
Second MAFA Variant Causing a Phosphorylation Defect in the Transactivation Domain and Familial Insulinomatosis

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
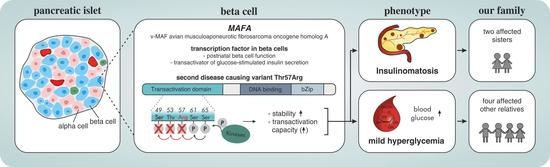
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Routine Laboratory Analyses
2.3. Histopathology and Immunohistochemistry
2.4. DNA Extraction and Sequencing
2.5. Bioinformatics Analyses
3. Results
3.1. Case Report
3.1.1. Clinical Case 1; Index Patient (Female, IV4, 48 Years Old at Time of Study)
3.1.2. Clinical Case 2; Sister of Clinical Case 1 (IV3, 57 Years Old at Time of Study)
3.2. Molecular Genetic Testing
3.2.1. Molecular Genetic Testing of the Two Patients and Classification of the Detected Variants According to the ACMG Criteria
3.2.2. Molecular Genetic Testing of Asymptomatic Family Members
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nessa, A.; Rahman, S.A.; Hussain, K. Hyperinsulinemic Hypoglycemia-The Molecular Mechanisms. Front. Endocrinol. 2016, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Yamada, Y.; Kitayama, K.; Oyachi, M.; Higuchi, S.; Kawakita, R.; Kanamori, Y.; Yorifuji, T. Nationwide survey of endogenous hyperinsulinemic hypoglycemia in Japan (2017–2018): Congenital hyperinsulinism, insulinoma, non-insulinoma pancreatogenous hypoglycemia syndrome and insulin autoimmune syndrome (Hirata’s disease). J. Diabetes Investig. 2020, 11, 554–563. [Google Scholar] [CrossRef]
- Anlauf, M.; Schlenger, R.; Perren, A.; Bauersfeld, J.; Koch, C.A.; Dralle, H.; Raffel, A.; Knoefel, W.T.; Weihe, E.; Ruszniewski, P.; et al. Microadenomatosis of the endocrine pancreas in patients with and without the multiple endocrine neoplasia type 1 syndrome. Am. J. Surg. Pathol. 2006, 30, 560–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Service, F.J.; Natt, N.; Thompson, G.B.; Grant, C.S.; van Heerden, J.A.; Andrews, J.C.; Lorenz, E.; Terzic, A.; Lloyd, R.V. Noninsulinoma pancreatogenous hypoglycemia: A novel syndrome of hyperinsulinemic hypoglycemia in adults independent of mutations in Kir6.2 and SUR1 genes. J. Clin. Endocrinol. Metab. 1999, 84, 1582–1589. [Google Scholar] [CrossRef] [PubMed]
- Klöppel, G.; Anlauf, M.; Raffel, A.; Perren, A.; Knoefel, W.T. Adult diffuse nesidioblastosis: Genetically or environmentally induced? Hum. Pathol. 2008, 39, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Dravecka, I.; Lazurova, I. Nesidioblastosis in adults. Neoplasma 2014, 61, 252–256. [Google Scholar] [CrossRef] [Green Version]
- García-Santos, E.P.; del Carmen Manzanares-Campillo, M.; Padilla-Valverde, D.; Villarejo-Campos, P.; Gil-Rendo, A.; Muñoz-Atienza, V.; Sánchez-García, S.; Puig-Rullán, A.M.; Rodríguez-Peralto, J.L.; Martín-Fernández, J. Nesidioblastosis. A case of hyperplasia of the islets of Langerhans in the adult. Pancreatology 2013, 13, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Bernard, B.; Kline, G.A.; Service, F.J. Hypoglycaemia following upper gastrointestinal surgery: Case report and review of the literature. BMC Gastroenterol. 2010, 10, 77. [Google Scholar] [CrossRef] [Green Version]
- Mala, T. Postprandial hyperinsulinemic hypoglycemia after gastric bypass surgical treatment. Surg. Obes. Relat. Dis. 2014, 10, 1220–1225. [Google Scholar] [CrossRef] [PubMed]
- Anlauf, M.; Bauersfeld, J.; Raffel, A.; Koch, C.A.; Henopp, T.; Alkatout, I.; Schmitt, A.; Weber, A.; Kruse, M.L.; Braunstein, S.; et al. Insulinomatosis: A multicentric insulinoma disease that frequently causes early recurrent hyperinsulinemic hypoglycemia. Am. J. Surg. Pathol. 2009, 33, 339–346. [Google Scholar] [CrossRef] [PubMed]
- Tragl, K.H.; Mayr, W.R. Familial islet-cell adenomatosis. Lancet 1977, 2, 426–428. [Google Scholar] [CrossRef]
- Iacovazzo, D.; Flanagan, S.E.; Walker, E.; Quezado, R.; de Sousa Barros, F.A.; Caswell, R.; Johnson, M.B.; Wakeling, M.; Brändle, M.; Guo, M.; et al. MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc. Natl. Acad. Sci. USA 2018, 115, 1027–1032. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Moriguchi, T.; Kajihara, M.; Esaki, R.; Harada, A.; Shimohata, H.; Oishi, H.; Hamada, M.; Morito, N.; Hasegawa, K.; et al. MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol. 2005, 25, 4969–4976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishimura, W.; Kondo, T.; Salameh, T.; El Khattabi, I.; Dodge, R.; Bonner-Weir, S.; Sharma, A. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev. Biol. 2006, 293, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artner, I.; Hang, Y.; Guo, M.; Gu, G.; Stein, R. MafA is a dedicated activator of the insulin gene in vivo. J. Endocrinol. 2008, 198, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artner, I.; Hang, Y.; Mazur, M.; Yamamoto, T.; Guo, M.; Lindner, J.; Magnuson, M.A.; Stein, R. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes 2010, 59, 2530–2539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hang, Y.; Stein, R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol. Metab. 2011, 22, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barco, S.; Sollfrank, S.; Trinchero, A.; Adenaeuer, A.; Abolghasemi, H.; Conti, L.; Häuser, F.; Hovinga, J.A.K.; Lackner, K.J.; Loewecke, F.; et al. Severe plasma prekallikrein deficiency: Clinical characteristics, novel KLKB1 mutations, and estimated prevalence. J. Thromb. Haemost. 2020, 18, 1598–1617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wenzel, J.J.; Rossmann, H.; Fottner, C.; Neuwirth, S.; Neukirch, C.; Lohse, P.; Bickmann, J.K.; Minnemann, T.; Musholt, T.J.; Schneider-Rätzke, B.; et al. Identification and prevention of genotyping errors caused by G-quadruplex- and i-motif-like sequences. Clin. Chem. 2009, 55, 1361–1371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bickmann, J.K.; Kamin, W.; Wiebel, M.; Häuser, F.; Wenzel, J.J.; Neukirch, C.; Stuhrmann, M.; Lackner, K.J.; Rossmann, H. A novel approach to CFTR mutation testing by pyrosequencing-based assay panels adapted to ethnicities. Clin. Chem. 2009, 55, 1083–1091. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Functional analysis of large MAF transcription factors and elucidation of their relationships with human diseases. Exp. Anim. 2021, 70, 264–271. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-I.; Aramata, S.; Yasuda, K.; Kataoka, K. MafA stability in pancreatic beta cells is regulated by glucose and is dependent on its constitutive phosphorylation at multiple sites by glycogen synthase kinase 3. Mol. Cell. Biol. 2007, 27, 6593–6605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niceta, M.; Stellacci, E.; Gripp, K.W.; Zampino, G.; Kousi, M.; Anselmi, M.; Traversa, A.; Ciolfi, A.; Stabley, D.; Bruselles, A.; et al. Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies. Am. J. Hum. Genet. 2015, 96, 816–825. [Google Scholar] [CrossRef] [Green Version]
- Han, S.-I.; Tsunekage, Y.; Kataoka, K. Phosphorylation of MafA enhances interaction with Beta2/NeuroD1. Acta Diabetol. 2016, 53, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Guanga, G.P.; Wan, C.; Rose, R.B. A novel DNA binding mechanism for maf basic region-leucine zipper factors inferred from a MafA-DNA complex structure and binding specificities. Biochemistry 2012, 51, 9706–9717. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, K.; Han, S.-I.; Shioda, S.; Hirai, M.; Nishizawa, M.; Handa, H. MafA is a glucose-regulated and pancreatic beta-cell-specific transcriptional activator for the insulin gene. J. Biol. Chem. 2002, 277, 49903–49910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocques, N.; Zeid, N.A.; Sii-Felice, K.; Lecoin, L.; Felder-Schmittbuhl, M.-P.; Eychène, A.; Pouponnot, C. GSK-3-mediated phosphorylation enhances Maf-transforming activity. Mol. Cell 2007, 28, 584–597. [Google Scholar] [CrossRef] [PubMed]
- Aramata, S.; Han, S.-I.; Kataoka, K. Roles and regulation of transcription factor MafA in islet beta-cells. Endocr. J. 2007, 54, 659–666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aramata, S.; Han, S.-I.; Yasuda, K.; Kataoka, K. Synergistic activation of the insulin gene promoter by the beta-cell enriched transcription factors MafA, Beta2, and Pdx1. Biochim. Biophys. Acta 2005, 1730, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Burnette, R.; Zhao, L.; Vanderford, N.L.; Poitout, V.; Hagman, D.K.; Henderson, E.; Ozcan, S.; Wadzinski, B.E.; Stein, R. The stability and transactivation potential of the mammalian MafA transcription factor are regulated by serine 65 phosphorylation. J. Biol. Chem. 2009, 284, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zankl, A.; Duncan, E.L.; Leo, P.J.; Clark, G.R.; Glazov, E.A.; Addor, M.-C.; Herlin, T.; Kim, C.A.; Leheup, B.P.; McGill, J.; et al. Multicentric carpotarsal osteolysis is caused by mutations clustering in the amino-terminal transcriptional activation domain of MAFB. Am. J. Hum. Genet. 2012, 90, 494–501. [Google Scholar] [CrossRef] [Green Version]
- Lecompte, S.; Pasquetti, G.; Hermant, X.; Grenier-Boley, B.; Gonzalez-Gross, M.; de Henauw, S.; Molnar, D.; Stehle, P.; Béghin, L.; Moreno, L.A.; et al. Genetic and molecular insights into the role of PROX1 in glucose metabolism. Diabetes 2013, 62, 1738–1745. [Google Scholar] [CrossRef] [Green Version]
- Benkhelifa, S.; Provot, S.; Nabais, E.; Eychène, A.; Calothy, G.; Felder-Schmittbuhl, M.P. Phosphorylation of MafA is essential for its transcriptional and biological properties. Mol. Cell. Biol. 2001, 21, 4441–4452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizawa, M.; Kataoka, K.; Vogt, P.K. MafA has strong cell transforming ability but is a weak transactivator. Oncogene 2003, 22, 7882–7890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eychène, A.; Rocques, N.; Pouponnot, C. A new MAFia in cancer. Nat. Rev. Cancer 2008, 8, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, Q.; Zhou, Z.; Ikeda, Y. PDX1, Neurogenin-3, and MAFA: Critical transcription regulators for beta cell development and regeneration. Stem. Cell Res. Ther. 2017, 8, 240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamze, Z.; Vercherat, C.; Bernigaud-Lacheretz, A.; Bazzi, W.; Bonnavion, R.; Lu, J.; Calender, A.; Pouponnot, C.; Bertolino, P.; Roche, C.; et al. Altered MENIN expression disrupts the MAFA differentiation pathway in insulinoma. Endocr. Relat. Cancer 2013, 20, 833–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huopio, H.; Otonkoski, T.; Vauhkonen, I.; Reimann, F.; Ashcroft, F.M.; Laakso, M. A new subtype of autosomal dominant diabetes attributable to a mutation in the gene for sulfonylurea receptor 1. Lancet 2003, 361, 301–307. [Google Scholar] [CrossRef]
- Yuchi, Y.; Cai, Y.; Legein, B.; de Groef, S.; Leuckx, G.; Coppens, V.; van Overmeire, E.; Staels, W.; de Leu, N.; Martens, G.; et al. Estrogen Receptor α Regulates β-Cell Formation During Pancreas Development and Following Injury. Diabetes 2015, 64, 3218–3228. [Google Scholar] [CrossRef] [Green Version]
- Eto, K.; Nishimura, W.; Oishi, H.; Udagawa, H.; Kawaguchi, M.; Hiramoto, M.; Fujiwara, T.; Takahashi, S.; Yasuda, K. MafA is required for postnatal proliferation of pancreatic β-cells. PLoS ONE 2014, 9, e104184. [Google Scholar] [CrossRef] [PubMed]
- Antwi, K.; Hepprich, M.; Müller, N.A.; Reubi, J.C.; Fani, M.; Rottenburger, C.; Nicolas, G.; Kaul, F.; Christ, E.R.; Wild, D. Pitfalls in the Detection of Insulinomas With Glucagon-Like Peptide-1 Receptor Imaging. Clin. Nucl. Med. 2020, 45, e386–e392. [Google Scholar] [CrossRef] [PubMed]
- Christ, E.; Antwi, K.; Fani, M.; Wild, D. Innovative imaging of insulinoma: The end of sampling? A review. Endocr. Relat. Cancer 2020, 27, R79–R92. [Google Scholar] [CrossRef] [Green Version]
- Kajiwara, K.; Yamagami, T.; Toyota, N.; Kakizawa, H.; Urashima, M.; Hieda, M.; Baba, Y.; Akita, T.; Tanaka, J.; Awai, K. New Diagnostic Criteria for the Localization of Insulinomas with the Selective Arterial Calcium Injection Test: Decision Tree Analysis. J. Vasc. Interv. Radiol. 2018, 29, 1749–1753. [Google Scholar] [CrossRef]
- Thompson, S.M.; Vella, A.; Thompson, G.B.; Rumilla, K.M.; Service, F.J.; Grant, C.S.; Andrews, J.C. Selective Arterial Calcium Stimulation With Hepatic Venous Sampling Differentiates Insulinoma From Nesidioblastosis. J. Clin. Endocrinol. Metab. 2015, 100, 4189–4197. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PROX1 | MAFA | |
|---|---|---|
| Genotype | GA | CG |
| Mutation Call | c.1180G>A | c.170C>G |
| Amino Acid Change | p.Ala394Thr | p.Thr57Arg |
| Exon | 1 | 1 |
| Coverage | 101 | 103 |
| Chromosome | 1 | 8 |
| RefSeq | NM_001270616.2 | NM_201589.4 |
| dbSNP | rs1217787927 | no entry |
| ClinVar | no entry | no entry |
| GnomAD | 1/251,260 alleles | no entry |
| Mutation Taster | disease causing | disease causing |
| PolyPhen2 | benign | probably damaging |
| SIFT | tolerated | damaging |
| PROVEAN | neutral | deleterious |
| OMIM Gene | 601546 | 610303 |
| OMIM Disease | no entry | 147630 |
| Inheritance (OMIM) | no entry | AD |
| ACMG criteria | likely benign | pathogenic |
| Reference | Patient IV3 | Patient IV4 | Patient V5 | Patient V3 | Patient V2 | Patient V4 | Patient III4 | |
|---|---|---|---|---|---|---|---|---|
| Sex/age | - | F/57 | F/48 | M/29 | M/38 | F/30 | M/31 | F/81 |
| BMI | 18.5–24.9 kg/m2 | 27.0 | 26.0 | 26.6 | 29.0 | 38.0 | 29.9 | 26.1 |
| Preexisting diagnosis | - | Insulinomatosis Diabetes Depression | Insulinomatosis Diabetes Hypothyroidism Crohn’s disease | Allergic asthma Impaired fasting glucose (IFG) | Allergic asthma Diabetes Rosacea | - | Allergic asthma Hypothyroidism Gastro-oesophageal reflux disease (GERD) | Hypertension Atrial fibrillations Cataract-operation Polyarthritis |
| Reported therapy | - | Duodenum-preserving resection of the pancreatic head (2010) | Left-sided pancreatectomy (2008) Tumor-enucleation of the pancreatic head (2009) Duodenum-preserving subtotal pancreatectomy (2010) | - | - | - | - | - |
| Medication | - | Pancreatic enzymes Pantoprazole | Insulin-therapy Pancreatic enzymes | Cetirizine | Formoterol/Beclomethasone | Oral contraceptives | Cetirizine Omalizumab Levothyroxine | Candesartan Bisoprolol Rivaroxaban |
| MAFA | c.170C>G, p.Thr57Arg | C/G | C/G | C/G | C/G | C/G | C/C | C/C |
| HbA1c | 4.1–5–6% | 6.9 ↑ | 7.5 (↑) | 5.9 (↑) | 6.6 ↑ | 5.8 (↑) | 5.4 | 5.4 |
| 21–38 mmol/mol Hb | 51 | 59 | 42 | 49 | 40 | 35 | 36 | |
| F-Glucose | 70–100 mg/dL | 143 ↑ | 128 ↑ | 129 ↑ | 125 ↑ | 95 | 94 | 85 |
| Insulin | 6–25 mU/L | 6.7 | 6.3 * | 24.3 ↑ | 26.5 ↑ | 15.7 | 9.3 | 4.5 |
| Pro-insulin | <9.4 pmol/L | 8.07 | <0.1 * | 2.42 | 7.50 | 11.2 ↑ | 5.86 | 3.10 |
| C-Peptide | 0.8–5.2 ng/mL | 2.0 | <0.01 * | 2.75 | 4.52 | 3.37 | 2.54 | 1.66 |
| OH-Butyrate | <74 µmol/L | 70 | 60 | 45 | 30 | 154 ↑ | 21 | 183 |
| CRP | <5 mg/L | 1.1 | 0.54 | 1.6 | 3.5 | 22 ↑ | 2.5 | 3.6 |
| TG | <150 mg/dL | 74 | 63 | 154 (↑) | 260 ↑ | 138 | 261 (↑) | 118 |
| Cholesterol | <200 mg/dL | 181 | 141 | 214 ↑ | 254 ↑ | 192 | 263 ↑ | 235 ↑ |
| HDL-C | >40 mg/dL | 81 | 75 | 55 | 45 | 71 | 56 | 82 |
| LDL-C | <160 mg/dL | 85 | 53 | 128 | 157 | 93 | 155 | 129 |
| AST | 5–31 U/L | 25 | 34 | 24 | 73 ↑ | 27 | 34 | 29 |
| ALT | <35 U/L | 21 | 27 | 40 | 171 ↑ | 19 | 59 ↑ | 14 |
| ALP | 37–111 U/L | 103 | 98 | 65 | 87 | 57 | 90 | 106 |
| GGT | 9–36 U/L | 24 | 14 | 29 | 78 ↑ | 11 | 59 | 19 |
| Bilirubin | 0.3–1.2 mg/dL | 0.58 | 0.42 | 0.49 | 0.45 | 0.38 | 0.59 | 0.54 |
| Albumin | 35–50 g/L | 40 | 38 | 40 | 41 | 37 | 46 | 41 |
| Lipase | <60 U/L | 12 | 4 | 38 | 36 | 29 | 30 | 41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fottner, C.; Sollfrank, S.; Ghiasi, M.; Adenaeuer, A.; Musholt, T.; Schad, A.; Miederer, M.; Schadmand-Fischer, S.; Weber, M.M.; Lackner, K.J.; et al. Second MAFA Variant Causing a Phosphorylation Defect in the Transactivation Domain and Familial Insulinomatosis. Cancers 2022, 14, 1798. https://doi.org/10.3390/cancers14071798
Fottner C, Sollfrank S, Ghiasi M, Adenaeuer A, Musholt T, Schad A, Miederer M, Schadmand-Fischer S, Weber MM, Lackner KJ, et al. Second MAFA Variant Causing a Phosphorylation Defect in the Transactivation Domain and Familial Insulinomatosis. Cancers. 2022; 14(7):1798. https://doi.org/10.3390/cancers14071798
Chicago/Turabian StyleFottner, Christian, Stefanie Sollfrank, Mursal Ghiasi, Anke Adenaeuer, Thomas Musholt, Arno Schad, Matthias Miederer, Simin Schadmand-Fischer, Matthias M. Weber, Karl J. Lackner, and et al. 2022. "Second MAFA Variant Causing a Phosphorylation Defect in the Transactivation Domain and Familial Insulinomatosis" Cancers 14, no. 7: 1798. https://doi.org/10.3390/cancers14071798