Article Text

Abstract
Decades of research on vascular endothelial growth factor (VEGF) have reached fruition with the recent development of intravitreal anti-VEGF treatments for exudative age-related macular degeneration. VEGF is a critical regulator of angiogenesis and vascular permeability with diverse roles, both pathological and physiological, during development and adulthood. The aim of this article is to review aspects of VEGF biology that may be relevant to the clinical use of anti-VEGF agents in ophthalmology: molecular characteristics and isoforms of VEGF; its roles in vasculogenesis, vascular maintenance and angiogenesis; systemic effects of VEGF inhibition; and properties of current anti-VEGF agents.
- AMD, age-related macular degeneration
- CNV, choroidal neovascularisation
- CRC, colorectal cancer
- ECM extracellular matrix,
- ITV, intravitreal
- mAB, monoclonal antibody
- PlGF, placental growth factor
- RPE, retinal pigment epithelium
- VEGF, vascular endothelial growth factor
- VEGFR, VEGF receptor
- VISION, VEGF Inhibition Study in Ocular Neovascularization
Statistics from Altmetric.com
- AMD, age-related macular degeneration
- CNV, choroidal neovascularisation
- CRC, colorectal cancer
- ECM extracellular matrix,
- ITV, intravitreal
- mAB, monoclonal antibody
- PlGF, placental growth factor
- RPE, retinal pigment epithelium
- VEGF, vascular endothelial growth factor
- VEGFR, VEGF receptor
- VISION, VEGF Inhibition Study in Ocular Neovascularization
The advent of anti-vascular endothelial growth factor (VEGF) treatments marks a major advancement in the treatment of angiogenic eye disease, first with the US Food and Drug Administration approval of pegaptanib sodium (Macugen; OSI/Eyetech Pharmaceuticals, New York, USA) in 2004, followed by recent positive clinical trial results for ranibizumab (Lucentis; Genentech) and the growing off-label use of intravitreal (ITV) bevacizumab (Avastin; Genentech, San Francisco, USA). Although it is clear from randomised clinical trials that ITV anti-VEGF treatments will have a fundamental effect for patients with neovascular age-related macular degeneration (AMD), ophthalmologists embarking on the use of these drugs are still faced with some unresolved issues. These include determining the ideal regimen and duration of treatment, the potential of combination treatments and safety concerns with long-term VEGF inhibition. Although some of these issues may be tackled by expanding clinical experience, a review of the relevant VEGF science can inform the clinical application of these novel treatments. The aim of this review is to consider the biology of the VEGF gene family and VEGF-A isoforms, the physiological role of VEGF-A in development and adulthood, the role of VEGF-A in the pathogenesis of ocular diseases, the differential therapeutic mechanisms of current and upcoming anti-VEGF agents and the safety considerations involved in VEGF inhibition.
VEGF BIOLOGY
VEGF is a homodimeric glycoprotein and is a growth factor specific for endothelial cells.1 It is a critical regulator of vasculogenesis and angiogenesis, as well as a potent inducer of vascular permeability.2–4 Additional VEGF functions under study include retinal leukostasis and neuroprotection.2,3,5–7 Three receptor tyrosine kinases have been identified for VEGF: VEGF receptor (VEGFR)1 (fms-like tyrosine kinase-1) has both positive and negative angiogenic effects; VEGFR2 (fetal liver kinase-1 and kinase insert domain-containing receptor) is the primary mediator of the mitogenic, angiogenic and vascular permeability effects of VEGF-A; and VEGFR3 mediates the angiogenic effects on lymphatic vessels.1,8 The pathological functions of VEGF-A have received the most attention, culminating in the development of a new class of drugs for neovascular eye disease and cancers. But VEGF-A and its receptors are also present in tissues and organ systems in normal adults, underlining the physiological role of VEGF-A.
VEGF gene family
The VEGF gene family consists of VEGF-A, VEGF-B, VEGF-C, VEGF-D and placental growth factor (PlGF), which have different binding affinities for the three VEGF receptors.9,10 VEGF-A is the best studied, has been most strongly associated with angiogenesis, and is the target of most current anti-VEGF treatments.11,12 VEGF-A signals through two receptor tyrosine kinases, VEGFR1 and VEGFR2, and is the only member of the VEGF gene family found to be induced by hypoxia.13 VEGF-B selectively binds VEGFR1 and has a role in the regulation of extracellular matrix degradation, cell adhesion and migration.9 Both VEGF-C and VEGF-D bind VEGFR2 and VEGFR3 and regulate lymphangiogenesis, and VEGF-C may also be involved in wound healing.9,10,14 PlGF selectively binds VEGFR19 and is the most abundantly expressed VEGF family member in endothelial cells.13 PlGF may potentiate VEGF-A-induced endothelial cell proliferation,13 but on its own PlGF exerts only weak mitogenicity.13
VEGF-A isoforms
Nine major VEGF-A isoforms have been identified in humans: VEGF121, VEGF145, VEGF148, VEGF162, VEGF165, VEGF165b (an endogenous inhibitory isoform that binds to VEGFR2 with similar affinity to VEGF165 but does not activate it), VEGF183, VEGF189, and VEGF206.15 These isoforms are produced by alternative exon splicing of the human VEGF-A gene on chromosome 6p21.3.1,15 VEGF165 and longer isoforms consist of two domains: a VEGF receptor-binding domain that is present in all VEGF-A isoforms and a heparin-binding domain that is absent in the shorter diffusible isoforms. Longer isoforms, such as VEGF189 and VEGF206, bind to the extracellular matrix (ECM) via the heparin-binding domain.1,16 The VEGF165 isoform is intermediate, and exists as both diffused and partly ECM-bound.17,18 Studies have established that VEGF165 is the most abundantly expressed VEGF-A isoform and has a vital role in angiogenesis.19,20 In addition, one study has found that VEGF121, although less abundant, is more mitogenic than VEGF165 or VEGF189.21 All VEGF-A isoforms except VEGF121 contain a plasmin cleavage site and theoretically may be cleaved by plasmin to generate the smaller VEGF110 form.15,19 VEGF110 can stimulate endothelial cell growth and induce vascular permeability in the Miles assay17; however, its mitogenic potency is less than that of VEGF165.22
VEGF121, VEGF165, VEGF183 and VEGF189 are distributed widely in tissue, with VEGF165 expression the most abundant. In contrast, VEGF145 and VEGF206 are less abundant. In mice the VEGF-A gene is highly conserved, with three splice variants, VEGF120, VEGF164 and VEGF188, equivalent to the human VEGF121, VEGF165 and VEGF189 isoforms, respectively.23
VEGF-A in vasculogenesis
Vascular formation can be defined in two distinct categories: vasculogenesis and angiogenesis. Vasculogenesis is the process of de novo blood vessel formation during embryogenesis,24 which begins with the differentiation, proliferation and migration of haemangiogenic stem cells under the possible influence of VEGF-A and PlGF.25 VEGF-A may also participate in endothelial cell survival and in capillary regression via synchronous apoptosis.26
Angiogenesis, which can occur in adulthood, is the process of neovascularisation from established blood vessels. Angiogenesis occurs in response to a variety of pathological stimuli including ischaemia, inflammation, wound healing and tumour formation. Angiogenesis can also be integral to tissue homoeostasis and normal processes such as the growth and maintenance of the ovarian follicle and corpus luteum during reproductive life in women.27 Thus, angiogenesis in adulthood can have both detrimental effects, such as neovascular AMD, rheumatoid arthritis and cancer,12 and beneficial effects, such as cardiac remodelling and wound healing.
VEGF-A is integral to blood vessel formation during both vasculogenesis and angiogenesis, but its role differs in development versus adulthood (fig 1). For instance, in developing mice, newly formed vessels require VEGF-A to persist through the fourth postnatal week, but after that time the vessels stabilise and no longer depend on VEGF-A for maintenance.28 VEGF deficiency is lethal during embryogenesis.11 Postnatal inhibition of the VEGF-A gene via conditional knockout technology increases mortality.28 Data from selective VEGF-A isoform knockout mice suggest that the absence of VEGF164 and VEGF188 results in a variety of vascular defects, including abnormal pulmonary vascular development.23

The role of VEGF-A and the effect of VEGF inhibition in embryogenesis, postnatal development and adulthood under normal and pathological conditions, particularly in the ocular system. AMD, age-related macular degeneration; CNV, choroidal neovascularisation; VEGF, vascular endothelial growth factor.
VEGF-A in ocular development
During ocular development, vasculogenesis occurs firstly in the choroid and hyaloid systems, where VEGF-A may have a role. VEGF-A expression has been implicated in the abnormal persistence of the hyaloid vasculature in a mouse model of persistent fetal vasculature.29 In VEGFrpe−/− mice, the lack of VEGF-A expression relegated specifically to the retinal pigment epithelium (RPE) results in failure of the choriocapillaris to form.30 Vascularisation of the retina occurs later in embryogenesis and is not completed until the final postnatal period,31 with VEGF-A controlling vessel sprouting and migration in the postnatal retina.32 VEGF-A expressed in the neural retina is correlated temporally and spatially with normal vasculogenesis in both humans24 and rats.33 VEGF-A appears to be induced by physiological hypoxia as part of normal retinal development, and VEGF-A suppression inhibits vascular formation.31
The function of the individual VEGF-A isoforms may differ during ocular development. Mice expressing only VEGF164 have normal retinal angiogenesis and vasculature, whereas mice expressing only VEGF120 or VEGF188 have aberrant vascular outgrowth and arterial development.34 VEGF164 shows the greatest upregulation during rat ocular development, in the presence of VEGF120, VEGF164 and VEGF188.35
VEGF-A in the maintenance of adult vasculature
In addition to vasculogenesis and angiogenesis, VEGF-A may participate in the maintenance of certain vascular systems in the adult. Specific binding of VEGF-A is associated with mature vessels in various adult rat tissues, such as the heart, kidney and brain, suggesting that VEGF-A has a function in the maintenance of quiescent vascular endothelium.36 VEGF-A maintains normal tracheal vasculature in adult mice, as shown by capillary loss after broad VEGF inhibition, an effect not seen with selective VEGF164 inhibition.37 In addition, chronic VEGF inhibition in adult rats results in lung alveolar septal abnormalities.38
VEGF-A in the maintenance of adult ocular vasculature
Little is known of the role of VEGF-A in the maintenance of adult ocular vasculature. VEGF-A is produced by human differentiated RPE cells both in vivo and in vitro, and may be involved in the paracrine signalling between the RPE and choriocapillaris.39 In normal adult monkey and mouse eyes, the VEGF121 and VEGF165 isoforms are most abundantly expressed in the choroid, RPE, retina and iris tissue, consistent with VEGF-A acting as a vascular survival factor in the adult eye.40 However, in adult mice treated systemically with vatalanib, a broad inhibitor of VEGF-A isoforms and platelet-derived growth factor, no effects are seen in the retinal vasculature.41 To date, clinical trials of the VEGF-A inhibitors pegaptanib and ranibizumab in humans have not shown adverse effects on normal retinal or choroidal vasculature (Rosenfeld et al41a; Brown et al41b).42
VEGF-A in ocular pathogenesis
The central role of VEGF-A is well established in ocular neovascular diseases.43–46 In humans, high levels of VEGF-A expression are found in choroidal neovascularisation (CNV) tissue excised from patients with AMD.43,47 Intraocular VEGF-A levels correlate with blood vessel formation in patients with diabetic retinopathy and other retinal disorders.44,48
Different VEGF-A isoforms may have different functions in ocular diseases. VEGF164 is the predominant isoform expressed at the time of maximal preretinal neovascularisation in a neonatal rat model35 and is the primary proinflammatory isoform in the retina of rats with diabetes.49,50 Levels of both VEGF121 and VEGF165 are increased in monkeys after laser-induced retinal vein occlusion.51 VEGF120 is the main isoform expressed in mouse CNV membranes, and inhibition of VEGF120 results in reduction of CNV in mice.43,52 Both VEGF121 and VEGF165 isoforms are found in CNV tissue excised from patients with AMD.43
VEGF-A INHIBITION
Anti-VEGF agents
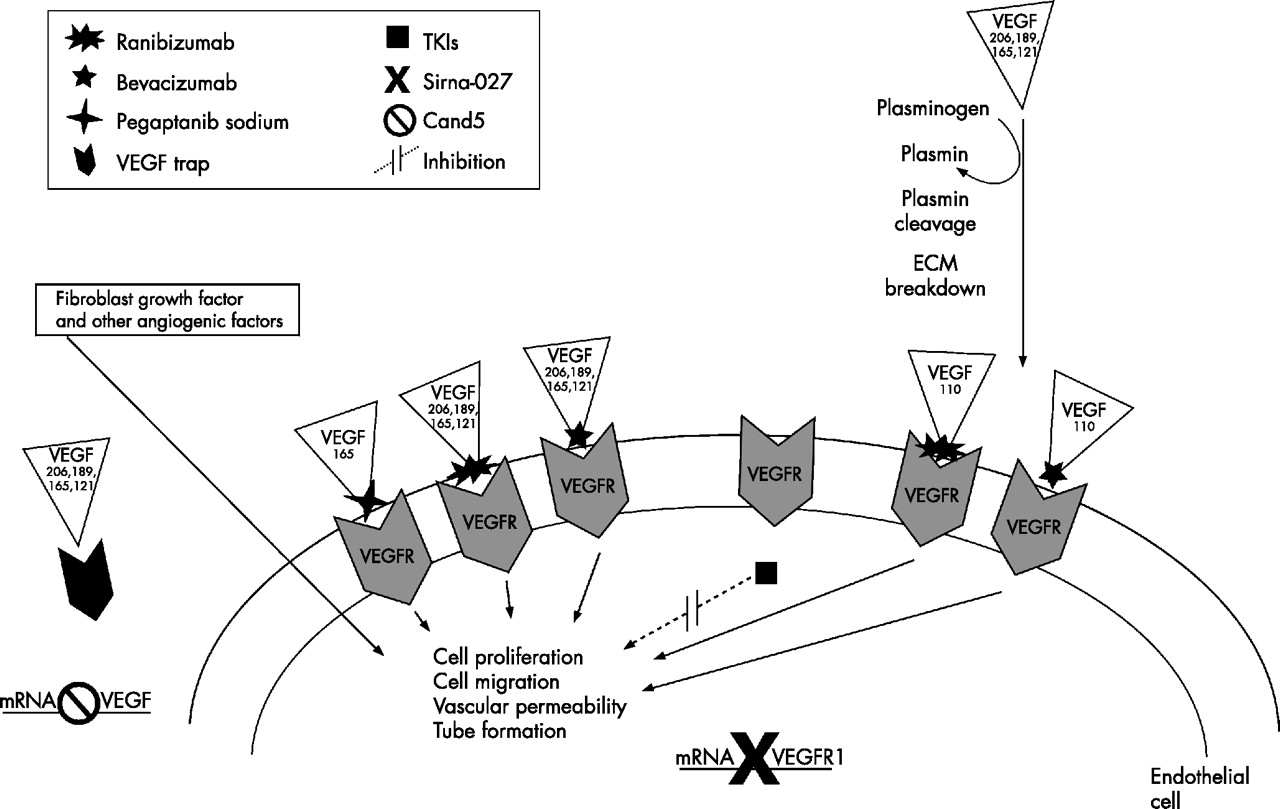
Currently available and emerging anti-VEGF treatments inhibit VEGF-A isoforms in different ways (summarised in fig 2). Pegaptanib sodium is a pegylated ribonucleic acid oligonucleotide ligand (or aptamer). It has a molecular mass of 50 kDa and is produced synthetically to be specific for the human VEGF164 isoform, binding at its heparin-binding domain.16 Theoretically, it may bind the longer VEGF-A isoforms that also contain the heparin-binding domain.

{kind=link}
{kind=link}
Key angiogenesis activators and inhibitors. Note that only the four major vascular endothelial growth factor (VEGF)-A isoforms are depicted in this schematic representation. ECM, extracellular matrix; TKIs, tyrosine kinase inhibitors; VEGFR, vascular endothelial growth factor receptor.
Ranibizumab is a humanised antigen-binding fragment of a murine full-length monoclonal antibody (mAB) directed against human VEGF-A. It is produced in Escherichia coli using recombinant DNA technology and has a molecular mass of 48 kDa. Ranibizumab has been shown to inhibit VEGF165, VEGF121 and VEGF110 (Lowe et al, unpublished data), and theoretically should inhibit all VEGF-A isoforms and their active degradation products, although it is technically difficult to evaluate the activity and binding of longer VEGF-A isoforms because they are sequestered in the ECM. Ranibizumab fully penetrates the retinal layers to the outer retina and inner choroid in rabbits (Gaudreault et al, unpublished data).
Bevacizumab is a 149-kDa full-length humanised mAB against all VEGF-A isoforms and their active degradation products, approved as intravenous infusion in combination with chemotherapy for metastatic colorectal cancer. Off-label use of ITV bevacizumab for neovascular AMD in small retrospective studies has shown benefits for vision and reduced macular thickening.53,54 The long-term benefits and safety of ITV bevacizumab remain to be investigated.
Several other anti-VEGF treatments are in clinical development for ophthalmology. VEGF Trap (Regeneron Pharmaceuticals, New York, USA) is a fusion protein containing the binding sites of VEGFR1 and VEGFR2, which inhibits all VEGF-A isoforms and PlGF.55 Receptor tyrosine kinase inhibitors include vatalanib (PTK787; Novartis AG) and AG-013958 (Pfizer, New York, USA); these target the VEGF receptor tyrosine kinases and inhibit all VEGF-A isoforms,56 although AG-013958 also inhibits platelet-derived growth factor receptors. RNA interference-based treatments are also being explored in neovascular AMD. Cand5 (Acuity Pharmaceuticals, Philadelphia, USA) prevents the production of all VEGF-A isoforms by degrading VEGF-A mRNA. Sirna-027 (Sirna Therapeutics, Boulder, USA) targets production of VEGFR1 mRNA.57
Therapeutic outcomes
In the phase III VGEF Inhibition Study in Ocular Neovascularization (VISION) clinical trials, pegaptanib has been shown to be beneficial for all subtypes of CNV secondary to AMD. At 12 months, considerably more pegaptanib-treated patients lost <15 letters than patients who received sham injections (70% v 55%) and more patients gained ⩾3 lines of visual acuity (6% v 2%)42; benefits were maintained at 24 months. Rates of visual gain might be enhanced for treatment of small and early CNV lesions, as suggested by a subanalysis of a small group of patients from the VISION trial.58 For patients with diabetic macular oedema, a phase II controlled clinical trial showed a considerably higher rate of visual gain of ⩾10 letters of visual acuity for pegaptanib-treated patients compared with sham-injected patients (34% v 10%).59
In the phase III MARINA trial of patients with occult or minimally classic subfoveal CNV secondary to AMD, at month 12 considerably more ranibizumab-treated patients lost <15 letters than sham-injected patients (95% v 62%). Of the ranibizumab-treated patients, 25% and 34% (0.3 and 0.5 mg, respectively) gained ⩾15 letters compared with 5% of sham-injected patients, a significant difference (Rosenfeld et al41). In the phase III ANCHOR trial of patients with predominantly classic subfoveal CNV secondary to AMD, 94% and 96% of ranibizumab-treated patients (0.3 and 0.5 mg, respectively) lost <15 letters of visual acuity compared with 64% of photodynamic therapy-treated patients in month 12 (Brown et al41a). Of the ranibizumab-treated patients, 36% and 40% (0.3 and 0.5 mg, respectively) gained ⩾15 letters compared with 6% of photodynamic therapy-treated patients, a significant difference (Brown et al41b).
Serious ocular adverse events were rare for both pegaptanib42 and ranibizumab (Rosenfeld et al41a; Brown et al41b), and were predominantly related to the ITV injection procedure itself, including presumed endophthalmitis, traumatic lens injury and retinal detachment.42 Serious uveitis occurred at a rate of 0–1% in pegaptanib-treated patients in the VISION trial, and at a rate of 0.7–0.8% in ranibizumab-treated patients in the MARINA and ANCHOR trials (Rosenfeld et al41a; Brown et al41b). There is a theoretical possibility of untoward effects with long-term VEGF-A inhibition, particularly on normal choroidal and retinal vasculature. However, such toxicities have not been observed to this point in human clinical trials of ocular anti-VEGF treatments.
SYSTEMIC RISKS WITH VEGF-A INHIBITION
Intravenous infusion
For patients with cancer, adjuvant bevacizumab treatment in phase III clinical trials improves median survival in patients with metastatic colorectal cancer (CRC)60 and improves response rate (but not progression-free survival) in patients with metastatic breast cancer.61 In these patients with late-stage cancer receiving serial intravenous infusion of bevacizumab at doses of 5–15 mg/kg, there were increased risks of systemic adverse events. Compared with chemotherapy alone, bevacizumab in combination with chemotherapy is associated with an increased incidence of hypertension, bleeding and proteinuria in patients with CRC62 and an increased rate of thromboembolic events, gastrointestinal perforations, myocardial infarction and death in patients with breast cancer and CRC.61,62
A different serious adverse-event profile has been seen in the limited number of patients without cancer studied with intravenous bevacizumab used as monotherapy. In a small study of off-label intravenous infusion of bevacizumab in patients with AMD, seven of nine patients experienced hypertension controllable with drugs, but no serious ocular or systemic adverse events were reported.63 Similarly, no ocular or systemic adverse events were reported in two patients with pathological myopia treated off-label with intravenous infusion of bevacizumab.64
Intravitreal administration
Given the risks of intravenous adjuvant bevacizumab treatment seen in patients with cancer, systemic risks also theoretically exist with ITV administration of anti-VEGF treatments. Although the dosages in ocular therapy are several orders of magnitude lower, ITV injection does lead to detectable levels in the serum. In monkeys the vitreous half life (t1/2) of ranibizumab is 3 days, and after ITV injection of 0.5 mg, the maximum serum level is 150 ng/ml, with a serum half life of 3.5 days.65 In human studies, the mean (SD) serum concentration of ranibizumab 1 h after ITV administration (0.3 mg) was 1.01 (1.35) ng/ml, and after 28 days serum concentrations were <0.300 ng/ml in 96% of the patients.66 Such levels of serum ranibizumab are below the approximately 10 ng/ml threshold estimated to affect VEGF-A-related activity in humans.66,67
The serum half life of pegaptanib after ITV administration (3 mg) in humans is 10 days, with a mean maximum serum concentration of pegaptanib of 80 ng/ml.68 In contrast, the serum half life of the full-length anti-VEGF antibody bevacizumab is 17–21 days in humans; mABs tend to have relatively longer half lives than antigen-binding fragments.69
The various VEGF-A isoforms are distributed widely in tissue; theoretically, broad inhibition of VEGF-A isoforms confers higher non-specificity and systemic risk, as does prolonged serum half life. To date, more than 2000 patients have received serial ITV injections in the controlled clinical trials of pegaptanib and ranibizumab without showing increased risk of systemic adverse events for either agent. The systemic safety of ITV bevacizumab is not established, but no known serious adverse events have been reported in uncontrolled studies to date.53,54,70,71 As the clinical use of ITV VEGF inhibition expands, ongoing monitoring for systemic adverse events may be warranted.
CONCLUSIONS
Anti-VEGF agents are the first biological compounds to be developed specifically for retinal therapeutics. The retinal community is currently dealing with aspects of these novel drugs in the management of patients with neovascular AMD. Unresolved clinical issues include: optimal injection frequency; duration of the treatment regimen; possible combination treatments; and potential applications for retinal diseases beyond AMD. In light of the role of VEGF-A in diverse organ systems in normal adults, long-term VEGF-A suppression raises theoretical ocular and systemic safety issues; however, no evidence of such increased risks has been observed in controlled clinical trials of pegaptanib or ranibizumab for patients with neovascular AMD. VEGF-A is perhaps the most widely researched molecule in ophthalmology; as such, VEGF science may be valuable to doctors in refining the clinical application of this new class of drugs.
Acknowledgments
This project was supported by “That Man May See Foundation”. Writing and research assistance was supported by Genentech.
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 41a.↵
- 41b.↵
- 42.↵
- 43.↵
- 44.↵
- 45.
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
Footnotes
-
Competing interests: None declared.