
Abstract
Background
Patient registries are organized systems that use observational methods to collect uniform data on specified outcomes in a population defined by a particular disease, condition, or exposure. Data collected in registries often coincide with data that could support clinical trials. Integrating clinical trials within registries to create registry-embedded clinical trials offers opportunities to reduce duplicative data collection, identify and recruit patients more efficiently, decrease time to database lock, accelerate time to regulatory decision-making, and reduce clinical trial costs. This article describes a project of the Clinical Trials Transformation Initiative (CTTI) intended to help clinical trials researchers determine when a registry could potentially serve as the platform for the conduct of a clinical trial.
Methods
Through a review of registry-embedded clinical trials and commentaries, semi-structured interviews with experts, and a multi-stakeholder expert meeting, the project team addressed how to identify and describe essential registry characteristics, practices, and processes required to for conducting embedded clinical trials intended for regulatory submissions in the United States.
Results
Recommendations, suggested practices, and decision trees that facilitate the assessment of whether a registry is suitable for embedding clinical trials were developed, as well as considerations for the design of new registries. Essential registry characteristics include relevancy, robustness, reliability, and assurance of patient protections.
Conclusions
The project identifies a clear role for registries in creating a sustainable and reusable infrastructure to conduct clinical trials. Adoption of these recommendations will facilitate the ability to perform high-quality and efficient prospective registry-based clinical trials.
Similar content being viewed by others


Background
For decades, well-conducted clinical trials have helped assure the safety and effectiveness of drugs, biologics, and devices entering the marketplace. However, the evolution of clinical trial science and regulatory oversight has resulted in a substantial increase in the cost and complexity of clinical trials and concerns about lack of generalizability to typical clinical practice. Many believe that alternative approaches to the design and execution of clinical trials should be considered [1,2,3,4].
A registry is an organized system that uses observational methods to collect uniform data on specified outcomes in a population defined by a particular disease, condition, or exposure. At their core, registries are data collection tools created for one or more predetermined scientific, clinical, or policy purposes. Entry in a registry is generally defined either by diagnosis of a disease (disease registry) or prescription of a drug, device, or other treatments (exposure registry) [5, 6]. Registries are often used to identify and understand trends in incidence and prevalence of diseases or to observe how patients are treated in the real world, including the identification of practice changes over time [7,8,9].
Use of registry data for clinical trial planning is common, such as hypothesis generation, refining eligibility criteria, estimating sample size, and predicting performance of a clinical trial site. [7, 10,11,12,13,14,15,16,17,18,19] However, the ability to infer causal relations between treatments and outcomes with observational registry analyses is limited due to risks of selection bias and confounding [20,21,22,23]. Registry-embedded clinical trials offer the ability to combine the strengths of conventional clinical trials and large registries [23, 24].
The CTTI Registry Trials Project was initiated to provide recommendations for the assessment and design of registries that could be suitable for conducting registry-embedded clinical trials. The primary goal of the project was to identify and describe the essential characteristics, practices, and processes required to embed and conduct registry-based clinical trials to support regulatory decision-making. The scope of the project included the conduct of trials of drugs, devices, biologics, and procedures within the context of appropriate registries. Use of other datasets (e.g., electronic health records and claims databases) to facilitate clinical trials was outside the scope of this project. This article describes the resulting resources intended to help investigators to either (1) determine if an existing registry is suitable for conducting an embedded clinical trial or (2) design a new registry with the intention of embedding clinical trials within the registry.
Materials and Methods
CTTI is a public–private partnership founded in 2007 by the Duke Clinical Research Institute and the US FDA. Its mission is to develop and drive adoption of practices that will increase the quality and efficiency of clinical trials. CTTI projects utilize multi-stakeholder project teams that follow an evidence-based methodology to identify impediments to research, gather evidence to identify gaps and barriers, explore results by analyzing and interpreting findings, and finalize solutions by developing recommendations and tools [25]. The CTTI Registry Trials Project Team consisted of stakeholders representing academia, pharmaceutical and device industries, government agencies, patient representatives, and patient advocacy organizations (https://www.ctti-clinicaltrials.org/projects/registry-trials). Evidence gathered in the execution of this project included a reviewing published registry-based clinical trials and commentaries, a series of interviews with subject matter experts, and the output of a multi-stakeholder expert meeting. The protocol for the interviews was reviewed by the Duke University Health System IRB (Protocol ID: Pro00064484) and declared exempt from IRB review (45CFR46.101(b)).
Interviews with Subject Matter Experts
Interviews were conducted to gather expert opinions regarding, but not limited to, barriers to and potential solutions for using clinical registries for prospective clinical trials. A semi-structured interview guide was created and refined in collaboration with Research Triangle Institute (RTI) (Research Triangle Park, NC). Thirty-seven experts with knowledge and experience on the use of registry data in clinical trials were identified and invited to participate. Of the 29 respondents, 25 were prioritized by the project team and RTI to achieve the widest variety of perspectives. All 25 experts gave verbal consent to be interviewed, have their interviews digitally recorded, and be listed as interviewees within the report. To summarize responses and identify recurrent themes for each question, the responses were coded in an iterative manner. The full report is provided in the Appendix.
Expert Meeting
After completion and assessment of the interviews, an expert meeting was held on March 30, 2016, in Silver Spring, MD, which included 42 stakeholders from industry, academia, patient advocacy organizations, and government agencies. A summary of published registry-based clinical trials, findings from the expert interviews, and case examples of previously conducted randomized registry trials were presented. The attendees were asked to provide feedback on potential benefits and existing barriers to the use of registries in clinical trials and to reach a consensus on best practices to encourage the adoption of the use of clinical trials within registries. An executive summary, list of meeting participants, the agenda, and presentations can be accessed at https://www.ctti-clinicaltrials.org/briefing-room/meetings/brave-new-world-registry-based-clinical-trials.
Following the expert meeting, the project team used the data from the evidence-gathering activities, information provided in the draft guidance on the use of real-world evidence to support regulatory decision-making for medical devices [26], and input from the expert meeting to create project recommendations and tools. Finally, the CTTI Executive Committee reviewed and approved the resources.
Results
Published registry-embedded clinical trials and commentaries and the results of the expert interviews reinforced the advantages of the combined methodology in controlling for confounding factors while also to enrolling a large and generalizable patient sample [20, 23, 24]. Registry-embedded clinical trials offer opportunities to identify highly qualified sites, reduce duplicative data collection and site workload, identify and recruit patients more efficiently, reduce patients lost to follow-up, decrease time to database lock, accelerate time to regulatory decision-making, and reduce clinical trial costs [20, 24, 27,28,29]. Registry type and characteristics are important for determining appropriateness for conducting clinical trials. Designing or modifying registries to accommodate clinical trials involves a number of key dimensions, including, but not limited to, informed consent, governance, interoperability, connectivity, flexibility, sustainability, data quality, regulatory, privacy, and business considerations. The need for guidance on how to assess existing registries appropriateness for, or design a new high-quality registry capable of, conducting clinical trials emerged as a recurring theme. Therefore, the project team created the following recommendations, divided into those applying to existing registries and those intended for new registries.
Recommendations
To determine if an existing registry is appropriate for embedding clinical trials:
-
(1)
Assess whether the historical evidence generated by an existing registry has demonstrated the relevancy, robustness, and reliability necessary to provide a platform for collecting data in an embedded clinical trial to support regulatory decision-making, with assurance of patient protections (see Fig. 1 and Table 1).
Figure 1. 
Decision tree 1, existing registry—historical assessment.
Table 1. Existing registry—historical assessment. -
(2)
Assess if an existing registry contains the elements needed to support a randomized clinical trial. Satisfaction of all the following requirements suggests that the existing registry, together with any appropriate configurable elements, may provide high-quality evidence suitable for regulatory decision-making (see Fig. 2 and Table 2):
-
a.
Are the data previously generated by the baseline registry historically regarded as robust and reliable (i.e., high-quality data)?
-
b.
Can the baseline registry and its dataset provide the core data needed to answer the question at hand (i.e., relevant or fit-for-purpose)?
-
c.
Can any processes or data not provided by the baseline registry be added or the registry reconfigured to accommodate these needs (e.g., programming to allow identification of suitable trial participants or documentation of informed consent, modular add-on datasets or linkages to other databases, and appropriate data accessibility with maintenance of patient and data privacy)?
Figure 2. 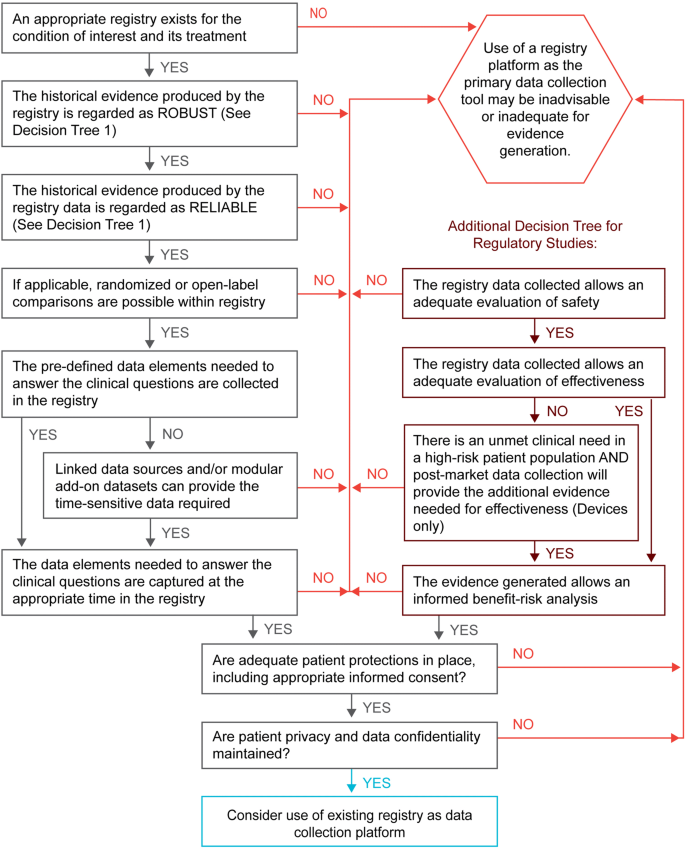
Decision tree 2, existing registry—suitability assessment.
Table 2. Existing registry—suitability assessment. -
a.
-
(3)
To design a new registry suitable for embedding clinical trials, we recommend following software industry guidelines, as well as guidance documents provided by regulatory agencies, to assure that the registry complies with both industry and regulatory standards (see Table 3).
Table 3. Designing a new registry with the capability of embedding a clinical trial.


Discussion
Embedding clinical trials into registries can contribute to the transformation of the clinical research enterprise to facilitate lower-cost, high-quality evidence generation. We report the results of a project about registry-based clinical trials including a literature search, in-depth interviews, a convened meeting of experts, and collaborative discussions of a multi-stakeholder project team. We have compiled these findings and developed recommendations for determining the suitability of an existing registry, or designing a new registry, for the purposes of conducting registry-based clinical trials. We have determined that registries can be well suited to facilitate clinical trials if they are relevant, robust, reliable, and respectful of patient privacy and data confidentiality.
The recommendations and tools presented here are intended to facilitate the path forward to the effective and efficient use of registries as reusable platforms for evidence generation and to encourage their use, as an alternative to creating de novo case report forms and databases for each new clinical trial. The recommendations are meant to identify key best practices and principles. Of note, these recommendations are not intended to be either a mandatory or exhaustive checklist. We recognize that some registries will not be suitable for conducting embedded clinical trials, but should continue to be used as successful tools to facilitate clinical trials through activities such as identifying and recruiting patients, conducting trial feasibility assessments, and reducing the amount of baseline and/or follow-up data that need to be collected for a clinical trial.
Collecting most or all data needed for a clinical trial from a registry is possible in some cases, i.e., registry is the primary data collection tool. However, a review of randomized registry-based randomized trials found that half used more than one registry [23]. Linkage of registries for registry-embedded trials is commonly conducted in countries where unique patient identifiers are used in national health registries [24, 30,31,32,33]. However, in situations where additional data sources are not available or the available registry data lack sufficient granularity (e.g., depth, definitional uncertainty, or insufficient temporal assessment) for clinical or regulatory purposes, targeted modular add-on data can be designed for use within the registry or as a separate case report form, to collect the additional essential data needed to reach a clinical or regulatory decision. This combined registry and case report form strategy was used in both the Consortium of Rheumatology Researchers of North America (CORRONA) Registry Treat to Target (T2T) and Study of Access site For Enhancement of Percutaneous Coronary Intervention for Women (SAFE-PCI for Women) trials. [29, 34].
Regulatory agencies have long accepted registry and other post-marketing data sources to collect safety information, such as surveillance for adverse events and conduct of post-approval studies [35,36,37,38,39]. Furthermore, data from registries can be used to accelerate expansion of patient access to an intervention, and generate evidence to identify potential new label indications [14, 40,41,42]. Recently, the United States (US) Food and Drug Administration (FDA) has signaled its commitment to develop policies regarding use of real-world data (RWD) sources—including registries—to support efficacy claims [43,44,45,46,47]. Guidance for devices, and draft guidance for drugs and biologics, regarding use of RWD and real-world evidence for regulatory purposes have been released [26, 48]. These recommendations were designed to assist in assessing the acceptability of data and evidence from registries that would meet the standard of acceptability for regulatory submissions in the United States. Researchers and those engaged with registry-embedded trial development are encouraged to interact with regulators early in study planning to ensure that planned data collection will be acceptable for regulatory submission.
The following limitations apply to the project and recommendations. The scope of the project was registries only. However, registries can be populated from other data sources such as electronic health records or claims data [49, 50]. The recommendations provided, particularly for assessment of historical evidence (Table 1), are applicable to assessment of other sources of real-world data whether they are being used to populate a registry, as an additional linkage to a registry, or alone [51]. These recommendations do not explore the costs of using registries for clinical trials, as they focus on acceptability. Although per-patient costs have shown to be lower in registry-embedded trials, there are costs associated with access to the data, building add-ons for a trial, and technical work required to establish connections to other datasets [13, 20, 28, 52]. These costs are an additional consideration when assessing registry-embedded trial feasibility. Greater efficiency and cost savings are possible when a registry or group of registries may be reused for multiple trials. Finally, these recommendations apply to the suitability of registry-embedded trial data for regulatory decision-making in the United States. Recently, the European Network for Health Technology Assessment (HTA) released a tool and vision paper to assess the quality of registry data and acceptability for HTA and regulatory purposes [53].
Conclusions
The CTTI Registry Trials Project has taken an evidence-based and highly collaborative approach to accomplishing its goal of providing recommendations for registry assessment and design regarding their suitability for conducting embedded clinical trials. We anticipate that these recommendations will encourage clinical trial stakeholders to collaborate effectively to increase utilization of prospective patient registries to facilitate high quality, efficient, registry-embedded clinical trials.
References
DiMasi JA, Hansen RW, Grabowski HG. The price of innovation: new estimates of drug development costs. J Health Econ. 2003;22(2):151–85.
Weisfeld N, English R, Claiborne AB. In: Envisioning a Transformed Clinical Trials Enterprise in the United States: Establishing An Agenda for 2020: Workshop Summary. Washington (DC); 2012.
Morgan S, Grootendorst P, Lexchin J, Cunningham C, Greyson D. The cost of drug development: a systematic review. Health Policy. 2011;100(1):4–17.
Corrigan-Curay J, Sacks L, Woodcock J. Real-world evidence and real-world data for evaluating drug safety and effectiveness. JAMA. 2018;320(9):867–8.
Gliklich RE, Dreyer N, Leavy MB. Registries for evaluating patient outcomes: a user’s guide. In: Gliklich RE, Dreyer NA, Leavy MB, editors. Registries for Evaluating Patient Outcomes: A User’s Guide. Rockville, MD: Agency for Healthcare Research and Quality; 2014.
European Medicines Agency. Scientific Guidance on Post-authorisation Efficacy Studies, 2015. London: European Medicines Agency; 2015.
Ashley L, Jones H, Thomas J, et al. Integrating cancer survivors’ experiences into UK cancer registries: design and development of the ePOCS system (electronic Patient-reported Outcomes from Cancer Survivors). Br J Cancer. 2011;105(Suppl 1):S74–81.
Ahmed B, Piper WD, Malenka D, et al. Significantly improved vascular complications among women undergoing percutaneous coronary intervention: a report from the Northern New England Percutaneous Coronary Intervention Registry. Circ Cardiovasc Interv. 2009;2(5):423–9.
Dauerman HL, Rao SV, Resnic FS, Applegate RJ. Bleeding avoidance strategies. Consensus and controversy. J Am Coll Cardiol. 2011;58(1):1–10.
Peelen L, Peek N, de Jonge E, Scheffer GJ, de Keizer NF. The use of a registry database in clinical trial design: assessing the influence of entry criteria on statistical power and number of eligible patients. Int J Med Inform. 2007;76(2–3):176–83.
Craig JC, Webster AC, Mitchell A, Irwig L. Expanding the evidence base in transplantation: more and better randomized trials, and extending the value of observational data. Transplantation. 2008;86(1):32–5.
Curtis JR, Wright NC, Xie F, et al. Use of health plan combined with registry data to predict clinical trial recruitment. Clin Trials. 2014;11(1):96–101.
Rao SV, Hess CN, Barham B, et al. A registry-based randomized trial comparing radial and femoral approaches in women undergoing percutaneous coronary intervention: the SAFE-PCI for Women (Study of Access Site for Enhancement of PCI for Women) trial. JACC Cardiovasc Interv. 2014;7(8):857–67.
Carroll JD, Edwards FH, Marinac-Dabic D, et al. The STS-ACC transcatheter valve therapy national registry: a new partnership and infrastructure for the introduction and surveillance of medical devices and therapies. J Am Coll Cardiol. 2013;62(11):1026–34.
Dowling NM, Olson N, Mish T, Kaprakattu P, Gleason C. A model for the design and implementation of a participant recruitment registry for clinical studies of older adults. Clin Trials. 2012;9(2):204–14.
Hansen VJ, Greene ME, Bragdon MA, et al. Registries collecting level-I through IV Data: institutional and multicenter use: AAOS exhibit selection. J Bone Joint Surg. 2014;96(18):e160.
de Blieck EA, Augustine EF, Marshall FJ, et al. Methodology of clinical research in rare diseases: development of a research program in juvenile neuronal ceroid lipofuscinosis (JNCL) via creation of a patient registry and collaboration with patient advocates. Contemp Clin Trials. 2013;35(2):48–54.
Sabbagh MN, Tariot PN. Commentary on “a roadmap for the prevention of dementia II. Leon Thal Symposium 2008.” A national registry to identify a cohort for Alzheimer’s disease prevention studies. Alzheimers Dement. 2009;5(2):128–9.
Achiron A, Givon U, Magalashvili D, et al. Effect of Alfacalcidol on multiple sclerosis-related fatigue: a randomized, double-blind placebo-controlled study. Mult Scler. 2015;21(6):767–75.
James S, Rao SV, Granger CB. Registry-based randomized clinical trials–a new clinical trial paradigm. Nat Rev Cardiol. 2015;12(5):312–6.
Ieva F, Gale CP, Sharples LD. Contemporary roles of registries in clinical cardiology: when do we need randomized trials? Expert Rev Cardiovasc Ther. 2014;12(12):1383–6.
Sharma A, Ezekowitz JA. Similarities and differences in patient characteristics between heart failure registries versus clinical trials. Curr Heart Fail Rep. 2013;10(4):373–9.
Mathes T, Buehn S, Prengel P, Pieper D. Registry-based randomized controlled trials merged the strength of randomized controlled trails and observational studies and give rise to more pragmatic trials. J Clin Epidemiol. 2018;93:120–7.
Li G, Sajobi TT, Menon BK, et al. Registry-based randomized controlled trials-what are the advantages, challenges, and areas for future research? J Clin Epidemiol. 2016;80:16–24.
Corneli A, Hallinan Z, Hamre G, et al. The clinical trials transformation initiative: methodology supporting the mission. Clin Trials. 2018;15(1_suppl):13–8.
U.S. Food and Drug Administration. Use of Real-World Evidence to Support Regulatory Decision-Making for Medical Devices. https://www.fda.gov/media/99447/download. Published 2017. Accessed 25 Nov 2019.
Thuesen L, Jensen LO, Tilsted HH, et al. Event detection using population-based health care databases in randomized clinical trials: a novel research tool in interventional cardiology. Clin Epidemiol. 2013;5:357–61.
Lauer MS, D’Agostino RB Sr. The randomized registry trial–the next disruptive technology in clinical research? N Engl J Med. 2013;369(17):1579–81.
Hess CN, Rao SV, Kong DF, et al. Embedding a randomized clinical trial into an ongoing registry infrastructure: unique opportunities for efficiency in design of the Study of Access site For Enhancement of Percutaneous Coronary Intervention for Women (SAFE-PCI for Women). Am Heart J. 2013;166(3):421–8.
Frobert O, Lagerqvist B, Olivecrona GK, et al. Thrombus aspiration during ST-segment elevation myocardial infarction. N Engl J Med. 2013;369(17):1587–97.
Gotberg M, Christiansen EH, Gudmundsdottir IJ, et al. Instantaneous wave-free ratio versus fractional flow reserve to guide PCI. N Engl J Med. 2017;376(19):1813–23.
Hedberg S, Olbers T, Peltonen M, et al. BEST: bypass equipoise sleeve trial; rationale and design of a randomized, registry-based, multicenter trial comparing Roux-en-Y gastric bypass with sleeve gastrectomy. Contemp Clin Trials. 2019;84:105809.
Kaczorowski J, Chambers LW, Dolovich L, et al. Improving cardiovascular health at population level: 39 community cluster randomised trial of Cardiovascular Health Awareness Program (CHAP). BMJ. 2011;342:d442.
Harrold LR, Reed GW, Harrington JT, et al. The rheumatoid arthritis treat-to-target trial: a cluster randomized trial within the Corrona rheumatology network. BMC Musculoskelet Disord. 2014;15:389.
Mack MJ, Brennan JM, Brindis R, et al. Outcomes following transcatheter aortic valve replacement in the United States. JAMA. 2013;310(19):2069–77.
Mehta AB, Chandra P, Dalal J, et al. One-year clinical outcomes of BioMatrix™-Biolimus A9™ eluting stent: the e-BioMatrix multicenter post marketing surveillance registry in India. Indian Heart J. 2013;65(5):593–9.
Oza SR, Hunter TD, Biviano AB, et al. Acute safety of an open-irrigated ablation catheter with 56-hole porous tip for radiofrequency ablation of paroxysmal atrial fibrillation: analysis from 2 observational registry studies. J Cardiovasc Electrophysiol. 2014;25(8):852–8.
Green A, Ramey DR, Emneus M, et al. Incidence of cancer and mortality in patients from the Simvastatin and Ezetimibe in Aortic Stenosis (SEAS) trial. Am J Cardiol. 2014;114(10):1518–22.
Jankovic J, Adler CH, Charles PD, et al. Rationale and design of a prospective study: Cervical Dystonia Patient Registry for Observation of OnaBotulinumtoxinA Efficacy (CD PROBE). BMC Neurol. 2011;11:140.
Genentech. FDA Approves Expanded Indication for ACTEMRA® in Rheumatoid Arthritis. www.gene.com/media/press-releases/14187/2012-10-12/fda-approves-expanded-indication-for-act. Accessed 7 July 2016.
Matsukage T, Yoshimachi F, Masutani M, et al. A new 0.010-inch guidewire and compatible balloon catheter system: the IKATEN registry. Catheter Cardiovasc Interv. 2009;73(5):605–10.
Russell ME, Friedman MI, Mascioli SR, Stolz LE. Off-label use: an industry perspective on expanding use beyond approved indications. J Interv Cardiol. 2006;19(5):432–8.
Sherman RE. Real-world evidence—what is it and what can it tell us? N Engl J Med. 2016;375(23):2293–7.
Sherman RE, Davies KM, Robb MA, Hunter NL, Califf RM. Accelerating development of scientific evidence for medical products within the existing US regulatory framework. Nat Rev Drug Discov. 2017;16(5):297–8.
Shuren J, Califf RM. Need for a national evaluation system for health technology. JAMA. 2016;316(11):1153–4.
Faris O, Shuren J. An FDA viewpoint on unique considerations for medical-device clinical trials. N Engl J Med. 2017;376(14):1350–7.
U.S. Food and Drug Administration. Framework for FDA’s Real-World Evidence Program. https://www.fda.gov/media/120060/download. Published 2018. Accessed 22 May 2019.
U.S. Food and Drug Administration. Submitting Documents Using Real-World Data and Real-World Evidence to FDA for Drugs and Biologics. Guidance for Industry. https://www.fda.gov/media/124795/download. Published May 2019. Accessed 22 May 2020.
Platt R, Takvorian SU, Septimus E, et al. Cluster randomized trials in comparative effectiveness research: randomizing hospitals to test methods for prevention of healthcare-associated infections. Med Care. 2010;48(6):S52–7.
United States Department of Veterans Affairs. Corporate Data Warehouse (CDW). https://www.hsrd.research.va.gov/for_researchers/vinci/cdw.cfm. Accessed 27 Nov 2019.
Gliklich RE, Leavy MB. Assessing real-world data quality: the application of patient registry quality criteria to real-world data and real-world evidence. Ther Innov Regul Sci. 2019. https://doi.org/10.1007/s43441-019-00058-6.
Huang SS, Septimus E, Kleinman K, et al. Targeted versus universal decolonization to prevent ICU infection. New Eng J Med. 2013;368(24):2255–65.
National Institute for Health and Care Excellence NU, French National Authority for Health (Haute Autorité de Santé), (France). H. Vision Paper on the Sustainable Availability of the Proposed Registry Evaluation and Quality Standards Tool (REQueST). https://eunethta.eu/wp-content/uploads/2019/10/EUnetHTAJA3_Vision_paper-v.0.44-for-ZIN.pdf. Published 2019. Accessed 26 Nov 2019.
Acknowledgements
The authors would like to thank Teresa McNally, PhD, and Karyn Hede, MS, for editorial assistance in the development of this article. Both are employees of Whitsell Innovations, Inc., Chapel Hill, North Carolina, and received no compensation for their work on this article other than their usual salary.
Funding
This work was supported by the US Food and Drug Administration (Grant Number R18FD005292) awarded to Duke University as the host of CTTI. Views expressed in this manuscript do not necessarily reflect the official policies of the US Department of Health and Human Services, nor does any mention of trade names, commercial practices, or organization imply endorsement by the US government. Additional support is provided by CTTI member organizations, whose annual fees support CTTI infrastructure expenses and projects. https://www.ctti-clinicaltrials.org/membership.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article. Arlene Swern is an employee and stockholder for Celgene Corp. E. Dawn Flick is a current employee of Celgene Corp., and a past employee of Genentech, Inc. Nicolle Gatto was an employee and shareholder of Pfizer at the time of manuscript submission, she is a current employee of Aetion, Inc. John Laschinger was at the U.S. FDA at the time of the project, he is a current employee of W. L. Gore & Associates. Emily Zeitler was at the Duke Clinical Research institute at the time of the project, she is a current employee of the Geisel School of Medicine at Dartmouth College.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Mikita, J.S., Mitchel, J., Gatto, N.M. et al. Determining the Suitability of Registries for Embedding Clinical Trials in the United States: A Project of the Clinical Trials Transformation Initiative. Ther Innov Regul Sci 55, 6–18 (2021). https://doi.org/10.1007/s43441-020-00185-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43441-020-00185-5