B7/CD28 and CD40/gp39 costimulation pathways. J. Immunol. 1997, 159, 3104–3108. [Google Scholar]
- Daikh, D.I.; Wofsy, D. Cutting edge: Reversal of murine lupus nephritis with CTLA4Ig and cyclophosphamide. J. Immunol. 2001, 166, 2913–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schiffer, L.; Sinha, J.; Wang, X.; Huang, W.; von Gersdorff, G.; Schiffer, M.; Madaio, M.P.; Davidson, A. Short term administration of costimulatory blockade and cyclophosphamide induces remission of systemic lupus erythematosus nephritis in NZB/W F1 mice by a mechanism downstream of renal immune complex deposition. J. Immunol. 2003, 171, 489–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keating, G.M. Abatacept: A review of its use in the management of rheumatoid arthritis. Drugs 2013, 73, 1095–1119. [Google Scholar] [CrossRef] [PubMed]
- Gazeau, P.; Alegria, G.C.; Devauchelle-Pensec, V.; Jamin, C.; Lemerle, J.; Bendaoud, B.; Brooks, W.H.; Saraux, A.; Cornec, D.; Renaudineau, Y. Memory B Cells and Response to Abatacept in Rheumatoid Arthritis. Clin. Rev. Allergy Immunol. 2017, 53, 166–176. [Google Scholar] [CrossRef]
- Furie, R.; Nicholls, K.; Cheng, T.T.; Houssiau, F.; Burgos-Vargas, R.; Chen, S.L.; Hillson, J.L.; Meadows-Shropshire, S.; Kinaszczuk, M.; Merrill, J.T. Efficacy and safety of abatacept in lupus nephritis: A twelve-month, randomized, double-blind study. Arthritis Rheumatol. 2014, 66, 379–389. [Google Scholar] [CrossRef]
- Wofsy, D.; Hillson, J.L.; Diamond, B. Abatacept for lupus nephritis: Alternative definitions of complete response support conflicting conclusions. Arthritis Rheum. 2012, 64, 3660–3665. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.J.; Strand, V.; Merrill, J.T.; Popa, S.; Spindler, A.J.; Eimon, A.; Petri, M.; Smolen, J.S.; Wajdula, J.; Christensen, J.; et al. Efficacy and safety of an interleukin 6 monoclonal antibody for the treatment of systemic lupus erythematosus: A phase II dose-ranging randomised controlled trial. Ann. Rheum. Dis. 2017, 76, 534–542. [Google Scholar] [CrossRef] [Green Version]
- Heidt, S.; Roelen, D.L.; Eijsink, C.; Eikmans, M.; van Kooten, C.; Claas, F.H.; Mulder, A. Calcineurin inhibitors affect B cell antibody responses indirectly by interfering with T cell help. Clin. Exp. Immunol. 2010, 159, 199–207. [Google Scholar] [CrossRef]
- Traitanon, O.; Mathew, J.M.; La Monica, G.; Xu, L.; Mas, V.; Gallon, L. Differential Effects of Tacrolimus versus Sirolimus on the Proliferation, Activation and Differentiation of Human B Cells. PLoS ONE 2015, 10, e0129658. [Google Scholar] [CrossRef] [Green Version]
- Wallin, E.F.; Hill, D.L.; Linterman, M.A.; Wood, K.J. The Calcineurin Inhibitor Tacrolimus Specifically Suppresses Human T Follicular Helper Cells. Front. Immunol. 2018, 9, 1184. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Tang, X.; Liu, Q.; Chen, W.; Fu, P.; Liu, F.; Liao, Y.; Yang, Z.; Zhang, J.; Chen, J.; et al. Short-term outcomes of induction therapy with tacrolimus versus cyclophosphamide for active lupus nephritis: A multicenter randomized clinical trial. Am. J. Kidney Dis. 2011, 57, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Mok, C.C.; Ying, K.Y.; Yim, C.W.; Siu, Y.P.; Tong, K.H.; To, C.H.; Ng, W.L. Tacrolimus versus mycophenolate mofetil for induction therapy of lupus nephritis: A randomised controlled trial and long-term follow-up. Ann. Rheum. Dis. 2016, 75, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Bao, H.; Liu, Z.H.; Xie, H.L.; Hu, W.X.; Zhang, H.T.; Li, L.S. Successful treatment of class V+IV lupus nephritis with multitarget therapy. J. Am. Soc. Nephrol. 2008, 19, 2001–2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Zhang, H.; Liu, Z.; Xing, C.; Fu, P.; Ni, Z.; Chen, J.; Lin, H.; Liu, F.; He, Y.; et al. Multitarget therapy for induction treatment of lupus nephritis: A randomized trial. Ann. Intern. Med. 2015, 162, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Rovin, B.H.; Solomons, N.; Pendergraft, W.F.; Dooley, M.A.; Tumlin, J.; Romero-Diaz, J.; Lysenko, L.; Navarra, S.V.; Huizinga, R.B.; Aura-Lv Study Group. A randomized, controlled double-blind study comparing the efficacy and safety of dose-ranging voclosporin with placebo in achieving remission in patients with active lupus nephritis. Kidney Int. 2019, 95, 219–231. [Google Scholar] [CrossRef]
- Wu, C.; Fu, Q.; Guo, Q.; Chen, S.; Goswami, S.; Sun, S.; Li, T.; Cao, X.; Chu, F.; Chen, Z.; et al. Lupus-associated atypical memory B cells are mTORC1-hyperactivated and functionally dysregulated. Ann. Rheum. Dis. 2019, 78, 1090–1100. [Google Scholar] [CrossRef] [Green Version]
- Lui, S.L.; Yung, S.; Tsang, R.; Zhang, F.; Chan, K.W.; Tam, S.; Chan, T.M. Rapamycin prevents the development of nephritis in lupus-prone NZB/W F1 mice. Lupus 2008, 17, 305–313. [Google Scholar] [CrossRef]
- Lui, S.L.; Tsang, R.; Chan, K.W.; Zhang, F.; Tam, S.; Yung, S.; Chan, T.M. Rapamycin attenuates the severity of established nephritis in lupus-prone NZB/W F1 mice. Nephrol. Dial. Transplant. 2008, 23, 2768–2776. [Google Scholar] [CrossRef] [Green Version]
- Yap, D.Y.; Ma, M.K.; Tang, C.S.; Chan, T.M. Proliferation signal inhibitors in the treatment of lupus nephritis: Preliminary experience. Nephrology 2012, 17, 676–680. [Google Scholar] [CrossRef]
- Lai, Z.W.; Kelly, R.; Winans, T.; Marchena, I.; Shadakshari, A.; Yu, J.; Dawood, M.; Garcia, R.; Tily, H.; Francis, L.; et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: A single-arm, open-label, phase 1/2 trial. Lancet 2018, 391, 1186–1196. [Google Scholar] [CrossRef]
- Yap, D.Y.H.; Tang, C.; Chan, G.C.W.; Kwan, L.P.Y.; Ma, M.K.M.; Mok, M.M.Y.; Chan, T.M. Longterm Data on Sirolimus Treatment in Patients with Lupus Nephritis. J. Rheumatol. 2018, 45, 1663–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]


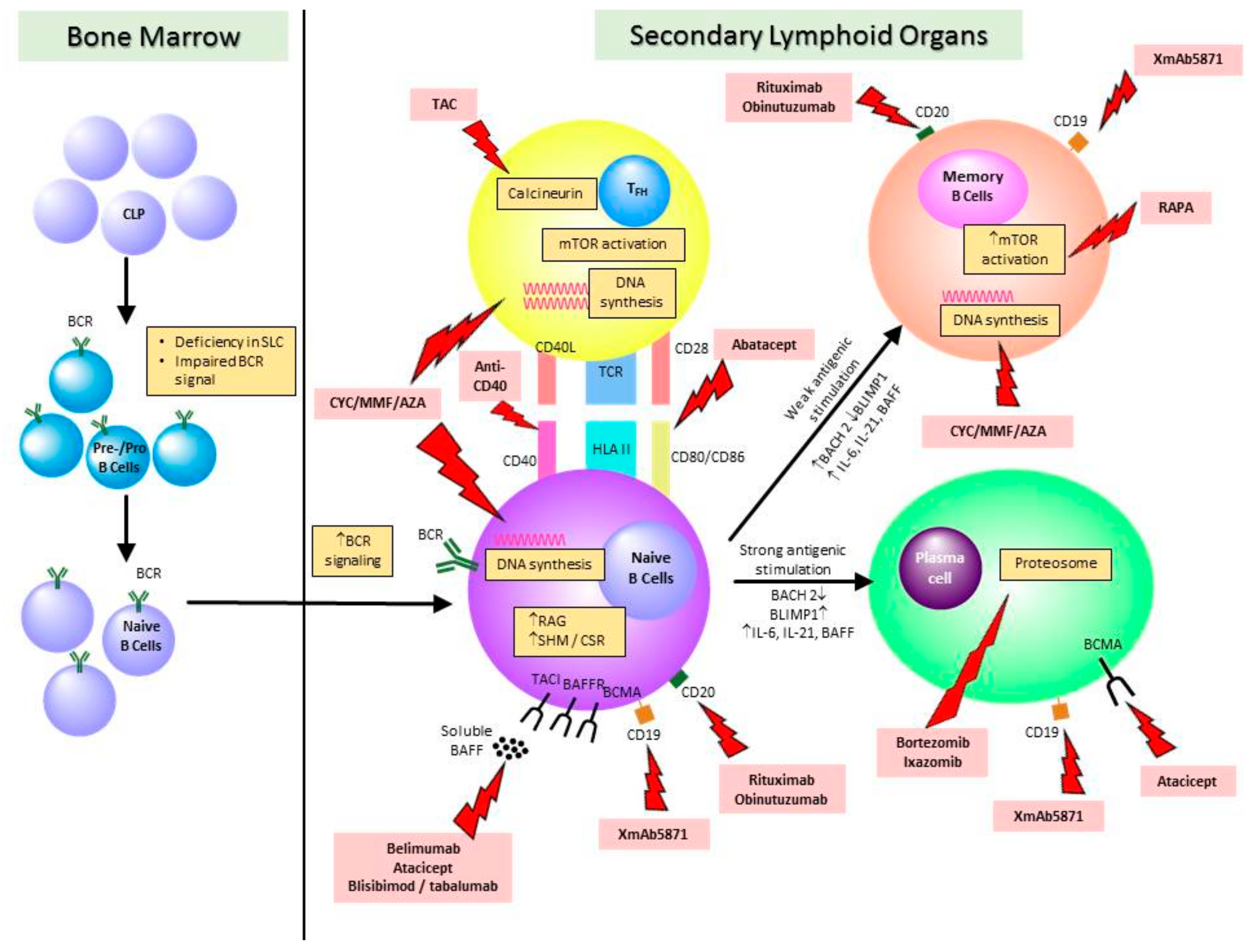{kind=link}
| Drugs | Mechanisms of Action | Effect on B Cells |
|---|---|---|
| CYC | Disrupts DNA replication and thus confers cytotoxic effect on actively proliferating cells including lymphocytes | Preferentially depletes less mature B cells (e.g., naïve B and pre-switched memory B cells) compared with MMF. Little effect on class-switched memory B cells |
| MMF | Inhibits IMPDH and therefore selectively blocks de novo purine synthesis in B and T lymphocytes | Earlier reduction of circulating plasmablasts compared with CYC but with little effect on class-switched memory B cells More potent than AZA in suppressing naïve and memory B cell proliferation |
| AZA | Converts to 6-mercaptopurine and interferes with DNA replication and purine synthesis in lymphocytes | Animal data shows higher AZA dose required to suppress humoral immunity than that required to suppress cellular immunity |
| TAC | Inhibits IL-2 production and thus T cell activation and proliferation | Inhibits TFH and GC formation, thereby impairs B cell maturation and antibody production |
| RAPA | Inhibits the activation of mTOR signals in lymphocytes | Suppresses proliferation of different B cell subsets (especially memory B cell with ↑mTORC1 activation). Blocks differentiation of B cells into plasma cells. ↓intra-renal B cell infiltration in murine LN models |
| Rituximab | Binds to CD20 on B cells, leading to ADCC, CDC and ↑apoptosis of B cells | Profoundly depletes different B subsets except plasma cells within 2 weeks. B cell reconstitution occurs at approximately 6–9 months |
| Belimumab | Inhibits BAFF and hence survival and maturation of B cells | Sustained reduction in naïve plasmacytoid B cells (80–90%), CD19+/CD20+ B cells (70–75%) and plasma cells (50–60%). |
| Abatacept | Interruption of co-stimulatory signal for B cell–T cell interaction | Preferentially suppresses memory B cells |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yap, D.Y.H.; Chan, T.M. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments. Int. J. Mol. Sci. 2019, 20, 6231. https://doi.org/10.3390/ijms20246231
Yap DYH, Chan TM. B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments. International Journal of Molecular Sciences. 2019; 20(24):6231. https://doi.org/10.3390/ijms20246231
Chicago/Turabian StyleYap, Desmond Y. H., and Tak Mao Chan. 2019. "B Cell Abnormalities in Systemic Lupus Erythematosus and Lupus Nephritis—Role in Pathogenesis and Effect of Immunosuppressive Treatments" International Journal of Molecular Sciences 20, no. 24: 6231. https://doi.org/10.3390/ijms20246231