
Abstract
SARS-CoV-2 infects cells via its spike protein binding to its surface receptor on target cells and results in acute symptoms involving especially the lungs known as COVID-19. However, increasing evidence indicates that many patients develop a chronic condition characterized by fatigue and neuropsychiatric symptoms, termed long-COVID. Most of the vaccines produced so far for COVID-19 direct mammalian cells via either mRNA or an adenovirus vector to express the spike protein, or administer recombinant spike protein, which is recognized by the immune system leading to the production of neutralizing antibodies. Recent publications provide new findings that may help decipher the pathogenesis of long-COVID. One paper reported perivascular inflammation in brains of deceased patients with COVID-19, while others showed that the spike protein could damage the endothelium in an animal model, that it could disrupt an in vitro model of the blood-brain barrier (BBB), and that it can cross the BBB resulting in perivascular inflammation. Moreover, the spike protein appears to share antigenic epitopes with human molecular chaperons resulting in autoimmunity and can activate toll-like receptors (TLRs), leading to release of inflammatory cytokines. Moreover, some antibodies produced against the spike protein may not be neutralizing, but may change its conformation rendering it more likely to bind to its receptor. As a result, one wonders whether the spike protein entering the brain or being expressed by brain cells could activate microglia, alone or together with inflammatory cytokines, since protective antibodies could not cross the BBB, leading to neuro-inflammation and contributing to long-COVID. Hence, there is urgent need to better understand the neurotoxic effects of the spike protein and to consider possible interventions to mitigate spike protein-related detrimental effects to the brain, possibly via use of small natural molecules, especially the flavonoids luteolin and quercetin.
Similar content being viewed by others
Introduction
The SARS-CoV-2 infects cells by first binding to its surface receptor, angiotensin converting enzyme 2 (ACE2), via its corona spike protein [1]. The S protein is trimeric and catalyzed fusion between the viral and host cell membrane; this “prefusion” trimer has three receptor-binding domains (RBD), while the post fusion structure expresses N-linked glycans that may serve to protect against immune responses [2]. Infection then leads to a complex immune response that involves the release of a “storm” [3, 4] of pro-inflammatory cytokines [3,4,5,6,7,8,9,10,11], especially IL-6 [12,13,14,15] and IL-1β [16, 17] leading to the development of COVID-19 [3, 18]. Most infected patients develop antibodies against the spike protein, but immune protection against SARS-CoV-2 may involve more than neutralizing antibodies [19].
A prospective study of more than 3,000 healthy members of the US Marines Corps concluded that those seropositive could still be infected but had only 20% the risk of subsequent re-infection as compared to those who were seronegative [20]. It is not known if individuals who get re-infected do not mount sufficient neutralizing antibodies or lack some other aspect of antiviral immunity. New data from immunized individuals indicate that the rate of re-infection varies depending on the type of vaccine used [21]. There is emerging evidence of reduced neutralization of some SARS-CoV-2 variants [22].
Hypothesis/Theory
Some of the damaging effects of SARS-CoV-2, especially in the brain, may be due to direct action of the Spike protein, acting alone or in conjunction with other mediators such as inflammatory cytokines, on target cells.
Long-COVID Syndrome
It is now recognized that many patients infected with SARS-CoV-2 develop a post-acute syndrome a few months after the initial infection known as “post-acute COVID” [23] or “long-COVID” [23,24,25,26]. Long-COVID occurs in 30–50% of COVID patients [23, 27,28,29,30] and is characterized by multisystem symptoms, primarily persistent fatigue and cognitive impairment [31] that varied considerably among patients [32] and were more common with increasing age and female sex [29]. These persistent symptoms should not be confused or misinterpreted as persistent infection that has been reported in immunocompromised hosts [33]. Nevertheless, patients with long-COVID have not recovered even by 7 months post infection and continue to suffer mostly from systemic and neurological symptoms [34].
Long-COVID is particularly associated with neurological [35,36,37,38,39,40,41,42,43], neurodegenerative [38, 44, 45], psychiatric [46,47,48,49,50,51,52], and cognitive [47,48,49,50,51,52,53,54,55,56,57] problems, especially brain fog [23, 25, 26, 46, 58,59,60,61,62]. In fact, over 90% of patients who were initially hospitalized for COVID-19 and had neurological symptoms had significantly worse outcome 6 months later [63]. Even though some of the mental fatigue experienced by long-haulers may be due to the perceived stress [64], the extent of this disability is unlike any other medical condition known.
In spite of early impressions that long-COVID may develop only in those patients who were hospitalized and intubated, increasing evidence indicates that long-COVID can develop regardless of the severity of the original symptoms [61, 65] and has been considered the “next health disaster” in the USA [66]. So far the duration of long-COVID symptoms is not known, but recent data indicate that it may depend on antigen persistence [67] and a sustained specific immune responses to SARS-CoV-2 [68].
The neurologic effects of COVID-19 may be due to SARS-CoV-2 entering the brain, but the pathways of such neurotropism are still unclear [69, 70]. One possibility is that the virus crosses or damages the blood-brain barrier (BBB) [71], accompanied by basement membrane disruption, in K18-hACE2 transgenic mice infected with SARS-CoV-2 [72]. Similar findings were reported independently, and it was also shown that the virus was detected in human cortical neurons [73]. In another study, a fragment specific to SARS-CoV-2 was amplified from cultures of a brain specimen from a deceased patient with COVID-19, and associated pathology showed neuronal necrosis and glial cell hyperplasia [74]. Alternatively, the virus could enter from the nose by crossing the neural-mucosal interface of the olfactory nerve [75] and enter the brain via the olfactory nerve tract [76]. Viral entry into the brain via gustatory-olfactory trigeminal pathway eventually compromising the BBB was recently reported in deer mice infected with SARS-CoV-2 [77]. It is interesting that single-cell RNA sequencing showed that ACE2 was not expressed by olfactory sensory or bulb neurons but instead was expressed by olfactory epithelium and pericytes [78].
The effect of SARS-CoV-2 to the brain is also not well understood. One paper showed the presence of megakaryocytes in cortical capillaries that could lead to brain ischemia [79] and subsequent cerebrovascular events [80,81,82]. In the autopsy report of an infant who died with COVID-19, there was evidence of cortical atrophy and severe neuronal loss, and findings were restricted to capillaries of the choroid plexus [83]. A recent paper did not document any molecular traces of SARS-CoV-2 in the brains of deceased patients with COVID-19, but detected choroid plexus perturbations associated with pathologic morphological changes in the microglia [84]. In addition to the evidence discussed above of neuronal damage due to SARS-CoV-2, a paper reported that the virus can enter a 3D human brain organoid and preferentially targets neurons resulting in their death [85]. Such pathology may be explained by the expression of the ACE2 receptor by human glial cells and neurons [86], exacerbated through the activation of the complement and kinin systems [87].
Increasing evidence indicates the involvement of neuro-inflammation [71, 88, 89] that may damage brain blood vessels [90, 91], as well as brain cells [88, 92, 93], possibly via activation of microglia [94, 95] and mast cells [96]. In fact, long-COVID could be considered a state of “brain autoimmunity” [22].
In summary, the effect of SARS-CoV-2 to the brain could be direct via invasion or indirect effect via damaging endothelial cells and pericytes or via activation of neuroimmune responses as has been invoked for neurologic complications following HIV [97].
Direct Effects of Spike Protein
An alternative explanation of the CNS effect of SARS-CoV-2 may be due to direct effects of the spike protein. The spike protein is made up of the S1 subunit containing a receptor-binding domain (RBD) that attaches to ACE2 and the S2 subunit containing a transmembrane anchor that mediates fusion of viral and host cell membranes [1]. Most infected patients develop antibodies that neutralize the spike protein to various extents. A recent paper reported that blood of patients infected with SARS-CoV-2 contained, in addition to antibodies against the RBD that were protective, also antibodies against the N-terminal domain (NTD) of the spike protein that induced the open conformation of the RBD enhancing its binding ability and infectivity in vitro using cultured cells [98]. A more recent study of molecular modeling using an antibody from a symptomatic COVID-19 patient concluded that there was higher NTD binding with the delta variant resulting in antibody-dependent enhancement (ADE) [99]. Such interactions, where antibodies can neutralize one serotype but are less potent at neutralizing another, are known to increase the chances of ADE to the new serotype [100]. Even though ADE remains controversial, a recent paper reported that virus-mimicking anti-idiotype antibodies present after infection or after vaccination may potentially explain long-COVD symptoms [101]. These findings may potentially explain why those vaccinated against the original Wuhan SARS-CoV-2 strain and then exposed to the Delta variant may still get infected. Al alternative or additional explanation may be the fact that immunity to vaccines has been reported to decrease over time [102, 103]
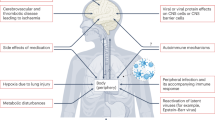
It is not yet known if the spike protein is released extracellularly after the SARS-CoV-2 infects its target cells. Given the absence of infection of the brain discussed above, the neuropathologic findings may be due to the SARS-C0V-2 spike protein. Indirect evidence of its presence within the CNS may be the detection of anti-SARS-CoV-2 antibodies in the CSF of two children who died with COVID-10 and had subacute neuropsychiatric symptoms [104], even though such antibodies may had crossed a disrupted BBB. Free spike protein could have a number of direct pathologic actions on different cell types (Fig. 1A). These include direct stimulation of peripheral nerves [105] and stimulation of release of pro-inflammatory and vasoactive mediators [106, 107], especially platelet-activating factor (PAF) [108, 109].

A Diagrammatic representation of how SARS-CoV-2 spike protein can stimulate different cell types and collectively contribute to the pathogenesis of long-COVID. B Diagrammatic representation of how SARS-CoV-2 can cross the blood-brain barrier (BBB) through endothelial cell gaps or how free spike protein can damage the integrity of the BBB and enter the brain.
A number of papers have reported direct pathologic effect of the spike protein by itself (without being part of the coronavirus). One paper reported that the spike protein could damage the endothelium in an animal model [110], while another paper showed that recombinant S1 RBD can damage mouse brain endothelial cells in vitro by inducing degradation of endothelial junction proteins, thus affecting endothelial barrier function [111]. A recent paper reported rapid internalization of S1 RBD and of the spike RBD active trimer by cultured human brain microvascular endothelial cells, followed by increased permeability of transferrin and dextran, as well as mitochondrial damage [112]. Another recent paper using a 3D-BBB microfluidic model showed that S1 upregulated ACE2 expression and triggered RhoA activation, a key molecule regulating endothelial cytoskeleton [113]. Yet, another paper reported that spike-transfected human epithelial cells showed increased senescence-associated secretory and inflammatory proteins [114].
Two other papers reported that the spike protein could disrupt the barrier function in an in vitro model of the blood-brain barrier (BBB) [115] and that the S1 protein can actually cross the BBB and enter the brain in mice [116] (Fig. 1A). Using transgenic mice expressing the human sigma protein, it was shown that intranasal infection with SARS-CoV-2 rapidly induced ischemic-like reactivity in brain pericytes and the S protein reached the brain of the mice [117].
In addition to direct damage, the spike protein appears to share antigenic epitopes with human molecular chaperons resulting in autoimmunity against endothelial cells [118]. Moreover, a recent paper showed that spike epitopes could form heterodimeric complexes with selected human glial proteins [119]. Interestingly, it was shown that three recombinant sigma protein peptides exhibited molecular interactions with acetylcholinesterase and antioxidant enzymes both in silico and in tad poles in vivo [120].
Interestingly, symptoms experienced by long-COVID patients, especially cognitive dysfunction [121,122,123], are similar [106] to those present in patients with mast cell activation syndrome (MCAS) [124, 125], in whom mast cells can be stimulated by environmental and stress triggers [126], including viruses [127] such as SARS-CoV-2 [107, 128]. Mast cells are located perivascularly in close proximity to neurons, especially in the hypothalamus [129, 130], where functional mast cell-neuron interactions have been documented [130, 131]. Mast cells also interact with microglia [132] leading to their activation [133] and neuro-inflammation [134].
SARS-CoV-2 binding may not be limited to the ACE2 receptor. New evidence indicates that the spike protein also binds to heparan sulfate (HS) molecules expressed on the surface of target cells, with mutant variants having higher binding affinity to HS [135]. This binding may be due to the fact that the SARS-CoV-2 spike protein contains four more positively charged and five fewer negatively charged residues than SARS-CoV, thus increasing the binding affinity of SARS-CoV-2 for HS [136]. Apparently, binding to HS allows the virus to reach the ACE2 receptor, and the RBD portion of the spike protein can engage both HS and ACE2 without dissociation of one or the other ligand [137]. The S1 subunit can also bind to the surface glycoprotein neuropilin-1 (NRP-1), thus increasing infectivity, but also dysregulating angiogenesis, immune responses, and neuronal development [138, 139]. Different coronavirus variants have evolved more efficient electrostatic interactions to allow them to bind to the ACE2 receptor [140]. SARS-CoV-2 also appears to become “pre-activated” by the proprotein convertase furin, thus bypassing the target cell proteases for entry [141].
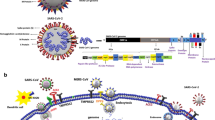
SARS-CoV-2 can do additional damage by activating toll-like receptors (TLRs), especially TLR2, leading to secretion of pro-inflammatory cytokines independent of viral entry [142, 143]. Such immune-mediating molecules could contribute to neurologic symptoms [144] as a result of or in addition to the action of the spike protein. Moreover, activating TLR4 increases expression of ACE2 [145] further enhancing viral infectivity in an autocrine loop. Activation of TLRs may not only involve activation of inflammasomes [146], but also activation of the mammalian target of rapamycin (mTOR) complex [147, 148], which is invoked in the pathogenesis of many neuropsychiatric diseases [149] (Fig. 2). Increased levels of a number of pro-inflammatory cytokines have been detected in the CSF of COVID-19 patients [150], especially IL-6 [150, 151]. In fact, use of an anti-IL-6 antibody or IL-6 receptor antibody reduced neuronal injury in a mouse model, accompanied by inflammation and neuronal death unrelated to hypoxia [152]. Integration of serum levels of IL-6 and heparin-binding protein were shown to have significant predictive value for severity of COVID-19 [153].

Diagrammatic representation of how SARS-CoV-2 spike protein can stimulate endothelial cells, mast cells, microglial cells, and neurons first by binding to the ACE2 receptor costimulated by binding to heparin sulfate, and then acted upon by a serine protease before entering the nucleus. SARS-CoV-2 can also stimulate Toll-like receptors (TLRs) and lead to the synthesis and release of pro-inflammatory cytokines via activation of the inflammasomes and or mTOR. The diagram also shows the targets of the inhibitory actions of luteolin, methoxyluteolin, and quercetin (green line), which may be used to prevent or treat the development of long-COVID.
A recent paper reported cloning and expressing 26 of the 29 proteins encoded by the SARS-CoV-2 genome and showed most proteins, especially non-structural protein (NSP) 2, 5, and 7, induced significant changes in endothelial permeability [154]. These findings imply that SARS-CoV2-associated proteins other than the spike protein may contribute to pathologic effects on their own, sequentially or synergistically with the structural sigma protein.
Lastly, a recent paper analyzed human fetal expression of six different S protein “interactors” and showed weak expression of ACE2 and TMPRSS2, but high expression of furin with peak expression 12–26 weeks post conception; moreover, using publically available single-cell RNA sequencing datasets, it was shown that these interactors showed higher co-expression with neurons [155]. This finding indicates that the spike protein can adversely affect the developing brain and potentially lead to neurologic complications in neonates of infected mothers [156], including autism spectrum disorder [157].
Discussion
A major unaddressed issue, especially with respect to the pathogenesis of long-COVID, is whether the spike protein that enters the brain or is expressed in neurons and glial cells can activate microglia directly or via stimulation of mast cells leading to neuro-inflammation [158]. This pathogenetic process would go on unhindered in the absence of any neutralizing antibodies since they do not cross the BBB, thus contributing to the pathogenesis of long-COVID. Moreover, such spike protein-induced neurocognitive damage could be worse in vulnerable populations like those with minimal cognitive impairment [159] or others suffering from traumatic brain injury [160].
There are presently no biologics that can block SARS-CoV-2 binding to its receptor(s). Certain biologics aimed at blocking IL-6 [161] or IL-1 [162] have been reported to improve clinical status of patients with COVID-19 However, a meta-analysis of clinical trials using IL-6 antagonists as an add-on to usual care did not reduce the risk of stroke [163], and a recent double-blind, randomized placebo-controlled study showed no benefit of an Il-6 blocker [164]. This conclusion may not be surprising as these humanized antibodies are not likely to cross the BBB unless it has already been disrupted. It is interesting that a main source of IL-6 is the mast cells [165,166,167], which have been reported to secrete it after stimulation with IL-1 [168] and acute stress [169]. Moreover, IL-6 can be constitutively released from human mast cells bearing the D816V-KIT mutation [170] and act on mast cell in an autocrine fashion to stimulate their proliferation [171].
This manuscript does not attempt to review and discuss all possible drugs, biologics, or natural molecules that could interfere with SARS-CoC-2 binding and its effects on target cells. Rather, it focuses on certain natural molecules for which there is sufficient basic and clinical evidence supporting their possible usefulness, both in prevention and treatment, especially in long-COVID. A number of recent reviews have discussed the potential use of natural molecules in that capacity [172,173,174]. Some simulation and in vitro studies have reported the potential benefit of small molecules found in Ginkgo biloba, such as the flavonoid quercetin discussed later. For instance, extracts from Ginkgo biloba leaves were identified as potential inhibitors of SARS-CoV-23CL(pro) using large-scale screening [175]. Another Ginkgo biloba extract was reported to block TNFα-induced reactive oxygen species from human aortic endothelial cells [176]. The Ginkgo biloba extract EGb 761 was beneficial in generalized anxiety disorder [177] and dementia [178], actions that may be useful for the neuropsychiatric aspects of long-COVID. Ginkgolic acid (GA) was shown to inhibit the fusion and synthesis of viral proteins [179]. Other studies have shown that green tea catechins could be useful in COVID-19 [180, 181], especially against entry of SARS-CoV-2 [182]. The broccoli extract sulforaphane inhibited expression of IL-6 and IL-8 induced by the SARS-CoV-2 spike protein in bronchial epithelial cells [183].
Certain natural flavonoids [184] have been proposed as prophylaxis or treatment against COVID-19 [185,186,187,188,189]. Such flavonoids are found in green plants and seeds and possess potent anti-oxidant, anti-inflammatory, and cytoprotective properties [184]. However, their consumption as part of the diet does not provide sufficient systemic levels. However, there are a number of sources of pharmaceutical-grade purity (>98%) using different biomasses such as Citrus limon, Cynara cardunculus (artichoke), oregano, and Saphora japonicum.
In particular, a number of studies using in silico approaches identified the flavonol quercetin and the structurally related flavone luteolin as a potential strong blockers of RBD [190,191,192]. Luteolin and some of its methylated analogues have a number of beneficial actions with respect to long-COVID: broad antiviral properties [193,194,195], inhibition of coronavirus entry [127, 196, 197], and inhibition of the serine protease required for spike protein processing [198, 199]. Furthermore, luteolin inhibits activation of both microglia [200,201,202,203] and mast cells [204, 205] via inhibition of signaling pathways involving the inflammasome [206, 207] and mTOR (Fig. 2) in both mast cells [205] and microglia [203]. The novel luteolin structural analogue tetramethoxyluteolin (methoxyluteolin) is an even more potent inhibitor than luteolin [203,204,205,206].
With respect to long-COVID especially, luteolin could prevent neuro-inflammation [208,209,210,211], is neuroprotective [208, 210, 212, 213], and reduces cognitive dysfunction [214,215,216,217,218], especially brain fog [58, 60, 62].
Quercetin has been discussed in a few recent studies [219, 220], including an open-label clinical study showing good tolerability and benefit [221]. A double-blind, placebo-controlled, randomized study using a liposomal preparation of luteolin (PureLut) in long-COVID patients is underway. Combining quercetin with luteolin may provide additional benefits, especially when formulated in olive pomace oil (FibroProtek) that increases oral absorption, that is otherwise quite limited (<10%) [222]. Moreover, olive pomace oil provides additional antiviral [223] and anti-inflammatory [224]. Such liposomal preparations are available [222] and have been successfully used in pilot clinical trials [225] and reduced neuropsychiatric symptoms and associated serum IL-6 levels [226].
Conclusion
Further studies are urgently needed to address the neuropathogenesis of SARS-CoV-2 infection [227, 228] or the long-term effects of COVID-19 especially in the brain [229]. COVID vaccines have been enormously helpful [230,231,232], but there have been reports of rare neurological complications including Guillain-Barre syndrome and Bell’s palsy [233]. These may be related to the recent finding that the spike protein expressed in response to mRNA vaccines was detected in the circulation as early as 1 day post vaccination and became undetectable by day 14 [234]. Hence, we should try to limit or prevent spike-related detrimental effects especially to the brain and their potential contribution to the development of long-COVID.
Availability of Data and Materials
Not applicable
References
Tai W, He L, Zhang X et al (2020) Characterization of the receptor-binding domain (RBD) of 2019 novel coronavirus: implication for development of RBD protein as a viral attachment inhibitor and vaccine. Cell Mol Immunol 17(6):613–620
Cai Y, Zhang J, Xiao T et al (2020) Distinct conformational states of SARS-CoV-2 spike protein. Science 369(6511):1586–1592
Canna SW, Cron RQ (2020) Highways to hell: mechanism-based management of cytokine storm syndromes. J Allergy Clin Immunol 146(5):949–959
Giamarellos-Bourboulis EJ, Netea MG, Rovina N et al (2020) Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe 27(6):992–1000
Ye Q, Wang B, Mao J (2020) The pathogenesis and treatment of the ‘cytokine storm’ in COVID-19. J Inf Secur 80(6):607–613
Chen G, Wu D, Guo W et al (2020) Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest 130(5):2620–2629
Conti P, Ronconi G, Caraffa A et al (2020) Induction of pro-inflammatory cytokines (IL-1 and IL-6) and lung inflammation by coronavirus-19 (COVI-19 or SARS-CoV-2): anti-inflammatory strategies. J Biol Regul Homeost Agents 34(2):327–331
Tang Y, Liu J, Zhang D, Xu Z, Ji J, Wen C (2020) Cytokine storm in COVID-19: the current evidence and treatment strategies. Front Immunol 11:1708
Paces J, Strizova Z, Smrz D, Cerny J (2020) COVID-19 and the immune system. Physiol Res 69(3):379–388
Ragab D, Salah EH, Taeimah M, Khattab R, Salem R (2020) The COVID-19 cytokine storm; what we know so far. Front Immunol 11:1446
Brodin P (2021) Immune determinants of COVID-19 disease presentation and severity. Nat Med 27(1):28–33
Herold T, Jurinovic V, Arnreich C et al (2020) Elevated levels of IL-6 and CRP predict the need for mechanical ventilation in COVID-19. J Allergy Clin Immunol 146(1):128–136
Han H, Ma Q, Li C et al (2020) Profiling serum cytokines in COVID-19 patients reveals IL-6 and IL-10 are disease severity predictors. Emerg Microbes Infect 9(1):1123–1130
Mazzoni A, Salvati L, Maggi L et al (2020) Impaired immune cell cytotoxicity in severe COVID-19 is IL-6 dependent. J Clin Invest 130(9):4694–4703
Liu F, Li L, Xu M et al (2020) Prognostic value of interleukin-6, C-reactive protein, and procalcitonin in patients with COVID-19. J Clin Virol 127:104370
Copaescu A, Smibert O, Gibson A, Phillips EJ, Trubiano JA (2020) The role of IL-6 and other mediators in the cytokine storm associated with SARS-CoV-2 infection. J Allergy Clin Immunol 146(3):518–534
Conti P, Caraffa A, Gallenga CE et al (2020) Coronavirus-19 (SARS-CoV-2) induces acute severe lung inflammation via IL-1 causing cytokine storm in COVID-19: a promising inhibitory strategy. J Biol Regul Homeost Agents 34(6):1971–1975
Moore JB, June CH (2020) Cytokine release syndrome in severe COVID-19. Science 368(6490):473–474
Sadarangani M, Marchant A, Kollmann TR (2021) Immunological mechanisms of vaccine-induced protection against COVID-19 in humans. Nat Rev Immunol 21(8):475–484
Letizia AG, Ge Y, Vangeti S et al (2021) SARS-CoV-2 seropositivity and subsequent infection risk in healthy young adults: a prospective cohort study. Lancet Respir Med 9(7):712–720
Puranik A, Lenehan PJ, Silvert E et al. Comparison of two highly-effective mRNA vaccines for COVID-19 during periods of Alpha and Delta variant prevalence. medRxiv 2021.
Harvey WT, Carabelli AM, Jackson B et al (2021) SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol 19(7):409–424
Nalbandian A, Sehgal K, Gupta A et al (2021) Post-acute COVID-19 syndrome. Nat Med 27(4):601–615
Baig AM. (2020) Chronic COVID syndrome: need for an appropriate medical terminology for long-COVID and COVID long-haulers. J Med Virol.
Higgins V, Sohaei D, Diamandis EP, Prassas I. (2020) COVID-19: from an acute to chronic disease? Potential long-term health consequences. Crit Rev Clin Lab Sci 1-23.
Huang C, Huang L, Wang Y et al (2021) 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet 397(10270):220–232
Moreno-Perez O, Merino E, Leon-Ramirez JM et al (2021) Post-acute COVID-19 syndrome. Incidence and risk factors: a Mediterranean cohort study. J Inf Secur 82(3):378–383
Montagne A, Nation DA, Sagare AP et al (2020) APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581(7806):71–76
Sudre CH, Murray B, Varsavsky T et al (2021) Attributes and predictors of long COVID. Nat Med 27(4):626–631
Dennis A, Wamil M, Alberts J et al (2021) Multiorgan impairment in low-risk individuals with post-COVID-19 syndrome: a prospective, community-based study. BMJ Open 11(3):e048391
de Erausquin GA, Snyder H, Carrillo M, Hosseini AA, Brugha TS, Seshadri S (2021) The chronic neuropsychiatric sequelae of COVID-19: the need for a prospective study of viral impact on brain functioning. Alzheimers Dement 17(6):1056–1065
Deer RR, Rock MA, Vasilevsky N et al (2021) Characterizing long COVID: deep phenotype of a complex condition. EBioMedicine 74:103722
Choi B, Choudhary MC, Regan J et al (2020) Persistence and evolution of SARS-CoV-2 in an immunocompromised host. N Engl J Med 383(23):2291–2293
Davis HE, Assaf GS, McCorkell L et al (2021) Characterizing long COVID in an international cohort: 7 months of symptoms and their impact. EClinicalMedicine 38:101019
Helms J, Kremer S, Merdji H et al (2020) Neurologic features in severe SARS-CoV-2 infection. N Engl J Med 382(23):2268–2270
Fotuhi M, Mian A, Meysami S, Raji CA (2020) Neurobiology of COVID-19. J Alzheimers Dis 76(1):3–19
Najjar S, Najjar A, Chong DJ et al (2020) Central nervous system complications associated with SARS-CoV-2 infection: integrative concepts of pathophysiology and case reports. J Neuroinflammation 17(1):231
Singh AK, Bhushan B, Maurya A, Mishra G, Singh SK, Awasthi R (2020) Novel coronavirus disease 2019 (COVID-19) and neurodegenerative disorders. Dermatol Ther 33(4):e13591
Liotta EM, Batra A, Clark JR et al (2020) Frequent neurologic manifestations and encephalopathy-associated morbidity in Covid-19 patients. Ann Clin Transl Neurol 7(11):2221–2230
Koralnik IJ, Tyler KL (2020) COVID-19: a global threat to the nervous system. Ann Neurol 88(1):1–11
Nepal G, Rehrig JH, Shrestha GS et al (2020) Neurological manifestations of COVID-19: a systematic review. Crit Care 24(1):421
Favas TT, Dev P, Chaurasia RN et al (2020) Neurological manifestations of COVID-19: a systematic review and meta-analysis of proportions. Neurol Sci 41(12):3437–3470
Nazari S, Azari JA, Mirmoeeni S et al (2021) Central nervous system manifestations in COVID-19 patients: a systematic review and meta-analysis. Brain Behav 11:e02025
Kempuraj D, Selvakumar GP, Ahmed ME et al (2020) COVID-19, mast cells, cytokine storm, psychological stress, and neuroinflammation. Neuroscientist 26(5-6):402–414
Levin SN, Venkatesh S, Nelson KE et al (2021) Manifestations and impact of the COVID-19 pandemic in neuroinflammatory diseases. Ann Clin Transl Neurol 8(4):918–928
Baig AM (2020) Deleterious outcomes in long-hauler COVID-19: the effects of SARS-CoV-2 on the CNS in chronic COVID syndrome. ACS Chem Neurosci 11(24):4017–4020
Ongur D, Perlis R, Goff D (2020) Psychiatry and COVID-19. JAMA 324(12):1149–1150
Vindegaard N, Benros ME (2020) COVID-19 pandemic and mental health consequences: systematic review of the current evidence. Brain Behav Immun 89:531–542
Pfefferbaum B, North CS (2020) Mental health and the Covid-19 pandemic. N Engl J Med 383(6):510–512
Xiang YT, Yang Y, Li W et al (2020) Timely mental health care for the 2019 novel coronavirus outbreak is urgently needed. Lancet Psychiatry 7(3):228–229
Gordon JA, Borja SE (2020) The COVID-19 pandemic: setting the mental health research agenda. Biol Psychiatry 88(2):130–131
Taquet M, Luciano S, Geddes JR, Harrison PJ (2021) Bidirectional associations between COVID-19 and psychiatric disorder: retrospective cohort studies of 62 354 COVID-19 cases in the USA. Lancet Psychiatry 8(2):130–140
Schirinzi T, Landi D, Liguori C (2020) COVID-19: dealing with a potential risk factor for chronic neurological disorders. J Neurol
Steardo L Jr, Steardo L, Verkhratsky A (2020) Psychiatric face of COVID-19. Transl Psychiatry 10(1):261
Shader RI (2020) COVID-19 and depression. Clin Ther 42(6):962–963
Smith CM, Komisar JR, Mourad A, Kincaid BR (2020) COVID-19-associated brief psychotic disorder. BMJ Case Rep 13(8)
Druss BG (2020) Addressing the COVID-19 pandemic in populations with serious mental illness. JAMA Psychiatry 77(9):891–892
Theoharides TC, Cholevas C, Polyzoidis K, Politis A (2021) Long-COVID syndrome-associated brain fog and chemofog: luteolin to the rescue. Biofactors 47(2):232–241
Graham EL, Clark JR, Orban ZS et al (2021) Persistent neurologic symptoms and cognitive dysfunction in non-hospitalized Covid-19 “long haulers”. Ann Clin Transl Neurol 8(5):1073–1085
Stefano GB, Buttiker P, Weissenberger S, Martin A, Ptacek R, Kream RM (2021) Editorial: The pathogenesis of long-term neuropsychiatric COVID-19 and the role of microglia, mitochondria, and persistent neuroinflammation: a hypothesis. Med Sci Monit 27:e933015
Bell ML, Catalfamo CJ, Farland LV et al (2021) Post-acute sequelae of COVID-19 in a non-hospitalized cohort: results from the Arizona CoVHORT. PLoS One 16(8):e0254347
Hugon J, Msika EF, Queneau M, Farid K, Paquet C (2021) Long COVID: cognitive complaints (brain fog) and dysfunction of the cingulate cortex. J Neurol Jun 18:1–3
Frontera JA, Yang D, Lewis A et al (2021) A prospective study of long-term outcomes among hospitalized COVID-19 patients with and without neurological complications. J Neurol Sci 426:117486
Podlesek A, Komidar L, Kavcic V (2021) The relationship between perceived stress and subjective cognitive decline during the COVID-19 epidemic. Front Psychol 12:647971
Townsend L, Dyer AH, Jones K et al (2020) Persistent fatigue following SARS-CoV-2 infection is common and independent of severity of initial infection. PLoS One 15(11):e0240784
Phillips S, Williams MA (2021) Confronting our next national health disaster - long-haul Covid. N Engl J Med 385(7):577–579
Gaebler C, Wang Z, Lorenzi JCC et al (2021) Evolution of antibody immunity to SARS-CoV-2. Nature 591(7851):639–644
Files JK, Sarkar S, Fram TR et al (2021) Duration of post-COVID-19 symptoms is associated with sustained SARS-CoV-2-specific immune responses. JCI. Insight 6(15)
McQuaid C, Brady M, Deane R (2021) SARS-CoV-2: is there neuroinvasion? Fluids Barriers CNS 18(1):32
Veleri S (2021) Neurotropism of SARS-CoV-2 and neurological diseases of the central nervous system in COVID-19 patients. Exp Brain Res
Welcome MO, Mastorakis NE (2021) Neuropathophysiology of coronavirus disease 2019: neuroinflammation and blood brain barrier disruption are critical pathophysiological processes that contribute to the clinical symptoms of SARS-CoV-2 infection. Inflammopharmacology 29(4):939–963
Zhang L, Zhou L, Bao L et al (2021) SARS-CoV-2 crosses the blood-brain barrier accompanied with basement membrane disruption without tight junctions alteration. Signal Transduct Target Ther 6(1):337
Song E, Zhang C, Israelow B et al (2021) Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med 218(3)
Xu J, Zhong S, Liu J et al (2005) Detection of severe acute respiratory syndrome coronavirus in the brain: potential role of the chemokine mig in pathogenesis. Clin Infect Dis 41(8):1089–1096
Meinhardt J, Radke J, Dittmayer C et al (2021) Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat Neurosci 24(2):168–175
Jiao L, Yang Y, Yu W et al (2021) The olfactory route is a potential way for SARS-CoV-2 to invade the central nervous system of rhesus monkeys. Signal Transduct Target Ther 6(1):169
Fagre A, Lewis J, Eckley M et al (2021) SARS-CoV-2 infection, neuropathogenesis and transmission among deer mice: implications for spillback to New World rodents. PLoS Pathog 17(5):e1009585
Brann DH, Tsukahara T, Weinreb C et al (2020) Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci Adv 6(31)
Nauen DW, Hooper JE, Stewart CM, Solomon IH (2021) Assessing brain capillaries in coronavirus disease 2019. JAMA Neurol 78(6):760–762
Tsivgoulis G, Palaiodimou L, Zand R et al (2020) COVID-19 and cerebrovascular diseases: a comprehensive overview. Ther Adv Neurol Disord 13:1756286420978004
Bass DI, Meyer RM, Barros G et al (2021) The impact of the COVID-19 pandemic on cerebrovascular disease. Semin Vasc Surg 34(2):20–27
Dhamoon MS, Thaler A, Gururangan K et al (2021) Acute cerebrovascular events with COVID-19 infection. Stroke 52(1):48–56
Gomes I, Karmirian K, Oliveira JT et al (2021) SARS-CoV-2 infection of the central nervous system in a 14-month-old child: a case report of a complete autopsy. Lancet Reg Health Am 2:100046
Yang AC, Kern F, Losada PM et al (2021) Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595(7868):565–571
Ramani A, Muller L, Ostermann PN et al (2020) SARS-CoV-2 targets neurons of 3D human brain organoids. EMBO J 39(20):e106230
Baig AM, Khaleeq A, Ali U, Syeda H (2020) Evidence of the COVID-19 virus targeting the CNS: tissue distribution, host-virus interaction, and proposed neurotropic mechanisms. ACS Chem Neurosci 11(7):995–998
Savitt AG, Manimala S, White T et al (2021) SARS-CoV-2 exacerbates COVID-19 pathology through activation of the complement and kinin systems. Front Immunol 12:767347
Karnik M, Beeraka NM, Uthaiah CA et al (2021) A review on SARS-CoV-2-induced neuroinflammation, neurodevelopmental complications, and recent updates on the vaccine development. Mol Neurobiol 5:1–29
Liberman AC, Trias E, da Silva CL et al (2018) Neuroimmune and inflammatory signals in complex disorders of the central nervous system. Neuroimmunomodulation 25(5-6):246–270
Lee MH, Perl DP, Nair G et al (2021) Microvascular injury in the brains of patients with Covid-19. N Engl J Med 384(5):481–483
Magro CM, Mulvey J, Kubiak J et al (2021) Severe COVID-19: a multifaceted viral vasculopathy syndrome. Ann Diagn Pathol 50:151645
Bodnar B, Patel K, Ho W, Luo JJ, Hu W (2021) Cellular mechanisms underlying neurological/neuropsychiatric manifestations of COVID-19. J Med Virol 93(4):1983–1998
Ng JH, Sun A, Je HS, Tan EK. (2021) Unravelling pathophysiology of neurological and psychiatric complications of COVID-19 using brain organoids. Neuroscientist 10738584211015136.
Tremblay ME, Madore C, Bordeleau M, Tian L, Verkhratsky A (2020) Neuropathobiology of COVID-19: the role for glia. Front Cell Neurosci 14:592214
McMahon CL, Staples H, Gazi M, Carrion R, Hsieh J (2021) SARS-CoV-2 targets glial cells in human cortical organoids. Stem Cell Rep 16(5):1156–1164
Theoharides TC (1990) Mast cells: the immune gate to the brain. Life Sci 46:607–617
Al-Harthi L, Campbell E, Schneider JA, Bennett DA (2021) What HIV in the brain can teach us about SARS-CoV-2 neurological complications? AIDS Res Hum Retrovir 37(4):255–265
Liu Y, Soh WT, Kishikawa JI et al (2021) An infectivity-enhancing site on the SARS-CoV-2 spike protein targeted by antibodies. Cell 9(7):712–720
Yahi N, Chahinian H, Fantini J. (2021) Infection-enhancing anti-SARS-CoV-2 antibodies recognize both the original Wuhan/D614G strain and Delta variants. A potential risk for mass vaccination? J Infect.
Wan Y, Shang J, Sun S et al (2020) Molecular mechanism for antibody-dependent enhancement of coronavirus entry. J Virol 94(5):e02015–e02019
Murphy WJ, Longo DL. (2021) A possible role for anti-idiotype antibodies in SARS-CoV-2 infection and vaccination. N Engl J Med.
Levin EG, Lustig Y, Cohen C et al. (2021) Waning immune humoral response to BNT162b2 Covid-19 vaccine over 6 months. N Engl J Med.
Goldberg Y, Mandel M, Bar-On YM et al. (2021) Waning immunity after the BNT162b2 vaccine in Israel. N Engl J Med.
Bartley CM, Johns C, Ngo TT et al. (2021) Anti-SARS-CoV-2 and autoantibody profiles in the cerebrospinal fluid of 3 teenaged patients with COVID-19 and subacute neuropsychiatric symptoms. JAMA Neurol.
Moosavi F, Hosseini R, Saso L, Firuzi O (2016) Modulation of neurotrophic signaling pathways by polyphenols. Drug Des Devel Ther 10:23–42
Theoharides TC, Conti P (2020) COVID-19 and multisystem inflammatory syndrome, or is it mast cell activation syndrome? J Biol Regul Homeost Agents 34(5):1633–1636
Theoharides TC (2020) Potential association of mast cells with COVID-19. Ann Allergy Asthma Immunol 126(3):217–218
Theoharides TC, Antonopoulou S, Demopoulos CA (2020) Coronavirus 2019, microthromboses, and platelet activating factor. Clin Ther 42(10):1850–1852
Demopoulos C, Antonopoulou S, Theoharides TC (2020) COVID-19, microthromboses, inflammation, and platelet activating factor. Biofactors 46(6):927–933
Lei Y, Zhang J, Schiavon CR et al (2021) SARS-CoV-2 spike protein impairs endothelial function via downregulation of ACE 2. Circ Res 128(9):1323–1326
Raghavan S, Kenchappa DB, Leo MD (2021) SARS-CoV-2 spike protein induces degradation of junctional proteins that maintain endothelial barrier integrity. Front Cardiovasc Med 8:687783
Kim ES, Jeon MT, Kim KS, Lee S, Kim S, Kim DG (2021) Spike proteins of SARS-CoV-2 induce pathological changes in molecular delivery and metabolic function in the brain endothelial cells. Viruses 13(10):2021
DeOre BJ, Tran KA, Andrews AM, Ramirez SH, Galie PA. (2021) SARS-CoV-2 spike protein disrupts blood-brain barrier integrity via rhoa activation. J Neuroimmune Pharmacol.
Meyer K, Patra T, Vijayamahantesh RR (2021) SARS-CoV-2 spike protein induces paracrine senescence and leukocyte adhesion in endothelial cells. J Virol 95(17):e0079421
Buzhdygan TP, DeOre BJ, Baldwin-Leclair A et al (2020) The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol Dis 146:105131
Rhea EM, Logsdon AF, Hansen KM et al (2021) The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nat Neurosci 24(3):368–378
Khaddaj-Mallat R, Aldib N, Bernard M et al (2021) SARS-CoV-2 deregulates the vascular and immune functions of brain pericytes via Spike protein. Neurobiol Dis 161:105561
Paladino L, Vitale AM, Caruso BC et al (2020) The role of molecular chaperones in virus infection and implications for understanding and treating COVID-19. J Clin Med 9(11):3518
Dasgupta S, Bandyopadhyay M (2021) Molecular docking of SARS-COV-2 spike epitope sequences identifies heterodimeric peptide-protein complex formation with human Zo-1, TLR8 and brain specific glial proteins. Med Hypotheses 157:110706
Charlie-Silva I, Araujo APC, Guimaraes ATB et al (2021) Toxicological insights of spike fragments SARS-CoV-2 by exposure environment: a threat to aquatic health? J Hazard Mater 419:126463
Zhang X, Dong H, Li N et al (2016) Activated brain mast cells contribute to postoperative cognitive dysfunction by evoking microglia activation and neuronal apoptosis. J Neuroinflammation 13(1):127
Moura DS, Sultan S, Georgin-Lavialle S et al (2012) Evidence for cognitive impairment in mastocytosis: prevalence, features and correlations to depression. PLoS One 7(6):e39468
Afrin LB, Pohlau D, Raithel M et al (2015) Mast cell activation disease: an underappreciated cause of neurologic and psychiatric symptoms and diseases. Brain Behav Immun 50:314–321
Akin C, Valent P, Metcalfe DD (2010) Mast cell activation syndrome: proposed diagnostic criteria. J Allergy Clin Immunol 126(6):1099–1104
Theoharides TC, Tsilioni I, Ren H (2019) Recent advances in our understanding of mast cell activation - or should it be mast cell mediator disorders? Expert Rev Clin Immunol 15(6):639–656
Theoharides TC, Valent P, Akin C (2015) Mast cells, mastocytosis, and related disorders. N Engl J Med 373(2):163–172
Marshall JS, Portales-Cervantes L, Leong E (2019) Mast cell responses to viruses and pathogen products. Int J Mol Sci 20(17):4241
Motta Junior JDS, Miggiolaro AFRD, Nagashima S et al (2020) Mast cells in alveolar septa of COVID-19 patients: a pathogenic pathway that may link interstitial edema to immunothrombosis. Front Immunol 11:574862
Theoharides TC, Konstantinidou A (2007) Corticotropin-releasing hormone and the blood-brain-barrier. Front Biosci 12:1615–1628
Rozniecki JJ, Dimitriadou V, Lambracht-Hall M, Pang X, Theoharides TC (1999) Morphological and functional demonstration of rat dura mast cell-neuron interactions in vitro and in vivo. Brain Res 849:1–15
Dimitriadou V, Rouleau A, Trung Tuong MD et al (1997) Functional relationships between sensory nerve fibers and mast cells of dura mater in normal and inflammatory conditions. Neuroscience 77:829–839
Hendriksen E, van BD, Oosting RS, Redegeld FA. (2017) Mast cells in neuroinflammation and brain disorders. Neurosci Biobehav Rev 79:119–133
Zhang X, Wang Y, Dong H, Xu Y, Zhang S (2016) Induction of microglial activation by mediators released from mast cells. Cell Physiol Biochem 38(4):1520–1531
Skaper SD, Facci L, Zusso M, Giusti P (2017) Neuroinflammation, mast cells, and glia: dangerous liaisons. Neuroscientist 23(5):478–498
Yue J, Jin W, Yang H et al (2021) Heparan Sulfate facilitates spike protein-mediated SARS-CoV-2 host cell invasion and contributes to increased infection of SARS-CoV-2 G614 mutant and in lung cancer. Front Mol Biosci 8:649575
Hassanzadeh K, Perez PH, Dragotto J et al (2020) Considerations around the SARS-CoV-2 spike protein with particular attention to COVID-19 brain infection and neurological symptoms. ACS Chem Neurosci 11(15):2361–2369
Liu L, Chopra P, Li X et al (2021) Heparan sulfate proteoglycans as attachment factor for SARS-CoV-2. ACS Cent Sci 7(6):1009–1018
Gudowska-Sawczuk M, Mroczko B (2021) The role of neuropilin-1 (NRP-1) in SARS-CoV-2 infection: review. J Clin Med 10(13):2772
Sarabipour S, Mac GF (2021) Targeting neuropilins as a viable SARS-CoV-2 treatment. FEBS J
Xie Y, Karki CB, Du D et al (2020) Spike proteins of SARS-CoV and SARS-CoV-2 utilize different mechanisms to bind with human ACE2. Front Mol Biosci 7:591873
Shang J, Wan Y, Luo C et al (2020) Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 117(21):11727–11734
Onofrio L, Caraglia M, Facchini G, Margherita V, Placido S, Buonerba C (2020) Toll-like receptors and COVID-19: a two-faced story with an exciting ending. Future Sci OA 6(8):FSO605
Sariol A, Perlman S (2021) SARS-CoV-2 takes its toll. Nat Immunol 22(7):801–802
Singh H, Singh A, Khan AA, Gupta V (2021) Immune mediating molecules and pathogenesis of COVID-19-associated neurological disease. Microb Pathog 158:105023
Aboudounya MM, Heads RJ (2021) COVID-19 and toll-like receptor 4 (TLR4): SARS-CoV-2 may bind and activate TLR4 to increase ACE2 expression, facilitating entry and causing hyperinflammation. Mediat Inflamm 2021:8874339
Yi YS, Yun M (2021) Editorial of Special Issue “Regulatory roles of inflammasomes in human diseases”. Int J Mol Sci 22(6):3008
Maiese K (2020) The mechanistic target of rapamycin (mTOR): novel considerations as an antiviral treatment. Curr Neurovasc Res 17(3):332–337
Zheng Y, Li R, Liu S (2020) Immunoregulation with mTOR inhibitors to prevent COVID-19 severity: a novel intervention strategy beyond vaccines and specific antiviral medicines. J Med Virol 92(9):1495–1500
Ryskalin L, Limanaqi F, Frati A, Busceti CL, Fornai F (2018) mTOR-related brain dysfunctions in neuropsychiatric disorders. Int J Mol Sci 19(8)
Espindola OM, Gomes YCP, Brandao CO et al (2021) Inflammatory cytokine patterns associated with neurological diseases in coronavirus disease 2019. Ann Neurol 89(5):1041–1045
Oka Y, Ueda A, Nakagawa T et al (2021) SARS-CoV-2-related progressive brain white matter lesion associated with an increased cerebrospinal fluid level of IL-6. Intern Med 60(19):3167–3170
Sparrow NA, Anwar F, Covarrubias AE et al (2021) IL-6 inhibition reduces neuronal injury in a murine model of ventilator-induced lung injury. Am J Respir Cell Mol Biol 65(4):403–412
Saridaki M, Metallidis S, Grigoropoulou S et al (2021) Integration of heparin-binding protein and interleukin-6 in the early prediction of respiratory failure and mortality in pneumonia by SARS-CoV-2 (COVID-19). Eur J Clin Microbiol Infect Dis 40(7):1405–1412
Rauti R, Shahoha M, Leichtmann-Bardoogo Y et al (2021) Effect of SARS-CoV-2 proteins on vascular permeability. Elife 10:e69314
Varma P, Lybrand ZR, Antopia MC, Hsieh J (2020) Novel targets of SARS-CoV-2 spike protein in human fetal brain development suggest early pregnancy vulnerability. Front Neurosci 14:614680
Vivanti AJ, Vauloup-Fellous C, Prevot S et al (2020) Transplacental transmission of SARS-CoV-2 infection. Nat Commun 11(1):3572
Theoharides TC (2021) Ways to address perinatal mast cell activation and focal brain inflammation, including response to SARS-CoV-2, in autism spectrum disorder. J Pers Med 11(9):860
Theoharides TC, Tsilioni I, Bawazeer M (2019) Mast cells, neuroinflammation and pain in fibromyalgia syndrome. Front Cell Neurosci 13:353
Rahman MA, Islam K, Rahman S, Alamin M (2021) Neurobiochemical cross-talk between COVID-19 and Alzheimer’s disease. Mol Neurobiol 58(3):1017–1023
Kempuraj D, Ahmed ME, Selvakumar GP et al (2019) Brain injury-mediated neuroinflammatory response and Alzheimer’s disease. Neuroscientist 26(2):134–155
(2021) WHO recommends life-saving interleukin-6 receptor blockers for COVID-19 and urges producers to join efforts to rapidly increase access. Saudi Med J 42(8):923
Kyriazopoulou E, Poulakou G, Milionis H et al (2021) Early treatment of COVID-19 with anakinra guided by soluble urokinase plasminogen receptor plasma levels: a double-blind, randomized controlled phase 3 trial. Nat Med 27(10):1752–1760
Sagris D, Florentin M, Tasoudis P et al (2021) Immunomodulation and reduction of thromboembolic risk in hospitalized COVID-19 patients: systematic review and meta-analysis of randomized trials. J Clin Med 10(22):5366
Rosas IO, Brau N, Waters M et al (2021) Tocilizumab in hospitalized patients with severe Covid-19 pneumonia. N Engl J Med 384(16):1503–1516
Theoharides TC, Boucher W, Spear K (2002) Serum interleukin-6 reflects disease severity and osteoporosis in mastocytosis patients. Int Arch Allergy Immunol 128:344–350
Brockow K, Akin C, Huber M, Metcalfe DD (2005) IL-6 levels predict disease variant and extent of organ involvement in patients with mastocytosis. Clin Immunol 115(2):216–223
Mayado A, Teodosio C, Garcia-Montero AC et al (2015) Increased IL6 plasma levels in indolent systemic mastocytosis patients are associated with high risk of disease progression. Leukemia 30(1):124–130
Kandere-Grzybowska K, Letourneau R, Kempuraj D et al (2003) IL-1 induces vesicular secretion of IL-6 without degranulation from human mast cells. J Immunol 171(9):4830–4836
Huang M, Pang X, Karalis K, Theoharides TC (2003) Stress-induced interleukin-6 release in mice is mast cell-dependent and more pronounced in Apolipoprotein E knockout mice. Cardiovasc Res 59(1):241–249
Tobio A, Alfonso A, Botana LM (2015) C-kit mutations determine dasatinib mechanism of action in HMC-1 neoplastic mast cells: dasatinib differently regulates PKCdelta translocation in HMC-1(560) and HMC-1(560,816) cell lines. Immunopharmacol Immunotoxicol 37(4):380–387
Desai A, Jung MY, Olivera A et al (2016) IL-6 promotes an increase in human mast cell numbers and reactivity through suppression of suppressor of cytokine signaling 3. J Allergy Clin Immunol 137(6):1863–1871
Peter AE, Sandeep BV, Rao BG, Kalpana VL (2020) Calming the storm: natural immunosuppressants as adjuvants to target the cytokine storm in COVID-19. Front Pharmacol 11:583777
Nouadi B, Ezaouine A, El MM, Blaghen M, Bennis F, Chegdani F (2021) Prediction of anti-COVID 19 therapeutic power of medicinal moroccan plants using molecular docking. Bioinform Biol Insights 15:11779322211009199
Alesci A, Aragona M, Cicero N, Lauriano ER. (2021) Can nutraceuticals assist treatment and improve covid-19 symptoms? Nat Prod Res 1-20.
Xiong Y, Zhu GH, Wang HN et al (2021) Discovery of naturally occurring inhibitors against SARS-CoV-2 3CL(pro) from Ginkgo biloba leaves via large-scale screening. Fitoterapia 152:104909
Chen JS, Chen YH, Huang PH et al (2012) Ginkgo biloba extract reduces high-glucose-induced endothelial adhesion by inhibiting the redox-dependent interleukin-6 pathways. Cardiovasc Diabetol 11:49
Woelk H, Arnoldt KH, Kieser M, Hoerr R (2007) Ginkgo biloba special extract EGb 761 in generalized anxiety disorder and adjustment disorder with anxious mood: a randomized, double-blind, placebo-controlled trial. J Psychiatr Res 41(6):472–480
Gauthier S, Schlaefke S (2014) Efficacy and tolerability of Ginkgo biloba extract EGb 761(R) in dementia: a systematic review and meta-analysis of randomized placebo-controlled trials. Clin Interv Aging 9:2065–2077
Ibrahim MA, Ramadan HH, Mohammed RN (2021) Evidence that Ginkgo biloba could use in the influenza and coronavirus COVID-19 infections. J Basic Clin Physiol Pharmacol 32(3):131–143
Jimenez-Avalos G, Vargas-Ruiz AP, Gado-Pease NE et al (2021) Comprehensive virtual screening of 4.8 k flavonoids reveals novel insights into allosteric inhibition of SARS-CoV-2 M(PRO). Sci Rep 11(1):15452
Wang YQ, Li QS, Zheng XQ, Lu JL, Liang YR (2021) Antiviral effects of green tea EGCG and its potential application against COVID-19. Molecules 26(13):3962
Mhatre S, Gurav N, Shah M, Patravale V (2021) Entry-inhibitory role of catechins against SARS-CoV-2 and its UK variant. Comput Biol Med 135:104560
Gasparello J, D'Aversa E, Papi C et al (2021) Sulforaphane inhibits the expression of interleukin-6 and interleukin-8 induced in bronchial epithelial IB3-1 cells by exposure to the SARS-CoV-2 Spike protein. Phytomedicine 87:153583
Middleton E Jr, Kandaswami C, Theoharides TC (2000) The effects of plant flavonoids on mammalian cells: implications for inflammation, heart disease, and cancer. Pharmacol Rev 52(4):673–751
Theoharides TC (2020) COVID-19, pulmonary mast cells, cytokine storms, and beneficial actions of luteolin. Biofactors 46(3):306–308
Richman S, Morris MC, Broderick G, Craddock TJA, Klimas NG, Fletcher MA (2019) Pharmaceutical interventions in chronic fatigue syndrome: a literature-based commentary. Clin Ther 41(5):798–805
Goris T, Perez-Valero A, Martinez I et al (2021) Repositioning microbial biotechnology against COVID-19: the case of microbial production of flavonoids. Microb Biotechnol 14(1):94–110
Gour A, Manhas D, Bag S, Gorain B, Nandi U (2021) Flavonoids as potential phytotherapeutics to combat cytokine storm in SARS-CoV-2. Phytother Res 35(8):4258–4283
Alzaabi MM, Hamdy R, Ashmawy NS et al. (2021) Flavonoids are promising safe therapy against COVID-19. Phytochem Rev 1-22.
Kumar B, Zaidi S, Haque S et al. (2020) In silico studies reveal antiviral effects of traditional Indian spices on COVID-19. Curr Pharm Des
Ali AM, Kunugi H (2021) Propolis, bee honey, and their components protect against coronavirus disease 2019 (COVID-19): a review of in silico, in vitro, and clinical studies. Molecules 26(5):1232
Stalin A, Lin D, Senthamarai KB et al. (2021) An in-silico approach to identify the potential hot spots in SARS-CoV-2 spike RBD to block the interaction with ACE2 receptor. J Biomol Struct Dyn 1-16.
Xu L, Su W, Jin J et al (2014) Identification of luteolin as enterovirus 71 and coxsackievirus A16 inhibitors through reporter viruses and cell viability-based screening. Viruses 6(7):2778–2795
Fan W, Qian S, Qian P, Li X (2016) Antiviral activity of luteolin against Japanese encephalitis virus. Virus Res 220:112–116
Yan H, Ma L, Wang H et al (2019) Luteolin decreases the yield of influenza A virus in vitro by interfering with the coat protein I complex expression. J Nat Med 73(3):487–496
Russo M, Moccia S, Spagnuolo C, Tedesco I, Russo GL (2020) Roles of flavonoids against coronavirus infection. Chem Biol Interact 328:109211
Derosa G, Maffioli P, D'Angelo A, Di PF (2020) A role for quercetin in coronavirus disease 2019 (COVID-19). Phytother Res 35(3):1230–1236
Jo S, Kim S, Shin DH, Kim MS (2020) Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem 35(1):145–151
Xue G, Gong L, Yuan C et al (2017) A structural mechanism of flavonoids in inhibiting serine proteases. Food Funct 8(7):2437–2443
Rezai-Zadeh K, Ehrhart J, Bai Y et al (2008) Apigenin and luteolin modulate microglial activation via inhibition of STAT1-induced CD40 expression. J Neuroinflammation 5:41
Jang S, Kelley KW, Johnson RW (2008) Luteolin reduces IL-6 production in microglia by inhibiting JNK phosphorylation and activation of AP-1. Proc Natl Acad Sci U S A 105(21):7534–7539
Burton MD, Rytych JL, Amin R, Johnson RW (2016) Dietary luteolin reduces proinflammatory microglia in the brain of senescent mice. Rejuvenation Res 19(4):286–292
Patel AB, Tsilioni I, Leeman SE, Theoharides TC (2016) Neurotensin stimulates sortilin and mTOR in human microglia inhibitable by methoxyluteolin, a potential therapeutic target for autism. Proc Natl Acad Sci U S A 113:E7049–E7058
Weng Z, Patel AB, Panagiotidou S, Theoharides TC (2015) The novel flavone tetramethoxyluteolin is a potent inhibitor of human mast cells. J Allergy Clin Immunol 135(4):1044–1052
Patel AB, Theoharides TC (2017) Methoxyluteolin inhibits neuropeptide-stimulated proinflammatory mediator release via mTOR activation from human mast cells. J Pharmacol Exp Ther 361(3):462–471
Taracanova A, Tsilioni I, Conti P, Norwitz ER, Leeman SE, Theoharides TC (2018) Substance P and IL-33 administered together stimulate a marked secretion of IL-1beta from human mast cells, inhibited by methoxyluteolin. Proc Natl Acad Sci U S A 115(40):E9381–E9390
Lee MN, Lee Y, Wu D, Pae M (2021) Luteolin inhibits NLRP3 inflammasome activation via blocking ASC oligomerization. J Nutr Biochem 92:108614
Ashaari Z, Hadjzadeh MA, Hassanzadeh G et al (2018) The flavone luteolin improves central nervous system disorders by different mechanisms: a review. J Mol Neurosci 65(4):491–506
Calis Z, Mogulkoc R, Baltaci AK (2020) The roles of flavonols/flavonoids in neurodegeneration and neuroinflammation. Mini-Rev Med Chem 20(15):1475–1488
Kempuraj D, Thangavel R, Kempuraj DD et al (2020) Neuroprotective effects of flavone luteolin in neuroinflammation and neurotrauma. Biofactors 47(2):190–197
Theoharides TC, Conti P, Economu M (2014) Brain inflammation, neuropsychiatric disorders, and immunoendocrine effects of luteolin. J Clin Psychopharmacol 34(2):187–189
Dajas F, Rivera-Megret F, Blasina F et al (2003) Neuroprotection by flavonoids. Braz J Med Biol Res 36(12):1613–1620
Lin TY, Lu CW, Wang SJ (2016) Luteolin protects the hippocampus against neuron impairments induced by kainic acid in rats. NeuroToxicol 55:48–57
Rezai-Zadeh K, Douglas SR, Bai Y et al (2009) Flavonoid-mediated presenilin-1 phosphorylation reduces Alzheimer’s disease beta-amyloid production. J Cell Mol Med 13(3):574–588
Theoharides TC, Stewart JM, Hatziagelaki E, Kolaitis G (2015) Brain “fog,” inflammation and obesity: key aspects of 2 neuropsychiatric disorders improved by luteolin. Front Neurosci 9:225
Yao ZH, Yao XL, Zhang Y, Zhang SF, Hu JC (2018) Luteolin could improve cognitive dysfunction by inhibiting neuroinflammation. Neurochem Res 43(4):806–820
Gratton G, Weaver SR, Burley CV et al (2020) Dietary flavanols improve cerebral cortical oxygenation and cognition in healthy adults. Sci Rep 10(1):19409
Devi SA, Chamoli A (2020) Polyphenols as an effective therapeutic intervention against cognitive decline during normal and pathological brain aging. Adv Exp Med Biol 1260:159–174
Manjunath SH, Thimmulappa RK. (2021) Antiviral, immunomodulatory, and anticoagulant effects of quercetin and its derivatives: potential role in prevention and management of COVID-19. J Pharm Anal.
Simsek Y, Baran SS, Aslim B (2021) In silico identification of SARS-CoV-2 cell entry inhibitors from selected natural antivirals. J Mol Graph Model 109:108038
Di PF, Derosa G, Maffioli P et al (2021) Possible therapeutic effects of adjuvant quercetin supplementation against early-stage COVID-19 infection: a prospective, randomized, controlled, and open-label study. Int J Gen Med 14:2359–2366
Theoharides TC (2020) Luteolin supplements: all that glitters is not gold. Biofactors 47(2):242–244
Hartvig P, Larsson BS, Lindberg BS et al (1986) Influence of monoamine oxidase inhibitors and a dopamine uptake blocker on the distribution of 11C-N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine, 11C-MPTP, in the head of the rhesus monkey. Acta Neurol Scand 74(1):10–16
Serreli G, Deiana M (2020) Extra virgin olive oil polyphenols: modulation of cellular pathways related to oxidant species and inflammation in aging. Cells 9(2):478
Taliou A, Zintzaras E, Lykouras L, Francis K (2013) An open-label pilot study of a formulation containing the anti-inflammatory flavonoid luteolin and its effects on behavior in children with autism spectrum disorders. Clin Ther 35(5):592–602
Tsilioni I, Taliou A, Francis K, Theoharides TC (2015) Children with autism spectrum disorders, who improved with a luteolin containing dietary formulation, show reduced serum levels of TNF and IL-6. Transl Psychiatry 5:e647
Khan S, Gomes J (2020) Neuropathogenesis of SARS-CoV-2 infection. Elife 9:e59136
Farhadian SF, Seilhean D, Spudich S (2021) Neuropathogenesis of acute coronavirus disease 2019. Curr Opin Neurol 34(3):417–422
Ellul MA, Benjamin L, Singh B et al (2020) Neurological associations of COVID-19. Lancet Neurol 19(9):767–783
Dai L, Gao GF (2021) Viral targets for vaccines against COVID-19. Nat Rev Immunol 21(2):73–82
Theoharides TC, Lauritano D, Ronconi G, Calvisi V, Conti P (2021) Antibodies for COVID-19 - which, when and how long? J Biol Regul Homeost Agents 35(2):417–422
Escobedo RA, Singh DK, Kaushal D (2021) Understanding COVID-19: from dysregulated immunity to vaccination status quo. Front Immunol 12:765349
Kneussl MP, Richardson JB (1978) Alpha-adrenergic receptors in human and canine tracheal and bronchial smooth muscle. J Appl Physiol Respir Environ Exerc Physiol 45(2):307–311
Ogata AF, Cheng CA, Desjardins M et al. (2021) Circulating SARS-CoV-2 vaccine antigen detected in the plasma of mRNA-1273 vaccine recipients. Clin Infect Dis.
Author information
Authors and Affiliations
Contributions
Single author
Corresponding author
Ethics declarations
Ethics Approval
Not applicable
Consent to Participate
Not applicable
Consent for Publication
Not applicable
Competing Interests
The author is Scientific Director of Algonot LLC that develops flavonoid-containing dietary supplements.
Research Involving Human Participants and/or Animals
Not applicable
Informed Consent
Not applicable
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Theoharides, T.C. Could SARS-CoV-2 Spike Protein Be Responsible for Long-COVID Syndrome?. Mol Neurobiol 59, 1850–1861 (2022). https://doi.org/10.1007/s12035-021-02696-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02696-0