
Abstract
Elevated blood pressure is closely related to increased circulatory fluid volume and peripheral vascular resistance. Patients with diabetes mellitus experience increased peripheral artery resistance caused by vascular remodeling and increased body fluid volume associated with insulin resistance-induced hyperinsulinemia and hyperglycemia. Both of these mechanisms elevate systemic blood pressure. Thus, fully understanding the pathophysiology of hypertension in diabetes mellitus requires knowing the natural history of type 2 diabetes. Patients exhibit hyperinsulinemia with insulin resistance due to impaired glucose tolerance and early-stage diabetes. Hypertension occurs because of increased body fluid volume. After reaching mid-stage diabetes the vascular remodeling has progressed and peripheral vascular resistance also contributes to hypertension. Moreover, vascular remodeling strongly influences diabetic complications. Specifically, afferent arteriolar remodeling during diabetic nephropathy leads to increased glomerular pressure. Thus, treatment with a renin-angiotensin system inhibitor that promotes renal damage regression is critical to lowering the systemic blood pressure and dilating efferent arterioles to reduce glomerular pressure.
Similar content being viewed by others
Introduction
Hypertension is a well-known complication of diabetes mellitus and diabetes is a well-known complication of hypertension [1]. In Japan, there are approximately 43 million patients with hypertension and 20.5 million patients with diabetes mellitus, including subjects with impaired glucose tolerance. Patients with diabetes can have elevated blood pressure and 40–60% of diabetes cases exhibit high blood pressure. Either diabetes or hypertension can present various complications without symptoms. The interaction between hypertension and diabetes can lead to the development of stroke and myocardial infarction, which represent major causes of death in Japan due to the progression of arteriosclerosis. Diabetic nephropathy also progresses rapidly as a result of blood pressure elevation. These findings suggest that considering the physiology and pathology of diabetes and hypertension are critical.
Pathology and physiology of blood pressure elevation
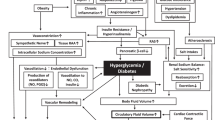
Figure 1 presents the current understanding of hypertension pathophysiology. Briefly, blood pressure is controlled by the relationship between circulatory fluid volume and peripheral vascular resistance [2]. The circulatory fluid volume is regulated by blood fluid volume and cardiac contractile force. The blood fluid volume is influenced by the sodium store/excretion balance (reflecting salt sensitivity and sodium intake). The cardiac contractile force is controlled by both sympathetic nervous activity and cardiac function. Peripheral vascular resistance is regulated by vascular tone, which is influenced by both vascular remodeling and vasoactive agents including the renin-angiotensin system.

Hypertension pathophysiology
High salt intake with salt sensitivity can lead to elevated blood pressure. This relationship can be understood by examining Guyton’s pressure–natriuresis relationship curve [3], which shows that sodium excretion is promoted at a blood pressure level higher than the threshold in the kidney. Under strongly salt-sensitive conditions a higher blood pressure is required to excrete sodium via urine. Moreover, hypertension itself shifts the threshold to a higher level. Salt sensitivity is influenced by salt excreting dysfunctions that are common in aging patients, chronic kidney disease cases, and by salt accumulation observed in obesity, diabetes, and menopause. A diet with high salt intake combined with salt sensitivity reportedly leads to nocturnal hypertension [4], and this condition overlaps with morning hypertension in patients with chronic kidney disease [5]. Notably, patients with diabetes and nocturnal high blood pressure show a greater than 16-fold higher risk of cardiovascular events [6].
Resistance vessel narrowing via vascular remodeling promotes increased vascular resistance and blood pressure elevation. The endothelial dysfunction and vascular remodeling precede an increase in blood pressure. The development of arteriosclerosis involves oxidation stress, inflammation, vasoactive materials, cytokines, chemokines, and growth factors that can affect each other. If the functional vasoconstriction remains at a constant level, then there is blood pressure elevation observed in resistance vessels that undergo vascular remodeling. A cycle can develop and advanced vascular endothelial dysfunction promotes progression of vascular remodeling. The presence of coexisting risk factors (e.g., hypertension, diabetes, and dyslipidemia) promotes more advanced vascular endothelial dysfunction and ultimately produces damage to various organs [7].
The relationship between insulin and blood pressure elevation
Insulin lowers plasma glucose levels and is a key hormone in diabetes mellitus development. Insulin has several functions, including the following: facilitates glucose uptake by organs, promotes glycogen storage in liver and muscle tissue, controls the breakdown of stored glycogen, promotes adipose tissue development, and controls fat resolution [8]. Additionally, the insulin receptor is part of the receptor tyrosine kinase family that includes platelet-derived growth factor receptor and heparin-binding epidermal growth factor-like receptor. Thus, insulin also stimulates vascular smooth muscle cell migration and proliferation [9].
Insulin translocates Na+/K+-ATPase from the cytoplasm to the cell membrane to open the Na+/H+ channel that passively transports hydrogen ions out of the cell and sodium ions into the cell. This process also increases the cellular calcium ion concentration and decreases pH [10]. The Na+/H+ exchange transporter opens following the reduction of intracellular sodium, and this is caused by an insulin-induced increase of Na+/K+-ATPase. The action of Na+/K+-ATPase leads to sodium ion transport into blood vessels through renal tubule cells [11]. When an insulin deficiency leads to diabetic ketoacidosis the Na+/K+-ATPase activity decreases, which increases the transport of sodium and hydrogen into the cell and potassium out of the cell. These changes increase the cellular sodium ion density and lead to symptoms of high serum potassium [12]. However, when insulin resistance induces hyperinsulinemia the sodium reabsorption from renal tubules is increased and leads to high blood pressure [13].
The circulatory fluid volume can also increase relative to hyperglycemia-induced hyperosmolarity [14]. The prolonged high plasma glucose levels in diabetes cases alter the extracellular osmotic pressure on the side with higher glucose concentration, which increases relative to the intracellular osmotic pressure. Water exits the tissue (into the vasculature) to reduce the difference between the intracellular and extracellular osmotic pressure and this flow increases the extracellular amount of body fluid and blood (i.e., circulatory blood volume). Therefore, hyperglycemia also leads to systemic blood pressure elevation by increasing the circulatory fluid volume.
Hyperinsulinemia stimulates sympathetic nervous activity and increases renin excretion [15]. The increase in renin activates the sympathetic nervous system and increases cardiac output and peripheral vascular resistance. These changes ultimately elevate blood pressure by increasing both the circulatory fluid volume and peripheral vascular resistance. Moreover, insulin stimulates obesity through fat accumulation and this leads to obesity-induced hypertension in association with type 2 diabetes mellitus [16].
Hypertension and natural history of diabetes mellitus
It is critical to understand the natural history of diabetes and the pathophysiology that gives rise to this disease [17]. Type 2 diabetes is a complex metabolic disorder characterized by elevated blood glucose and a markedly increased risk of cardiovascular disease due to a cluster of metabolic and vascular abnormalities that include hyperglycemia, dyslipidemia, and hypertension. Tissue insulin resistance is an important part of the pathophysiology of type 2 diabetes and is often tied to obesity and/or adiposity. Insulin resistance and defective insulin secretion (relative insulin deficiency) are considered the key factors that promote hyperglycemia, which is a characteristic of type 2 diabetes [18].
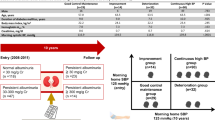
In the natural history of type 2 diabetes, insulin resistance prompts β-cells to secrete additional insulin, which initially increases the insulin levels (Fig. 2). However, the increased insulin secretion actually represents a relative insulin deficiency because β-cell function begins to deteriorate during the early course of type 2 diabetes. Impaired β-cell function is both necessary and sufficient for the development of pre-diabetes and diabetes onset. At the onset of diabetes, the level of insulin secretion no longer keeps pace with insulin resistance and β-cell function is already significantly reduced. According to the UKPDS, diabetes is often not clinically diagnosed until 10 years after its onset and at this time β-cell function may be reduced by over 50% [19]. It is now recognized that β-cell impairment arises and worsens several years before diabetes onset and the initial manifestation of postprandial hyperglycemia [20].

Natural history of type 2 diabetes mellitus. Source: Ref. [18]
It is interesting that the pathophysiology of patients with hypertension and type 2 diabetes involves the natural history of type 2 diabetes and the above-described mechanisms of blood pressure elevation. The period from the initial impairment of glucose tolerance to diabetes onset is characterized by both hyperglycemia and hyperinsulinemia with insulin resistance. During this period the main pathologies causing blood pressure elevation are the promotion of sodium reabsorption due to higher insulin levels and excessive body fluid due to hyperglycemia-induced osmolar adjustment.
In the early stage of diabetes, both hyperglycemia and hyperinsulinemia can promote vascular remodeling. The gradual progression of vascular remodeling leads to increased peripheral artery resistance and eventually contributes to hypertension. The stages involving advanced vascular remodeling lead to pancreatic β-cell loss and attenuation of insulin secretion with a corresponding reduction of sodium reabsorption by insulin. At this time, the main mechanism causing blood pressure elevation is increased peripheral artery resistance because the increase of body fluid is stable with only osmolar adjustment due to hyperglycemia.
Vascular remodeling proceeds through all stages of diabetes and treatment with renin-angiotensin system inhibitors should be administered early in the disease course to prevent vascular remodeling [2, 21]. From a pathophysiological viewpoint, both sodium restriction and thiazide diuretics are useful treatments during early-stage diabetes involving hyperinsulinemia. Additionally, calcium antagonist treatment is advisable in the middle-to-late stage of diabetes when insulin production is lower. It is preferable to manage diabetes without insulin elevation to avoid increased fluid retention and vascular remodeling [22].
Hypertension and diabetic complications
The diabetic complications of macroangiopathy [23] and microangiopathy [24] are largely caused by vascular remodeling and are influenced by various components of metabolic syndrome including hypertension, diabetes, dyslipidemia, and obesity. The coexistence of hypertension with diabetes is associated with a 6-fold increase in the risk of cardiovascular events compared to in healthy subjects [25]. White coat and masked hypertension also increase cardiovascular risk in patients with diabetes and chronic kidney diseases [26].
Hypertension influences the development of diabetic nephropathy [27], which pathologically consists of the following: thickening of the glomerulus and renal tubule basement membrane, mesangial and interstitial proliferation, endothelium denaturation, and interstitial changes involving exudative lesions in small vessels [28, 29]. Diabetic nephropathy also involves smooth muscle cell proliferation with medial and intimal thickening of afferent arterioles [30]. There are also proinflammatory reactions such as T lymphocyte permeation and small vessel hyperplasia [31]. These changes are caused by hyperglycemia-induced glomerular circulation and renin-angiotensin system stimulation [32], glycation [33], and oxidative stress [34]. There are also intracellular metabolic abnormalities such as polyol and protein kinase activation [35], microinflammation, cytokine production [36], and extracellular matrix production or resolution [37]. Blood pressure elevation influences these mechanisms and diabetic nephropathy is worsened by glomerular hypertension due to afferent arteriole medial and intimal thickening with systemic hypertension [38].
Hyperglycemia and hyperinsulinemia induce medial and intimal thickening in afferent arterioles, which causes dysfunction of contraction and dilation and leads to failure of optimal control of glomerular pressure. In healthy individuals without diabetes or obesity the afferent arterioles contract to maintain glomerular pressure at 50 mmHg against systemic hypertension. However, diabetes-induced afferent arteriole dysfunction can allow glomerular pressure to increase to over 70 mmHg. The high glomerular pressure promotes hyperfiltration and lowers the serum creatinine concentration and leads to albuminuria. Continuous high glomerular pressure results in more dominant proteinuria and ultimately leads to decreased glomerular filtration due to mesangial denaturation or glomerular necrosis, which can progress to end-stage renal damage [39].
The reduction of glomerular high pressure is one possible means of avoiding end-stage renal failure. However, maintenance of optimal glomerular pressure requires strict systemic blood pressure control and dilation of efferent arterioles via renin-angiotensin inhibition [40]. Proper glomerular pressure control can reduce the hyperfiltration and glomerular filtration rate and increase serum creatinine. These changes should be monitored to achieve glomerular pressure adjustment without worsening renal function. The guidelines of the Japanese Society of Hypertension suggest that patients with diabetic nephropathy and hypertension should be treated with angiotensin receptor antagonists or an angiotensin-converting enzyme inhibitor to maintain a blood pressure of below 130/80 mmHg [2].
References
Lee SW, Kim HC, Lee JM, Yun YM, Lee JY, Suh I. Association between changes in systolic blood pressure and incident diabetes in a community-based cohort study in Korea. Hypertens Res. 2017;40:710–6.
Shimamoto K, Ando K, Fujita T, Hasebe N, Higaki J, Horiuchi M, Imai Y, Imaizumi T, Ishimitsu T, Ito M, Ito S, Itoh H, Iwao H, Kai H, Kario K, Kashihara N, Kawano Y, Kim-Mitsuyama S, Kimura G, Kohara K, Komuro I, Kumagai H, Matsuura H, Miura K, Morishita R, Naruse M, Node K, Ohya Y, Rakugi H, Saito I, Saitoh S, Shimada K, Shimosawa T, Suzuki H, Tamura K, Tanahashi N, Tsuchihashi T, Uchiyama M, Ueda S, Umemura S, Japanese Society of Hypertension Committee for Guidelines for the Management of Hypertension. The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH 2014). Hypertens Res. 2014;37:253–390.
Guyton AC, Manning RD Jr, Hall JE, Norman RA Jr, Young DB, Pan YJ. The pathogenic role of the kidney. J Cardiovasc Pharmacol. 1984;6:S151–61.
Uzu T, Ishikawa K, Fujii T, Nakamura S, Inenaga T, Kimura G. Sodium restriction shifts circadian rhythm of blood pressure from nondipper to dipper in essential hypertension. Circulation. 1997;96:1859–62.
Mizuno M, Fukuda M, Miura T, Wakamatsu T, Naito T, Sato R, Togawa H, Sasakawa Y, Tomonari T, Ono M, Kato Y, Ichikawa T, Shirasawa Y, Ito A, Yoshida A, Kimura G. Morning hypertension in chronic kidney disease is sustained type, but not surge type. Blood Press Monit. 2012;17:20–3.
Eguchi K, Pickering TG, Hoshide S, Ishikawa J, Ishikawa S, Schwartz JE, Shimada K, Kario K. Ambulatory blood pressure is a better marker than clinic blood pressure in predicting cardiovascular events in patients with/without type 2 diabetes. Am J Hypertens. 2008;21:443–50.
Joffe D, Yanagisawa RT. Metabolic syndrome and type 2 diabetes: can we stop the weight gain with diabetes? Med Clin North Am. 2007;91:1107–23.
Saltiel AR. Insulin signaling in the control of glucose and lipid homeostasis. Handb Exp Pharmacol. 2016;233:51–71.
Arnqvist HJ, Bornfeldt KE, Chen Y, Lindström T. The insulin-like growth factor system in vascular smooth muscle: interaction with insulin and growth factors. Metabolism. 1995;44:58–66.
Cleland SJ, Petrie JR, Ueda S, Elliott HL, Connell JM. Insulin as a vascular hormone: implications for the pathophysiology of cardiovascular disease. Clin Exp Pharmacol Physiol. 1998;25:175–84.
Young A. Renal effects. Adv Pharmacol. 2005;52:251–68.
Sweeney G, Klip A. Regulation of the Na+/K+-ATPase by insulin: why and how? Mol Cell Biochem. 1998;182:121–33.
Martínez FJ, Sancho-Rof JM. Epidemiology of high blood pressure and obesity. Drugs. 1993;46(Suppl 2):160–4.
Kawasoe S, Maruguchi Y, Kajiya S, Uenomachi H, Miyata M, Kawasoe M, Kubozono T, Ohishi M. Mechanism of the blood pressure-lowering effect of sodium-glucose cotransporter 2 inhibitors in obese patients with type 2 diabetes. BMC Pharmacol Toxicol. 2017;18:23.
Seravalle G, Grassi G. Sympathetic nervous system, hypertension, obesity and metabolic syndrome. High Blood Press Cardiovasc Prev. 2016;23:175–9.
Kishida K, Funahashi T, Shimomura I. Clinical importance of assessment of type 2 diabetes mellitus with visceral obesity: a Japanese perspective. Curr Diabetes Rev. 2012;8:84–91.
Kendall DM, Cuddihy RM, Bergenstal RM. Clinical application of incretin-based therapy: therapeutic potential, patient selection and clinical use. Am J Med. 2009;122:S37–50.
Toft-Nielsen MB, Damholt MB, Madsbad S, Hilsted LM, Hughes TE, Michelsen BK, Holst JJ. Determinants of the impaired secretion of glucagon-like peptide-1 in type 2 diabetic patients. J Clin Endocrinol Metab. 2001;86:3717–23.
U.K. Prospective Diabetes Study Group. U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. Diabetes. 1995;44:1249–58.
Ferrannini E, Gastaldelli A, Miyazaki Y, Matsuda M, Mari A, DeFronzo RA. β-cell function in subjects spanning the range from normal glucose tolerance to overt diabetes: a new analysis. J Clin Endocrinol Metab. 2005;90:493–500.
Strawn WB, Ferrario CM. Mechanisms linking angiotensin II and atherogenesis. Curr Opin Lipidol. 2002;13:505–12.
Reed JW. Impact of sodium-glucose cotransporter 2 inhibitors on blood pressure. Vasc Health Risk Manag. 2016;12:393–405.
Gärtner V, Eigentler TK. Pathogenesis of diabetic macro- and microangiopathy. Clin Nephrol. 2008;70:1–9.
Rizzoni D, Rosei EA. Small artery remodeling in diabetes mellitus. Nutr Metab Cardiovasc Dis. 2009;19:587–92.
Ninomiya T, Kubo M, Doi Y, Yonemoto K, Tanizaki Y, Rahman M, Arima H, Tsuryuya K, Iida M, Kiyohara Y. Impact of metabolic syndrome on the development of cardiovascular disease in a general Japanese population: the Hisayama Study. Stroke. 2007;38:2063–9.
Kushiro T, Kario K, Saito I, Teramukai S, Sato Y, Okuda Y, Shimada K. Increased cardiovascular risk of treated white coat and masked hypertension in patients with diabetes and chronic kidney disease: the HONEST Study. Hypertens Res. 2017;40:87–95.
Ahmad J. Management of diabetic nephropathy: recent progress and future perspective. Diabetes Metab Syndr. 2015;9:343–58.
Qi C, Mao X, Zhang Z, Wu H. Classification and differential diagnosis of diabetic nephropathy. J Diabetes Res. 2017;2017:8637138.
Najafian B, Alpers CE, Fogo AB. Pathology of human diabetic nephropathy. Contrib Nephrol. 2011;170:36–47.
Thomas MC. Pathogenesis and progression of proteinuria. Contrib Nephrol. 2011;170:48–56.
Zheng Z, Zheng F. Immune cells and inflammation in diabetic nephropathy. J Diabetes Res. 2016;2016:1841690.
Hollenberg NK. Review: the renin-angiotensin-aldosterone system, blockade and diabetic nephropathy. J Renin Angiotensin Aldosterone Syst. 2001;2:S185–7.
Fukami K, Taguchi K, Yamagishi S, Okuda S. Receptor for advanced glycation endproducts and progressive kidney disease. Curr Opin Nephrol Hypertens. 2015;24:54–60.
Kashihara N, Haruna Y, Kondeti VK, Kanwar YS. Oxidative stress in diabetic nephropathy. Curr Med Chem. 2010;17:4256–69.
Kitada M, Zhang Z, Mima A, King GL. Molecular mechanisms of diabetic vascular complications. J Diabetes Investig. 2010;1:77–89.
Shikata K, Makino H. Microinflammation in the pathogenesis of diabetic nephropathy. J Diabetes Investig. 2013;4:142–9.
Bhattacharjee N, Barma S, Konwar N, Dewanjee S, Manna P. Mechanistic insight of diabetic nephropathy and its pharmacotherapeutic targets: an update. Eur J Pharmacol. 2016;791:8–24.
Tonneijck L, Muskiet MH, Smits MM, van Bommel EJ, Heerspink HJ, van Raalte DH, Joles JA. Glomerular hyperfiltration in diabetes: mechanisms, clinical significance, and treatment. J Am Soc Nephrol. 2017;28:1023–39.
Anderson S, Meyer TW, Rennke HG, Brenner BM. Control of glomerular hypertension limits glomerular injury in rats with reduced renal mass. J Clin Invest. 1985;76:612–9.
Parving HH, Andersen S, Jacobsen P, Christensen PK, Rossing K, Hovind P, Rossing P, Tarnow L. Angiotensin receptor blockers in diabetic nephropathy: renal and cardiovascular end points. Semin Nephrol. 2004;24:147–57.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MO has received financial support for research from Daiichi Sankyo Company, Boehringer Ingelheim, Takeda Pharmaceutical Company, MSD KK, Kyowa Hakko Kirin, Kowa Pharmaceutical Company, Teijin Home Healthcare, Mitsubishi Tanabe Pharma Corporation, Pfizer Japan, Bristol-Myers Squibb, Sumitomo Dainippon Pharma, Mochida Pharmaceutical, Actelion Pharmaceuticals Japan, Otsuka Pharmaceutical, Teijin Pharma, and Genzyme Japan KK.
Rights and permissions
About this article

Cite this article
Ohishi, M. Hypertension with diabetes mellitus: physiology and pathology. Hypertens Res 41, 389–393 (2018). https://doi.org/10.1038/s41440-018-0034-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41440-018-0034-4
This article is cited by
-
Association between triglyceride glucose index and all-cause mortality in patients with cerebrovascular disease: a retrospective study
Diabetology & Metabolic Syndrome (2024)
-
The impact of GLP-1 receptor agonist liraglutide on blood pressure profile, hydration, natriuresis in diabetic patients with severely impaired kidney function
Scientific Reports (2024)
-
Significance of plasma relaxin-2 levels in patients with primary hypertension and type 2 diabetes mellitus
Wiener Medizinische Wochenschrift (2024)
-
Uric acid is associated with type 2 diabetes: data mining approaches
Diabetology International (2024)
-
Assessment the awareness toward hypertension and diabetes mellitus: Syrian cross sectional study
BMC Public Health (2023)
