
Abstract
In the last decades, evidence suggesting the direct or indirect involvement of B cells on multiple sclerosis (MS) pathogenesis has accumulated. The increased amount of data on the efficacy and safety of B-cell-depleting therapies from several studies has suggested the addition of these drugs as treatment options to the current armamentarium of disease modifying therapies (DMTs) for MS. Particularly, rituximab (RTX), a chimeric monoclonal antibody directed at CD20 positive B lymphocytes resulting in cell-mediated apoptosis, has been demonstrated to reduce inflammatory activity, incidence of relapses and new brain lesions on magnetic resonance imaging (MRI) in patients with relapsing–remitting MS (RRMS). Additional evidence also demonstrated that patients with progressive MS (PMS) may benefit from RTX, which also showed to be well tolerated, with acceptable safety risks and favorable cost-effectiveness profile.
Despite these encouraging results, RTX is currently approved for non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, several forms of vasculitis and rheumatoid arthritis, while it can only be administered off-label for MS treatment. Between Northern European countries exist different rules for using not licensed drug for treating MS. The Sweden MS register reports a high rate (53.5%) of off-label RTX prescriptions in relation to other annually started DMTs to treat MS patients, while Danish and Norwegian neurologists have to use other anti-CD20 drugs, as ocrelizumab, in most of the cases.
In this paper, we review the pharmacokinetics, pharmacodynamics, clinical efficacy, safety profile and cost effectiveness aspects of RTX for the treatment of MS. Particularly, with the approval of new anti-CD20 DMTs, the recent worldwide COVID-19 emergency and the possible increased risk of infection with this class of drugs, this review sheds light on the use of RTX as an alternative treatment option for MS management, while commenting the gaps of knowledge regarding this drug.
Similar content being viewed by others


Introduction
Multiple sclerosis (MS) has been historically considered as an autoimmune disease of the central nervous system (CNS) mediated by CD4+ T cells reactive to myelin antigens [1]. According to this model, the autoimmunity processes directed to the CNS are induced by the imbalance between CNS-reactive effector T cells of the helper-1 (Th1) and Th17 type and regulatory T cells (Treg) [2, 3].
Although T cells have been considered the major contributors to the inflammatory activity in MS, growing evidences shed light on B-cell role [4,5,6]. Indeed, it has been demonstrated that B cells are present in MS lesions, meninges and cerebrospinal fluid (CSF) and can contribute to disease progression through several antibody-dependent (i.e., secreting intrathecal IgG) and -independent mechanisms [7]. In addition to their capability to produce antibodies after differentiating into plasma cells B cells can also stimulate T cells activity through antigen presentation [8], production of soluble neurotoxic factors [9] and switch to memory cells, the latter stimulating self-proliferation of CD4+ T cells [10].
The findings of cerebrospinal fluid oligoclonal IgG bands (OCBs), as well as the meningeal-based ectopic B-cell follicles adjacent to areas of focal cortical demyelination, suggest a more central role for B cells in MS [11,12,13,14,15,16]. In particular, OCBs, one of the hallmarks of MS, are shown to persist in the CSF in approximately 90% of patients [17]. OCBs produced by intrathecal B cells in the CSF contribute to the inflammation and destruction of the myelin sheath [18, 19]. In vivo and in vitro models showed that IgG leads to demyelination and axonal damage in a complement-dependent manner [20]. These data are indirectly confirmed by the efficacy of plasmapheresis and immunoadsorption in treating steroid-resistant MS relapses [21]. In addition, different antibody targets, such as myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), neurofilament, sperm-associated antigen 16 (SPAG16), coronin-1a, heat shock proteins, etc., have been demonstrated in MS patients [22, 23].
Moreover, B cells are able to migrate to the CNS using surface markers such as C–X–C motif receptor 3 (CXCR3), CXCR5, and CC chemokine receptor 5 (CCR5). In the meninges, these migrated B cells form ectopic B-cell follicular-like structures [24].
B cells are also able to induce antigen-specific T-cell expansion, memory formation and cytokine production, via a highly effective and selective antigen presentation [25]. B cells expressing co-stimulatory molecules, such as CD80, CD86, and CD40, contribute to T-cell activation. Particularly in MS patients, B-cell expression of CD80 and CD86 is higher than in controls [26].
Moreover, B-cell activation factor (BAFF), an important survival factor balancing pro-inflammatory and regulatory B-cell subtypes, is upregulated in MS lesions [27].
In addition, B cells from untreated MS patients (compared to healthy controls) have been demonstrated to secrete more pro-inflammatory interleukines-IL (such as IL-6) and less regulatory ones (such as IL-10) [16, 28].
Finally, B cells host Epstein–Barr virus (EBV), which is strongly related to MS in epidemiological analyses [29, 30].
In light of these findings, several B-cell-targeted therapies have been developed. To date, available B-cell-depleting monoclonal antibodies target specific Fab domains of CD20+ or CD19+ B lymphocytes, selectively allowing the depletion of the circulating B-cell population, apart from the mature antibody-secreting plasma cells [31, 32]. CD20 is a transmembrane, non-glycosylated phosphoprotein expressed on the surface of cells of the human B-cell lineage from pre-B cells to naïve and memory B cells. It is involved in the generation of T-cell-independent antibody responses [33]. The most effective therapies employ monoclonal antibodies depleting B cells through NK cell-mediated antibody-dependent cellular cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC), and antibody-triggered apoptosis [34, 35].
Anti-CD20 monoclonal antibodies currently in use for the treatment of MS are Rituximab (RTX), Ocrelizumab (OCR), Ofatumumab (OFA) and Ublituximab (UTX).
Rituximab (RTX) is a chimeric monoclonal B-cell-depleting anti-CD20 antibody and it was the first anti-CD20 drug licensed for the treatment of B-cell lymphomas, refractory rheumatoid arthritis (RA) and antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis [36]. Two randomized placebo-controlled phase 2 trials, the “Helping to Evaluate Rituxan in Relapsing–Remitting Multiple Sclerosis (HERMES)” and “A Study to Evaluate the Safety and Efficacy of Rituximab in Adults With Primary Progressive Multiple Sclerosis (OLYMPUS)”, have demonstrated the efficacy of RTX for the treatment of relapsing–remitting MS (RRMS) and primary progressive MS (PPMS), respectively [31, 37].
In the last years, several observational studies confirmed the high efficacy, relatively benign safety/tolerance profile, low cost and convenient administration regimen contributing to make RTX an interesting option for MS treatment, attracting increasing attention as an escalation and first-line therapy [38].
In this review, we discuss current evidence on pharmacokinetics, mechanisms of action, clinical efficacy, safety profile and cost effectiveness of RTX for the treatment of relapsing MS. With the approval of new anti-CD20 DMTs, the recent worldwide COronaVIrus Disease 19 (COVID-19) pandemic emergency and the possible increased risk of infection with this class of drugs, this review raises considerations regarding the use of RTX as a valid alterative treatment option for MS management.
Methods
A search of the relevant literature (up to November 2020) was conducted on MEDLINE (PubMed), PubMed Central, EMBASE and Cochrane Library, applying the medical subject headings (MeSH) terms “multiple sclerosis” and “rituximab” and “efficacy” and “safety” and “cost analysis” and “COronaVIrus Disease 19”. The Prisma flow diagram is illustrated in Fig. 1. If publications were not available via open or institutional access, the authors of the papers were contacted.

PRISMA diagram of the literature search
From the web-based search, we selected peer-reviewed, full-text and English language manuscripts. Randomized controlled trials (RCTs) with their extension trials and sub-studies, prospective studies, non-randomized clinical trials, retrospective studies, post hoc analyses, meta-analyses, reviews, and studies made from registries were included. We excluded single case studies, pediatric studies, and non-peer-reviewed publications. Each selected paper was preliminarily examined by both the authors FP and SA (via abstract reading), downloaded and summarized.
Pharmacokinetics
Considering that RTX was widely studied in hematological diseases, most pharmacokinetic data do not come from studies concerning MS. Historically, apart from hematological diseases, RTX was studied in other immune-mediated diseases, like RA [39]. It has been demonstrated that the average half-life for standard intravenous administration of RTX (2 × 1000 mg 2 weeks apart) is nearly 20 days, but it can vary depending on sex, body mass index (BMI) and renal clearance [40]. A smaller study in MS reproduced comparable results [41]. It has been supposed that RXT could negatively interfere with the activity of meningeal B-cell follicles driving the inflammatory cascade behind a closed blood brain barrier (BBB) [42]. However, big molecules (e.g., antibodies like RXT) do not easily overcome the BBB. This was confirmed by a study evaluating three RRMS patients treated with RTX, whereby the Positron Emission Tomography Computed Tomography (PET-CT) showed very low levels of RTX inside the CNS [43]. Another paper reported that RXT peak concentration was 400- to 1000-fold lower compared to serum concentrations until 4 weeks after intravenous administration [44]. Some studies also examined the administration of RTX through lumbar puncture (1–25 mg) or intraventricular catheter, demonstrating a rapid CSF clearance of the drug, probably depending on the Fc-receptor-mediated immunoglobulin efflux [45,46,47]. In a study involving 27 secondary progressive MS (SPMS) patients, intrathecal RTX administration showed a 20-fold higher bioavailability compared to intravenous infusion (2 vs 0.1% after intravenous administration) [44, 45, 48]. The authors concluded that the intrathecal RTX administration might be effective on intrathecal B cells and it could be adopted to reduce systemic doses, thus reducing risks. In accordance with these data, it has been highlighted that a low-dose intrathecal administration of RTX led to a profound peripheral B-cell depletion for up to 12 months, supporting the hypothesis that lower intravenous RTX doses might be sufficient to proficiently control peripheral B-cell levels [45, 49].
Pharmacodynamics
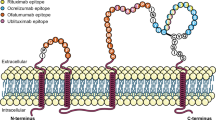
The RTX mechanism of action consists of binding a specific overlapping core epitope (170ANPS173) on the extracellular CD20 loop. The adjacent amino acids also contribute to binding. Consequently, the bond stability may vary [50]. RTX induces cell death through apoptosis, ADCC, antibody-dependent cell-mediated phagocytosis, and CDC mechanisms [50]. Compared to OCR, RTX binds weaker to the low-affinity variant of FcƔRIIIa, which is present in over 80% of MS patients. These data may explain why RTX induces more CDC (and less ADCC) [50,51,52,53,54] and displays a lower incidence of infusion-related reactions (IRR) when compared to OCR [37]. As showed in pharmacokinetics studies, the intravenous administration of RTX causes rapid and complete depletion of B cells both in blood [55] and, with a lesser degree, in the CSF [56]. The exact mechanism by which the depletion of B and T cells contrasts the inflammatory activity in MS patients is not fully understood. It has been speculated that it could be linked to the indirect effects depending on B cells, such as cytokine production (i.e., inflammation-driving granulocyte–macrophage colony-stimulating factor—GM-CSF—or modulators of T-cell activity) [57, 58]. RTX also induces apoptosis in small subgroups of pro-inflammatory CD3+ T cells expressing CD20 [59, 60]. Moreover, another hypothesis sheds light on Epstein–Barr virus (EBV) that, according to several studies, may be considered implicated in MS development [61]. With this regard, some effects of anti-CD20 drugs could be related to the clearing of the viral pool (Fig. 2) [62].

Rituximab mechanisms of action. MS multiple sclerosis, MAC membrane attack complex
Clinical efficacy
Phase I and pivotal studies
The preliminary safety, tolerability and efficacy profiles of RTX were initially established in a phase I open-label study of RRMS patients receiving a double course (2 weeks apart) of 1000 mg of RTX at baseline and after 6 months, with a follow-up of a total of 72 weeks [41]. Main clinical outcomes of efficacy included the proportion of patients experiencing a confirmed relapse and the number of relapses per patient during the study. MRI imaging outcomes were the total number of new gadolinium (Gd)-enhancing T1 lesions, of new T2 lesions and the cumulative volume of T2 brain lesions. This study demonstrated that in active MS patients, peripheral B-cell depletion was associated with sustained reductions in the number of relapses (annualized relapse rates [ARR] 0.18 on week 72 compared to 1.27 in the year before the study), while the majority of the subjects (80.8%) remained relapse free. Concomitantly, Gd-enhancing lesions were completely suppressed by week 72 (from a mean number of lesions of 1.31 per patient at baseline) and the mean number of new T2 lesions decreased over the course of the study (from 0.92 at week 4 to 0 at week 72) [41].
In 2008, a pivotal randomized double-blind placebo-controlled multicentre phase 2 clinical trial (HERMES) was carried out to evaluate the effects of RTX in a cohort of 104 RRMS patients over 48 weeks. A total of 69 patients were randomized to receive 1000 mg of intravenous RTX, while 35 patients were assigned to placebo on days 1 and 15, respectively. In the RTX group, the proportion of patients reporting clinical relapses was significantly lower compared to placebo at week 24 (14.5 vs. 34.3%, p = 0.02) and week 48 (20.3 vs. 40.0%, p = 0.04). Moreover, patients who received RTX had a pronounced reduction (91%) in the cumulative number of Gd-enhancing lesions at weeks 12, 16, 20, and 24 (p < 0.001), and these results were sustained at week 48 (p < 0.001) [31].
Phase II/III studies
RTX was also evaluated in a double-blind, placebo-controlled, multicentre phase II/III clinical trial investigating efficacy, safety and tolerability in 439 patients with PPMS (OLYMPUS) [37]. Patients were randomized to receive two intravenous infusions (2 weeks apart) of 1000 mg of RTX (n = 292) or placebo (n = 147) every 24 weeks, throughout 96 weeks. This study considered, as primary efficacy outcome measure, the time-to-confirmed disease progression, defined as a sustained Expanded Disability Status Scale (EDSS) increase of 1.0 point from baseline if the baseline EDSS was between 2.0 and 5.5 points, or an EDSS increase of 0.5 point if the baseline EDSS was > 5.5 points. MRI outcomes included the change in the volume of T2 lesions and in brain volume from baseline to week 96. Although this trial failed to demonstrate any significant effect on disease progression in patients with PPMS, sub-group analyses suggested a possible beneficial impact in younger patients (aged ≤ 51 years) with active inflammatory lesions, as denoted by Gd-enhancing lesions on cranial MRI. As for the secondary endpoint, patients treated with RTX had less increase in T2 lesion volume (p = 0.010), while the brain volume change was similar to placebo (p = 0.62).
In a Sweden class IV evidence study evaluating the safety and efficacy profile of RTX in a large multicentre cohort (n = 822), a remarkably low annualized relapse ratio of 0.044 and a constant median EDSS score over the follow-up period were observed in the RRMS population treated with a dose of 500–1000 mg of RTX every 6–12 months [36].
Based on these promising results, in 2010, another small MRI-blinded phase II trial for RTX was performed on 30 subjects with RRMS who had experienced a relapse within the past 18 months despite the use of an injectable DMT, and with at least 1 Gd-enhancing lesion on any of the 3 pre-treatment MRI scans [63]. The trial evaluated the safety, efficacy and tolerability of add-on RTX administered at a regimen of four times 375 mg/m2 intravenously (i.v.) weekly. Considering the primary endpoint concerning radiological disease activity, 74% of post-treatment MRI scans did not show any Gd-enhancing lesions compared with 26% at baseline (p < 0.0001). Importantly, the combination of RTX with standard DMTs was overall well tolerated, with few adverse effects. Several studies were performed to compare efficacy and safety profiles of RTX versus other DMTs.
Observational studies
The Swedish group has carried out a clinical comparison of RTX versus fingolimod (FTY), used as exit strategy in patients discontinuing natalizumab (NTZ) due to positive JCV status, in a multicentre, observational, cohort study of 256 RRMS patients at three MS centers in Sweden based upon the Swedish MS register [64]. This study demonstrated that patients who switched to RTX displayed significantly less MRI lesions (number of new Gd-enhancing lesions: 1% on RTX vs. 16% on FTY), clinical relapses (2% of patients relapsing on RTX vs 18% on FTY) and adverse events (5% on RTX vs 21% on FTY), along with a better overall drug survival compared with FTY, as a result of a reduced discontinuation rate and a better tolerability.
Efficacy and safety of RTX were also tested in an observational study from Southern Switzerland involving 453 patients, of which 82 (43 (52.4%) RRMS and 39 (47.6%) PPMS, undergoing a RTX induction regimen first and a maintenance regimen then [65]. Number of relapses, EDSS worsening, MRI lesion accrual and “evidence of disease activity” (EDA) status were the main outcomes. The most common DMTs used before RTX were NTZ and FTY and a comparison between therapies was performed. Compared with NTZ-treated patients, those treated with RTX showed reduced disease activity and a similar time to EDA (HR = 1.64, 95%CI 0.46–5.85, p = 0.44), further supporting a comparable efficacy between these two monoclonal antibodies. Moreover, no relapses occurred in patients who switched from NTZ to RTX because of positive JCV serology, as previously suggested in another study [64], highlighting that RTX may represent a valid treatment option in this context as well.
Moreover, in a recent real-world retrospective comparative Swedish study in newly diagnosed RRMS patients treated with a first line DMT, RTX showed to have a lower discontinuation rate and a better clinical efficacy compared to injectable DMTs and dimethyl-fumarate (DMF). In comparison with FTY and NTZ, relapse rates and Gd-enhancing lesions were numerically lower but did not reach statistical significance in all analyses [66].
Given the importance of starting treatment in RRMS patients as early as possible to reduce disability accumulation, RTX was evaluated as induction therapy in a study testing if RTX followed by glatiramer acetate (GA) monotherapy was more effective than GA alone for the treatment of active forms of RRMS [67]. The study results indicate that induction therapy with RTX followed by GA was superior to placebo induction and GA monotherapy in reaching NEDA (44.4% of participants in the R-GA arm vs 19.2% in the P-GA arm) in patients with active MS, although the effect appeared to be temporally limited.
Data about efficacy of RTX in MS population are summarized in Table 1.
Safety and tolerability
Infusion-related reactions (IRR)
The most common adverse events (AEs) described during the use of RTX in MS populations were the IRRs [38, 68]. In two randomized clinical trials, IRRs appeared in 67.1% (placebo: 23.1%) and 78.3% of patients (placebo: 40.0%) respectively, after the first infusion [31, 37]. IRRs levels decreased to those observed in placebo arms with subsequent infusions [31, 37]. Two smaller studies reported 25–26% of patients being affected by infusion reactions [68, 69]. The vast majority of these reactions are mild-to-moderate and include fever, rush, and chills. Other frequent IRRs include nausea, vomiting, pruritus, angioedema, throat irritation, bronchospasm, hypotension, rhinitis, urticaria, headache, myalgia, dizziness, and hypertension. The IRRs typically arise 30–120 min after initiating the first infusion and usually resolve with slow withdrawal, infusion discontinuation or symptomatic treatment. Premedication with paracetamol, prednisone and antihistaminic drugs may reduce the probability of infusion-related adverse effects [70, 71].
In a recent study, the incidence of IRRs was similar in RTX compared to OCR-treated patients, suggesting that switching between them is safe and that the mechanism behind the IRRs may be related at least in part to B-cell levels [72].
Allergic anaphylactic reactions are less commonly observed. The incidence of severe hypersensitivity reactions is < 10% in cancer patients treated with RTX [73] and they rarely necessitate treatment discontinuation. The risk can be reduced by pre-medication with corticosteroids, antihistamines and antipyretics [71].
Susceptibility to infections
In general, treatment with RTX, especially after longer treatment periods, is associated with an increased risk of infections [37, 74,75,76]. A recent register-based study in MS population reported that RTX treatment was associated with the highest rate of serious infections compared with NTZ, FTY, interferon beta, and GA [77]. In randomized clinical trials of RTX in PPMS, serious infections occurred in 4.5% of RTX-treated patients and in < 1.0% in the placebo [37], with no clear association to the number of infusions, which corroborates findings from large trials [78]. Other studies have reported an increased risk for different types of infections, mainly affecting respiratory and urinary systems [79,80,81,82,83].
Safety data of 56 patients treated with RTX for MS and neuromyelitis optica (NMO) did not report any signs of infection after 6 months from the last infusion in 53 patients (94.6%); a mild infection was reported in one patient and severe infections in two patients [82]. In particular, the infections/year per patient in this study was lower that one demonstrated in other neurological diseases [84].
Overall, there were similar incidences of infections in the placebo (71.4%) and RTX group (69.7%) in the first phase II study in MS; however, an increased incidence of urinary tract infections and sinusitis was found in RTX arm compared to placebo [31]. In a recent retrospective observational study of 84 relapsing MS treated with RTX, 36 infections were reported (among 53 non-infusion-related adverse events), of which four were serious, including a case of pneumonia with concomitant late-onset neutropenia [85].
Reactivation of tuberculosis, hepatitis and HIV have been reported in patients treated with anti-CD20 medications. Consequently, all patients should be screened for latent infections before starting treatment [86, 87]. Indeed, especially in endemically affected areas or populations, the risk of tuberculosis reactivation should be considered through specific prescreening and active surveillance with latent tuberculosis testing.
In patients treated with RTX for onco-hematological diseases, 67–85% of surface antigen of the hepatitis B virus (HBsAg)-positive patients not under antiviral therapy may experience a flare of hepatitis, due to reactivation of hepatitis B virus (HBV) in up to 25% of patients [88, 89]. Clinically, the reactivation of latent HBV infection can range from a subclinical increase of HBV DNA levels to elevated liver enzymes or more serious clinical pictures of acute severe hepatitis and liver failure, with a significant risk of death, ranging from 4 to 60% [89]. Hence, to prevent the risk of HBV reactivation, the U.S. Food and Drug Administration (FDA) has approved changes to the prescribing information of RTX and has added a new Boxed Warning information about the risk of reactivation of HBV infection [90]. FDA recommends to screen all patients for HBV infection before starting treatment with RTX by measuring HBsAg and hepatitis B core antibody (anti-HBc) [91]. These recommendations are not largely applied in all the European countries with some exceptions: for example, in Germany, before starting RTX, hepatitis testing is mandatory.
On the other hand, there are several case reports and retrospective investigations suggesting that RTX may induce viral replication in patients with HCV infection and onco-hematological diseases.
Although less frequently reported, other possibly RTX-associated infections include cytomegalovirus (CMV), herpes simplex virus (HSV), varicella zoster virus (VZV), and West Nile virus [76]. Particularly, CMV disease is an uncommon adverse event in RTX-treated patients, except in those with HIV infection or following allogenic transplant; indeed, there are several reports showing CMV disease in patients with hematologic malignancies treated with combined chemotherapy [92]. Cases of HSV and VZV reactivation have also been reported in patients administered RTX for lymphoma [76]. Cases of Pneumocystis jirovecii pneumonia has been reported among patients treated with RTX for aggressive B-cell lymphoma as part of specific chemotherapy regimens including other immunosuppressant such as cyclophosphamide [76, 93].
In the last years, the risk of progressive multifocal leukoencephalopathy (PML) by JCV has gained ever greater importance in the management of several of the current MS DMTs, in particular NTZ. PML cases have been reported in patients with lymphoma treated with RTX; however, the JC viral reactivation was due to the immunosuppression related to the disease [94]. Recent observational data from over 100,000 MS patients in the FDA Adverse Event Reporting System database have indicated that RTX-treated patients have an increased PML risk with an adjusted odds ratio = 3.22; 95% confidence interval (CI) =1.07–9.72 [95]. Recently, in the nationwide register-based cohort study conducted in Sweden, one case of RTX-related PML was described (the patient had switched from NTZ within 6 months before the diagnosis of PML). No deaths due to infections were recorded among the patients treated for MS [77].
The pathophysiology of RTX-associated PML remains unclear. Data about the JCV reactivation and development of PML in patients with congenital disorders of humoral immunity have suggested that B lymphocytes may play a role in the JCV immune responses [96]. However, the mechanisms underlying viral reactivation after RTX treatment could also involve the changes in T-lymphocyte activity after B-lymphocyte depletion due to the alteration of T-lymphocyte cytokine profiles [97]. In a review of PML cases among RTX-treated patients referring to the 1997–2008 FDA database, 52 patients with B-cell lymphoproliferative disorder, 2 patients with systemic lupus erythematosus, one patient with RA, one patient with an idiopathic autoimmune pancytopenia, and one patient with immune thrombocytopenia developed PML after RTX with a median time to death after PML diagnosis of 2.0 months and a mortality rate of 90% [94].
In MS settings, as showed in NTZ-treated patients, RTX-associated PML is more frequently seen in immune-compromised patients. A recent population-based Swedish study reported that after two decades of stable PML incidence of 0.026/100,000 person-years, the incidence has increased to 0.11 in 2011–2013, apparently related to the use of monoclonal antibodies therapies [98]. Considering that all patients treated with NTZ regardless of duration of therapy, the incidence of PML exceeded one in 250 (4.22/1000 with confidence intervals of 3.91–4.51/1000) [99]. The incidence of PML was very low in the first 12 months of infusion, though it has been observed within 8 months of drug initiation [100].
The incidence of PML due to RTX treatment is estimated to be one case per 32,000 [101]. However, even if there are no specific recommendations to screen patients for JCV prior to administration of RTX, it is important for clinicians to keep in mind that RTX may be associated to PML, and it is crucial to suspend therapy in the event of signs and symptoms suggestive of PML, and urgently carry out a specific workup in order to reduce morbidity and mortality.
Laboratory test abnormalities
One of the most reported laboratory values alterations in MS patients is hypogammaglobulinemia, especially with long-term RTX treatment [102]. The underlying mechanism for the development of hypogammaglobulinemia is unknown; however, it could be hypothesized that the diminished B-cell-secreted cytokines, such as BAFF [103] and interleukin 6 [104], may determine a reduction in the formation of plasma cells from precursors.
In an observational retrospective study on MS patients, 25 out of 822 patients (3%) had IgG levels below the lower normal reference value (< 6.2 g/L) at some point during treatment. There was, however, no difference in IgG levels between 500 and 1000 mg RTX [36]. In another study on NMO spectrum disorder (NMOSD) patients, 11 out of 15 patients (73%) developed hypogammaglobulinemia (IgG < 7 g/L) and three patients (20%) had severe hypogammaglobulinemia (< 4 g/L) [105]. In RA, 1.5–5.9% out of 1039 patients had hypogammaglobulinemia (IgG < 5 g/L) [106]. None of the studies assessing the safety profile of cumulative doses of RTX in RA demonstrated a higher risk of hypogammaglobulinemia with increasing numbers of RTX cycles [107, 108]. Moreover, low gamma-globulin baseline levels may be more relevant than treatment duration/cumulative RTX doses in predicting the development of hypogammaglobulinemia. Furthermore, sustained hypogammaglobulinemia (≥ 4 months) was associated with an increased risk for serious infections in open extension studies [105, 109]. For all these reasons, the measurement of total serum immunoglobulins before starting RTX and at least yearly during treatment is strongly recommended.
Late-onset neutropenia (LON) is defined as an absolute neutrophil count of < 1.5 × 10 to the power of 9/L occurring > 4 weeks following the last dose and was described as a rare complication during RTX treatment. About 5–27% of RTX-treated lymphoma patients may present LON [110]. A similar incidence is reported in patients with autoimmune diseases, with a higher infection rate during the neutropenic period [111]. Recently, it has been hypothesized that host-related factors, such as polymorphisms in FCGR3, may play a role in the development of LON [111].
The incidence of LON is likely to be much lower in MS patients. In a retrospective analysis of 385 patients treated with RTX for NMOSD, MOG-antibody-associated disease (MOGAD) and MS, LON was found in 10 (2.6%) patients; in particular, 16% were affected by MOGAD, 10% were NMOSD, and only 1.2% were MS patients [112].
Moreover, in an observational study, 1 of 90 MS patients developed agranulocytosis 3 months after the RTX infusion [113]. The mechanism by which RTX induces LON/agranulocytosis is still unknown; however, it is probably immune mediated, involving the anti-granulocyte antibody production and the neutrophil apoptosis by the large granular lymphocyte population [111].
Malignancies
Sporadic cases of malignancies in RTX-treated MS patients have been reported [83, 114]. In a large Swedish nationwide study, no higher risk of malignancies was found in RTX patients compared to the general population [115]. Moreover, in MS setting, a low frequency of all types of malignancies was reported, which did not differ significantly from the general population (26.6 [15.2–43.3] in RTX vs 28.9 [25.3–32.7] in the general population) [116].
Other AEs
Studies of RTX in MS and non-MS populations have reported several AEs involving cardiovascular system (i.e., angina pectoris, cardiac arrhythmias, heart failure and/or myocardial infarction), upper and lower airways (i.e., bronchospasm, chest pain, dyspnoea, cough, rhinitis), gastrointestinal system (i.e., vomiting, abdominal pain, dysphagia, stomatitis, constipation, dyspepsia, anorexia, reflux disease, abdominal pain, diarrhea, gastritis, pharyngolaryngeal pain), musculoskeletal and connective (i.e., myalgias, arthralgias, arthritis; hypertonia, pain), nervous system (paraesthesia, hypoesthesia, agitation, insomnia, vasodilatation, dizziness, anxiety, fatigue, neuropsychiatric disorders), skin (i.e., rash, itching, pruritus, alopecia) and endocrine system [31, 37, 45, 117,118,119,120,121,122,123,124,125].
Data about safety profile of RTX are summarized in Table 2.
Anti-drug antibodies (ADAs)
In recent years, as the evidence suggesting the high clinical efficacy of RTX have increased its off-label use in MS, several studies explored the degree of immunogenicity and, in particular, the possible interference of anti-drug antibodies (ADAs) with RTX efficacy. ADAs were identified in a third of MS patients treated with RTX, with a title higher than that recorded in other diseases treated with the same drug. Moreover, a lower frequency of ADAs was seen in PMS, compared to RRMS [69]. In two randomized trials, 24.6 and 29% of RRMS patients had developed ADAs at 48 weeks [31, 41]. In a study involving PPMS patients, 7.5% under RTX treatment versus 3.4% under placebo were found positive for ADAs 1 year after the last infusion [37]. Recently, a large cross-sectional real world using a more sensitive technique showed ADA in 38% of the RRMS patients and in 27% of PPMS [69].
However, the role of ADAs in treatment failure is uncertain. However, such a failure could relate to the higher level of immunological activity found in the earlier relapsing stage of the disease. Interestingly, a negative relationship found between the number of infusions and the frequency of ADAs suggests that the risk of developing ADAs and experiencing a lack of effect might diminish over time [69]. Nevertheless, outside of trials, the detection of ADAs could be technically difficult, poorly standardized and hard to apply in routine use.
Hence, although ADAs are present in a large proportion of RTX-treated patients, the existing evidence does not support a clinically relevant role for anti-RTX antibodies. Indeed, several studies have demonstrated that the presence of ADAs does not correlate with a higher incidence of infusion reactions, adverse events, or lack of clinical effect [31, 37, 41]. However, larger prospective studies are needed to confirm these data.
Vaccinations
Data from the oncology and rheumatology literature have shown that the response to vaccination may be ineffective in patients receiving RTX [126]. In particular, no protective serologic responses to a single-dose influenza A vaccination were achieved in lymphoma patients within 6 months of RTX treatment [127]. In another study evaluating the effects of RTX on the antibody and cellular responses to Streptococcus pneumonia polysaccharide vaccine and Haemophilus influenza type b (Hib) conjugate vaccine in patients with immune thrombocytopenia, antibody responses were impaired for at least 6 months after RTX treatment [128].
However, a study of patients with autoimmune blistering skin diseases previously treated with RTX reported a robust recall response to the seasonal influenza vaccination, comparable to healthy controls, both at a cellular and a serological level [129].
Notably, RTX is known to target all B cells, except the early precursor pro-B cells and long-lived plasma cells that do not express CD20, with little or no effect on pre-existing serum antibody titers produced by long-lived plasma cells, such as antibodies against childhood vaccines, including tetanus or meningitis [130]. Moreover, after the reduction of peripheral B-cell count typically lasting for 6–9 months, the recovery of total B-cell numbers generally occurs after 12 months; in particular, the repopulated B-cell compartment mostly includes naïve cells, while the depletion of memory B cells (MBCs) may persist in peripheral blood even 5 years after treatment [131].
According to these data, it is recommended to wait at least 6 months after RTX for vaccination, while patients should be advised to complete any required vaccinations at least 6 weeks prior to RTX initiation. Particularly, vaccinations for hepatitis B, pneumococcus, tetanus toxoid every 10 years and for influenza annually should be undertaken for patient considered for RTX therapy. Live-attenuated or live vaccines are not recommended during RTX treatment and until B-cell recovery since, currently, there are no sufficient data on the potential risk of vaccination with this kind of vaccines [132].
Pregnancy and breastfeeding
The European Medicines Agency (EMA) and the FDA recommend that pregnant women should not receive RTX infusion, unless the possible benefit outweighs the potential risk [133, 134]. Indeed, not enough data are available about B-cell levels in human neonates following maternal exposure to RTX. However, some infants born to mothers exposed to RTX during pregnancy presented transient B-cell depletion and lymphocytopenia [135].
Two cases of MS patients with a highly disabling disease form, and treated with RTX during pregnancy, were reported [136]. The first patient was treated with RTX during the third trimester, showing a clinical improvement over several weeks, while no complications were reported in her infant. The second patient received RTX in the early second trimester, also showing a clinical improvement and no complications. It has been suggested that RTX during pregnancy may be safe and effective when used in an appropriate context. Moreover, it has been demonstrated that RTX may be detected only in minimal concentrations in the breast milk of breastfeeding patients with MS (six samples of breast milk from four lactating patients) [137]. Thus, due to the lack of largest and longitudinal studies, the choice of resuming RTX treatment should be carefully evaluated in breastfeeding patients after risk/benefit considerations.
COVID-19 infection risk
Nowadays, the therapeutic management of MS patients during COVID-19 pandemic is one of the most relevant concerns, also raised by the possible role of white matter lesions as a virus reservoir, as for other corona-viruses [138].
MS patients are thought to be at higher risk than the normal population and the question of whether to continue or stop DMTs has been raised, in the absence of formal guidelines and a plethora of recommendations [139].
The severe pulmonary complications of COVID-19 infection, in particular the acute respiratory distress syndrome (ARDS), are demonstrated to be immune-mediated [140]. This is also confirmed by several data showing that immunosuppression (or at the least the moderate immunosuppression induced by DMTs), may exert a protective effect against the development of severe COVID-19 infection [141]. Indeed, the emerging knowledge of the biology of the ARDS to the COVID-19 and of the role of the immune mechanisms contributing to the disease have suggested that viral-specific CD8+ T-cell responses seem to eliminate the virus, while viral-specific antibodies could probably be able to prevent re-infection and create long-lasting immunity [142].
Nevertheless, data about the early phase of the pandemic highlighted that the most important independent risk factor associated with COVID-19 infection, aside from having close contact with people with upper respiratory symptoms, was the DMT category and in particular therapies with a B-cell depleting profile [143].
More recently, the Italian “Multiple Sclerosis and COVID-19” (MuSC-19) study also demonstrated an increased frequency of anti-CD20 treatments in COVID-19-infected MS patients compared with the expected frequency based on Italian data. Anti-CD20 were also associated with a more severe clinical course compared to other DMTs [144].
Accordingly, the Society of Italian Neurologists (SIN), the Association of British Neurologists (ABN) MS and Neuroimmunology Advisory Group practical guidance recommended to delay further infusions of anti-CD20 drugs, as that anti-CD20 therapies may probably increase the risk of COVID-19 infection [145].
Another study described the lethal disease course in two severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infected patients with hematological malignancies after RTX therapy. Complete B-cell depletion and the decrease of immunoglobulin G (IgG) level in both patients and the persistent viremia in blood samples could be correlated with increased morbidity, suggesting that B-cell function might be one important mechanism in resolving SARS-CoV-2 infection [146].
On the other hand, a case of complete recovery from COVID-19 has been reported in a MS patient treated with RTX for 3 years despite having a 0% B-lymphocyte count and not developing SARS-CoV-2 IgG antibodies. These observations shed light on possible immuno-mechanisms behind COVID-19 infection, considering that patients with mild disease symptoms have low antibody levels, whereby weak IgG responses have been associated with a faster virus clearance. [147].
Moreover, it was also hypothesized that while B-cell depletion may not necessarily expose people to severe SARS-CoV-2-related issues, it could inhibit protective immunity following infection and vaccination [148]. In fact, drug-induced B-cell subset inhibition would not influence innate and CD8 T-cell responses, which are central to SARS-CoV-2 elimination, nor the hypercoagulation and innate inflammation causing severe morbidity, but it would slow down the production of antibodies. The protective neutralizing antibody and vaccination responses are predicted to be blunted until naïve B cells repopulate, based on B-cell repopulation kinetics and vaccination responses from published RTX (NCT00676715) and unpublished OCR (NCT02545868) trial data [149].
Particularly referring to OCR, it has been recommended to consider the initiation of this drug only if a high-efficacy drug is required and the use of NTZ is contraindicated. During OCR treatment, it has been recommended to delay further infusions [145]. Despite these stringent recommendations, published data have contradicted the assumption that patients treated with immunosuppressive drugs could be at risk for severe complications of COVID-19. Indeed, a case report of an OCR-treated PPMS patient who developed COVID-19 showed that, despite complete B-cell depletion, the infection has been resolved in few days after hospitalization, and no new symptoms occurred after 14 days [141]. A very recent study analyzed the frequency and severity of COVID-19 in patients treated with anti-CD20 in a tertiary hospital in Madrid, Spain, one of the most affected countries by the COVID-19 pandemic [150]. In this study, COVID-19 infection was reported in 9 (15%) cases in the whole population, 7 (12.9%) in patients treated with RTX, and 2 (33.3%) in patients on OCR, with no apparent relationship with the time of the therapy administration. All patients reporting COVID-19 did not show serious complications and only one of them required hospital admission. Thus, the authors suggested that patients treated with anti-CD20 could not be particularly at risk for severe complications of COVID-19.
Cost-effectiveness
RTX used at a single dose of 500 or 1000 mg twice yearly results in lower treatment costs, even in comparison to platform MS therapies. However, the status of being an off-label drug, subjected to variable insurance regulations in countries other than Sweden, remains as a potential barrier for its use in MS patients [151].
A study showed that in 2015 the use of RTX for the treatment of RA at the dosage of two infusions of 1000 mg doses totally given 2 weeks apart every 6 months, costs about $30,000 annually [152]. Although costs may vary by region and may change over time, the current cost for the off-label use of RTX (1000 mg spread over 2 doses) in Italy is approximately €1,400. In Sweden, the cost of a yearly RTX course including two doses of 500 mg costs is about €2,400 [152, 153].
Thus, RTX costs seem to be well below the average wholesale acquisition costs of the standard DMTs, including the first-line drugs, which are currently estimated to cost approximately $70,000–$80,000 or more annually [16, 152].
An American pharmacoeconomic study demonstrated that the off-label use of RTX is less expensive than most of the currently available FDA-approved DMTs, confirming the results from randomized trials and observational studies showing that RTX infusions of 1–2 g annually are cost effective, with a Weighted Average Cost of Capital (WACC) of $16,704 per 1000 mg [151, 154].
In addition, even if the stated price of the recently approved OCR is within the range or less than other current approved DMTs with an annual cost of twice-a-year infusions of $65,000, it remains significantly more expensive than RTX [155].
Finally, with a price ranging from 15 to 30% lower than the originator molecule (MabThera®), the development of RTX biosimilars may also significantly contribute to cost savings for healthcare systems [156, 157].
Dosing regimens
Although the US Food and Drug Administration (FDA) approved RTX only for use in non-Hodgkin’s lymphoma (NHL) and RA, it is commonly used as off-label treatment for severe MS. However, due to the absence of formal head-to-head trials of therapy regimen comparisons for RTX in MS, there are neither consensus nor treatment guidelines on dosing regimens.
At the beginning, basic dosing and interval strategies for RTX in MS have been adopted from RTX usage in oncology and RA, giving 375 mg once weekly for 4 weeks or two infusions of 500–1000 mg given a fortnight apart [158]. Then, in the clinical trials RTX has been administered as 1000 mg i.v. twice 2 weeks apart in patients with RRMS [31] and as 1000 mg i.v. twice 2 weeks apart every 24th week in four cycles in patients with PMS [37]. Nowadays, in European countries (and for most of the neuro-immunological diseases including MS) RTX doses of 500 mg are typically administered every 6–9 months. Indeed, the currently used dosing strategy in Sweden consists of one i.v. dose of 500–1000 mg RTX every 6 months [66, 69, 159], since a similar degree of CD19+ B-cell suppression at 6 months after infusion has been observed with 500 and 1000 mg [60].
High-dose RTX therapy often results in B-cell depletion for approximately 12 months [160]. However, the development of PML in patients treated could be a fearful complication, so it remains unclear whether high doses of RTX are safe or necessary for sustained clinical efficacy in inflammatory diseases [161, 162]. In 2011, an open-label study investigated the effects of a low dose of RTX on B cells CD19 level in RRMS patients [163]. The study design was based on the treatment of 12 patients who were refractory to conventional DMTs with methylprednisolone 500 mg intravenously followed by a 100 mg infusion of RTX. The findings suggested that a single 100 mg infusion of RTX adequately depleted peripheral B cells for at least 6 weeks, with CD19+ lymphocytes recovering at levels above the 25% of baseline at 12 weeks (27%) and at 24 weeks (45%). During the follow-up, there was a reduction in the number of clinical relapses (21 in the year prior to first infusion vs 7 in the year following) and a fewer number of baseline cumulative Gd-enhanced lesions on brain MRI (23 at baseline vs 3,2, and 0 at weeks 12, 24, and 52, respectively). Thus, the authors raised the question whether the use of minimal doses of RTX could maintain the clinical effects in the treatment of RRMS through a sustained B-cell depletion [163].
Growing evidence supports the hypothesis that the progressive phase of MS might be associated with intrathecal compartmentalization of inflammatory cells; thus, several studies investigated the effect of intrathecal administration of immunosuppressants as a new therapeutic approach in MS [164]. A single case report of intrathecal use of RTX was performed in 2014 to evaluate the central and peripheral effects of repeated intrathecal administrations of RTX in a patient with severe PMS [165]. The investigators demonstrated the marked reduction of peripheral CD20+ B cells, several central pro-inflammatory cytokines and markers of neurodegeneration, with no effect on oligoclonal bands.
An open-label phase 1b study on the efficacy of intrathecal administration of RTX for the treatment of PMS was also conducted, which involved the monitoring B lymphocytes in peripheral blood and CSF up to 1 year post-treatment [166]. This study demonstrated that the intrathecal administration of ultra-low doses of RTX was able to completely deplete peripheral B lymphocytes, thus confirming the potential effects in both the CNS and systemic compartments. A randomized placebo-controlled phase II trial of combined IV and intrathecally administered RTX in patients with SPMS is currently ongoing (ClinicalTrials.gov Identifier: NCT01212094).
It has been shown that an almost complete B-cell depletion occurs within a fortnight of infusion, usually persisting for 6–12 months. For this reason, treatment courses have commonly been repeated at regular six-month intervals. However, the initial RTX dose required to achieve B-cell depletion and the time to B-cell repopulation may considerably vary [167] with a reported prolonged B-cell depletion lasting over 3 years following a single dose of RTX [168].
Dosing and interval strategies for RTX commonly used in onco-hematology setting and for RA treatment have been also applied in MS patients [158]. In randomized clinical trials, RTX has been administered as 1000 mg intravenous twice 2 weeks apart in RRMS [31] and 1000 mg intravenous twice 2 weeks apart every 24th week in four cycles in PPMS patients [37]. Two studies have measured blood CD19+ B cells to schedule the RTX re-infusion [113, 169]. It has been suggested that monitoring circulating memory B cells (CD19+ and CD27+) could be a viable strategy to control relapsing NMO [170], which may be similarly pertinent to MS, in order to schedule a personalized treatment regimen [32]. These considerations explain why in Sweden the most common approach consists of one intravenous dose of 500 mg RTX every 6 months [36, 64, 66, 69, 159], since it was demonstrated that this regimen can determine a CD19+ B-cell suppression at 6 months comparable to the 1000 mg dose one [60]. On the other side, the refill of B cells has a significant individual variability. In a study involving 439 PPMS patients, about 40% of them had recovered peripheral B cells 48 weeks after their last dose (in the 1000 mg intravenous twice 2 weeks apart regimen) [37]. In another study 26 RRMS patients had a reconstitution to a mean of 35% of baseline counts by week 72 (48 weeks after 2 × 1000 mg 2 weeks apart), in particular with a greater amount of naïve B cells rather than memory B cells [41], producing less pro-inflammatory and more regulatory cytokines [58]. Dosing regimens more frequently used in MS population are summarized in Table 3.
Biosimilars
RTX’s patent expired in Europe in February 2013 and in the US in September 2016 [171]. Since 2015, FDA and EMA have approved several biosimilars of RTX, such as while other biosimilars are to date in the pipeline [172, 173].
CT-P10 (Truxima®) is the first biosimilar approved for use in all indications reported for the originator RTX, including follicular lymphoma (FL), diffuse large B-cell non-Hodgkin’s lymphoma, chronic lymphocytic leukemia, RA, granulomatosis with polyangiitis and microscopic polyangiitis. CT-P10 shares similar physicochemical and pharmacodynamic characteristics, comparable tolerability, immunogenicity and safety profiles with reference RTX, and switching to CT-P10 has no impact on safety or efficacy [174].
Another bioequivalent, GP2013 (Rixathon®), has been compared to the originator RTX in patients with a diagnosis of FL and active RA and has shown high similarity from a biochemical point of view [175,176,177].
Since a biosimilar is not the exact copy of the originator, its efficacy and safety may significantly differ. For these reasons, a recent study tested the equivalence of the RTX biosimilar CT-P10 and its originator RTX used for MS treatment in terms of efficacy, safety, and tolerability [178]. Concerning efficacy, similar CD19+ lymphocyte depletion, relapse rate and evolution of MRI activity were observed between the two groups of treatment. Results suggest that CT-P10 could represent a relatively cheaper and safe therapeutic alternative and could improve access to a highly efficient therapy for MS in low- or middle-income countries. Recently, a prospective study demonstrated the equivalence of the RTX biosimilar Truxima® compared to its originator MabThera® in terms of efficacy, safety, and tolerability in a MS population [178].
Clinical use in other neurological disorders
RTX has been approved for the treatment of B-cell lymphomas (i.e., non-Hodgkin’s lymphoma, chronic lymphatic leukemia) in 1997. Nowadays, RTX is being deployed for a multitude of chronic inflammatory diseases apart from MS, representing an attractive alternative to conventional immunomodulatory medications because of growing evidence of its efficacy and tolerability.
A recent review has summarized indications and evidence for RTX use in neurological disorders depending on the type and course of the disease [179].
RTX is usually adopted as a second-line acute therapy in anti-NMDAR encephalitis [180] and in other autoimmune encephalitis [181] to maximise neurological recovery. The most commonly used dosing regimen is 375 mg/m2 weekly for four doses. Favorable outcomes as acute therapy have also been reported in small case series of patients with diagnosis of primary angiitis of the CNS [182].
Use of RTX in NMOSD is supported by numerous studies demonstrating consistent reductions in ARR [183, 184]. No consensus exists about the exact efficient dose. Usually, dosing regimen of 375 mg/m2 weekly for four cycles is adopted, but in small series of patients, NMOSD doses as low as 100 mg weekly for 3–4 weeks have been successfully used [185, 186].
Two randomized controlled trials have shown non-inferiority of RTX to cyclophosphamide in inducing remission in patients with ANCA-associated vasculitis [187]. In particular, RTX in association with steroids is recommended by the National Institute for Health and Care Excellence (NICE) as an option for inducing remission of severe disease when cyclophosphamide has failed or is contraindicated [188].
The treatment of immune-mediated peripheral neuropathies with RTX showed modest benefits; however, there may be circumstances in which RTX could be helpful, as in patients with diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) with an inadequate response to conventional therapy (corticosteroids, intravenous immunoglobulin and plasma exchange) [181].
Finally, evidences of benefits with RTX use in patients with refractory myasthenia gravis have been showed in some retrospective case series, with much greater efficacy in muscle-specific tyrosine kinase (MuSK)-associated than acetylcholine receptor (AChR)-associated forms [189, 190].
Discussion
An increasing body of evidence supports the high efficacy and the low drug discontinuation rate of B-cell-depleting anti-CD20-antibody RTX for the treatment of MS. Long-term observation of RTX therapy in RA and in NMOSD suggests that RTX is highly effective, safe and well tolerated [38, 41, 70, 81, 85]. Two large randomized clinical trials and results from large real-world studies have confirmed its safety and efficacy in both RRMS and PMS [31, 37]. Furthermore, the evidence from long-term use of RTX in other clinical conditions, such as RA, has supported its favorable safety profile. However, it should be noted that careful monitoring is needed, in particular considering the infection risk. For this reason, it is recommended to measure total serum immunoglobulins before starting RTX and during the follow-up to carry out specific vaccination at least 6 weeks before starting RTX treatment.
In addition to the efficacy and safety data, the off-label use of RTX is less expensive than most of the currently available FDA-approved DMTs, confirming the results from randomized trials and observational studies showing that RTX infusions of 1–2 g annually are cost-effective [151, 154]. However, although the annual cost of RTX is lower than that of most MS drugs, its access is not universal because its cost remains high for some patients and healthcare services. In that respect, the introduction of cheaper biosimilars may further reduce costs and also represents a highly efficient therapeutic option for MS in low- or middle-income countries. Moreover, it is unknown whether the introduction of new anti-CD20 monoclonal antibodies, particularly those SC administered such as ofatumumab, could also impact the price competition.
The encouraging results emerging from the trials and observational studies have raised the question if RTX may represent an acceptable and valid alternative to OCR. This was also highlighted by the significant disparity in cost between these two anti-CD20 drugs, with RTX being markedly less expensive. Notably, RTX use for MS is variably covered depending on the health system in the European countries and according to the insurance companies in USA. Nevertheless, it should be considered that RTX has a more pronounced immunogenicity with a higher production of ADAs compared to OCR, since the unique molecular structure of OCR allows eliminating B cells though a more direct path. Thus, in the absence of head-to-head trials, the choice of RTX or OCR should be made carefully on the basis of efficacy and safety issues.
A further knowledge gap is represented by the use of RTX for the treatment of PPMS; indeed, even if data showed that younger PPMS patients, particularly those with inflammatory lesions, may benefit from RTX, the effectiveness of RTX in PPMS needs to be further explored also taking into account specific clinical variables, such as age, disease duration, comorbidities and evidence of inflammatory activity defined by clinical relapses, progression rate and MRI data [37].
Another open issue is the lack of no formal dose-finding trials of different RTX therapy regimens. Thus, further studies are needed to optimize dosing regimen, to identify the administration interval, possibly individualized by adjustment to immunological parameters and disease activity. Moreover, in the light of the higher incidence of infections reported in the trials, future researches may investigate if a reduced dosing schedule may be able to reduce the risk of infections, preserving the efficacy and also the favorable safety profile [31, 37]. In our MS center of the University Hospital of Catania, MS and NMO patients are treated with 1000 mg i.v. twice 2 weeks apart and the RTX re-treatment is usually scheduled based on the refill of the CD20+ and CD19+ lymphocytes (over 1%). However, the B-cell repopulation may individually vary. Hence, dose-finding studies should be carried out to identify “early re-populators” at risk of disease relapse in order to retreat them before disease progression and avoid the overtreatment of patients with sustained B-cell depletion over time [167].
To date, a major concern during the COVID-19 pandemic has been represented by the use of immunosuppressive therapies for MS treatment. This was particularly true for Italy, as it was the first European country to face the COVID-19 pandemic. Several data have shown encouraging results, suggesting that immunosuppression, or at the least the moderate immunosuppression induced by DMTs, may have a protective effect against the development of severe COVID-19 infection [141].
This is not surprising, as ARDS are demonstrated to be immune mediated [140]. Besides, evidences on the immunopathogenesis of ARDS due to the COVID-19 infection have suggested that viral-specific CD8+ T-cell responses are involved in the virus clearance [142]. Notably, most MS-related DMTs, probably except alemtuzumab, do not seem to target the innate immune system, whereas only few of them have any significant long-term effect on CD8+ T-cell counts. More importantly, MS DMTs in general do not usually target the immature B-cell development, thus allowing antibody production preventing (re)infection, as well as response to vaccines. In the light of these findings, numerous trials with immunosuppressive therapies, such as FTY (NCT04280588), Tocilizumab (NCT04331795), Anakinra (NCT04341584) and Emapalumab (NCT04324021) are currently being tested as treatments for COVID-19-associated ARDS [191].
However, the recent Italian report of higher risk of COVID infection in MS patients treated with anti-CD20 drugs has risen some concerns about the use of these class of drugs [144]. In this study, no association between time to last infusion of OCR and COVID-19 risk was found, supporting the hypothesis that the immunological effects of these drugs may last longer than 6 months. Similar to the NTZ extended-dose strategy for reducing PML risk, this may suggest that different RTX schedules, reducing the frequency of dosing, or adjusting it according to the monitoring of B-cell refill, may maintain efficacy while limiting the risk of infection.
Conclusion
Despite the status of being an off-label drug, and therefore being subjected to variable regulations in countries other than Italy, RTX is a valid treatment option for MS patients considering the growing evidence about the high efficacy and the safety profile. In this time of pandemic crisis, the recent concern raised by the recent findings showed the higher risk of COVID-19 infection in patients treated with anti-CD20 drugs should not be ignored, especially for naïve patients, during treatment-decision making. This new clinical insight needs to be confirmed in other autoimmune diseases. The analysis of the increasing amount of real-world data being collected in several registries may shed new light on the pathophysiology of the COVID-19 infection and confirm or reject the hypothesis regarding the higher susceptibility to develop severe COVID-19 of MS patients treated with B-cell depleting therapies.
References
Segal BM (2019) The diversity of Encephalitogenic CD4+ T cells in multiple sclerosis and its animal models. J Clin Med 8(1):120. https://doi.org/10.3390/jcm8010120
Dendrou CA, Fugger L, Friese MA (2015) Immunopathology of multiple sclerosis. Nat Rev Immunol 15(9):545–558. https://doi.org/10.1038/nri3871
Garg N, Smith TW (2015) An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav 5(9):e00362. https://doi.org/10.1002/brb3.362
Wekerle H (2017) B cells in multiple sclerosis. Autoimmunity 50(1):57–60. https://doi.org/10.1080/08916934.2017.1281914
Krumbholz M, Derfuss T, Hohlfeld R, Meinl E (2012) B cells and antibodies in multiple sclerosis pathogenesis and therapy. Nat Rev Neurol 8(11):613–623. https://doi.org/10.1038/nrneurol.2012.203
Li R, Patterson KR, Bar-Or A (2018) Reassessing B cell contributions in multiple sclerosis. Nat Immunol 19(7):696–707. https://doi.org/10.1038/s41590-018-0135-x
Wanleenuwat P, Iwanowski P (2019) Role of B cells and antibodies in multiple sclerosis. Mult Scler Relat Disord 36:101416. https://doi.org/10.1016/j.msard.2019.101416
Lisak RP, Benjamins JA, Nedelkoska L, Barger JL, Ragheb S, Fan B, Ouamara N, Johnson TA, Rajasekharan S, Bar-Or A (2012) Secretory products of multiple sclerosis B cells are cytotoxic to oligodendroglia in vitro. J Neuroimmunol 246(1–2):85–95. https://doi.org/10.1016/j.jneuroim.2012.02.015
Lassmann H (2018) Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front Immunol 9:3116. https://doi.org/10.3389/fimmu.2018.03116
Jelcic I, Al Nimer F, Wang J, Lentsch V, Planas R, Jelcic I, Madjovski A, Ruhrmann S, Faigle W, Frauenknecht K, Pinilla C, Santos R, Hammer C, Ortiz Y, Opitz L, Gronlund H, Rogler G, Boyman O, Reynolds R, Lutterotti A, Khademi M, Olsson T, Piehl F, Sospedra M, Martin R (2018) Memory B cells activate brain-homing autoreactive CD4(+) T cells in multiple sclerosis. Cell 175(1):85–100. https://doi.org/10.1016/j.cell.2018.08.011
Lovato L, Willis SN, Rodig SJ, Caron T, Almendinger SE, Howell OW, Reynolds R, O’Connor KC, Hafler DA (2011) Related B cell clones populate the meninges and parenchyma of patients with multiple sclerosis. Brain 134(Pt 2):534–541. https://doi.org/10.1093/brain/awq350
Baranzini SE, Jeong MC, Butunoi C, Murray RS, Bernard CC, Oksenberg JR (1999) B cell repertoire diversity and clonal expansion in multiple sclerosis brain lesions. J Immunol 163(9):5133–5144
Owens GP, Ritchie AM, Burgoon MP, Williamson RA, Corboy JR, Gilden DH (2003) Single-cell repertoire analysis demonstrates that clonal expansion is a prominent feature of the B cell response in multiple sclerosis cerebrospinal fluid. J Immunol 171(5):2725–2733. https://doi.org/10.4049/jimmunol.171.5.2725
Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J (1998) Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Investig 102(5):1045–1050. https://doi.org/10.1172/JCI3568
Magliozzi R, Howell O, Vora A, Serafini B, Nicholas R, Puopolo M, Reynolds R, Aloisi F (2007) Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130(Pt 4):1089–1104. https://doi.org/10.1093/brain/awm038
Greenfield AL, Hauser SL (2018) B-cell therapy for multiple sclerosis: entering an era. Ann Neurol 83(1):13–26. https://doi.org/10.1002/ana.25119
Disanto G, Morahan JM, Barnett MH, Giovannoni G, Ramagopalan SV (2012) The evidence for a role of B cells in multiple sclerosis. Neurology 78(11):823–832. https://doi.org/10.1212/WNL.0b013e318249f6f0
Obermeier B, Lovato L, Mentele R, Bruck W, Forne I, Imhof A, Lottspeich F, Turk KW, Willis SN, Wekerle H, Hohlfeld R, Hafler DA, O’Connor KC, Dornmair K (2011) Related B cell clones that populate the CSF and CNS of patients with multiple sclerosis produce CSF immunoglobulin. J Neuroimmunol 233(1–2):245–248. https://doi.org/10.1016/j.jneuroim.2011.01.010
Bankoti J, Apeltsin L, Hauser SL, Allen S, Albertolle ME, Witkowska HE, von Budingen HC (2014) In multiple sclerosis, oligoclonal bands connect to peripheral B-cell responses. Ann Neurol 75(2):266–276. https://doi.org/10.1002/ana.24088
Elliott C, Lindner M, Arthur A, Brennan K, Jarius S, Hussey J, Chan A, Stroet A, Olsson T, Willison H, Barnett SC, Meinl E, Linington C (2012) Functional identification of pathogenic autoantibody responses in patients with multiple sclerosis. Brain 135(Pt 6):1819–1833. https://doi.org/10.1093/brain/aws105
Keegan M, Konig F, McClelland R, Bruck W, Morales Y, Bitsch A, Panitch H, Lassmann H, Weinshenker B, Rodriguez M, Parisi J, Lucchinetti CF (2005) Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 366(9485):579–582. https://doi.org/10.1016/S0140-6736(05)67102-4
Berger T, Rubner P, Schautzer F, Egg R, Ulmer H, Mayringer I, Dilitz E, Deisenhammer F, Reindl M (2003) Antimyelin antibodies as a predictor of clinically definite multiple sclerosis after a first demyelinating event. N Engl J Med 349(2):139–145. https://doi.org/10.1056/NEJMoa022328
Rouwette M, Somers K, Govarts C, De Deyn PP, Hupperts R, Van Wijmeersch B, De Jong BA, Verbeek MM, Van Pesch V, Sindic C, Villar LM, Alvarez-Cermeno JC, Stinissen P, Somers V (2012) Novel cerebrospinal fluid and serum autoantibody targets for clinically isolated syndrome. J Neurochem 123(4):568–577. https://doi.org/10.1111/jnc.12005
Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F (2004) Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 14(2):164–174. https://doi.org/10.1111/j.1750-3639.2004.tb00049.x
Crawford A, Macleod M, Schumacher T, Corlett L, Gray D (2006) Primary T cell expansion and differentiation in vivo requires antigen presentation by B cells. J Immunol 176(6):3498–3506. https://doi.org/10.4049/jimmunol.176.6.3498
Aung LL, Balashov KE (2015) Decreased Dicer expression is linked to increased expression of co-stimulatory molecule CD80 on B cells in multiple sclerosis. Mult Scler 21(9):1131–1138. https://doi.org/10.1177/1352458514560923
Krumbholz M, Meinl E (2014) B cells in MS and NMO: pathogenesis and therapy. Semin Immunopathol 36(3):339–350. https://doi.org/10.1007/s00281-014-0424-x
Ireland SJ, Guzman AA, O’Brien DE, Hughes S, Greenberg B, Flores A, Graves D, Remington G, Frohman EM, Davis LS, Monson NL (2014) The effect of glatiramer acetate therapy on functional properties of B cells from patients with relapsing-remitting multiple sclerosis. JAMA Neurol 71(11):1421–1428. https://doi.org/10.1001/jamaneurol.2014.1472
Lossius A, Johansen JN, Torkildsen O, Vartdal F, Holmoy T (2012) Epstein-Barr virus in systemic lupus erythematosus, rheumatoid arthritis and multiple sclerosis-association and causation. Viruses 4(12):3701–3730. https://doi.org/10.3390/v4123701
Endriz J, Ho PP, Steinman L (2017) Time correlation between mononucleosis and initial symptoms of MS. Neurol Neuroimmunol Neuroinflamm 4(3):e308. https://doi.org/10.1212/NXI.0000000000000308
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH, HERMES Trial Group (2008) B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358(7):676–688. https://doi.org/10.1056/NEJMoa0706383
Baker D, Marta M, Pryce G, Giovannoni G, Schmierer K (2017) Memory B cells are major targets for effective immunotherapy in relapsing multiple sclerosis. EBioMedicine 16:41–50. https://doi.org/10.1016/j.ebiom.2017.01.042
Cragg MS, Walshe CA, Ivanov AO, Glennie MJ (2005) The biology of CD20 and its potential as a target for mAb therapy. Curr Dir Autoimmun 8:140–174. https://doi.org/10.1159/000082102
Lehmann-Horn K, Kinzel S, Weber MS (2017) Deciphering the role of B cells in multiple sclerosis-towards specific targeting of pathogenic function. Int J Mol Sci 18(10):2048. https://doi.org/10.3390/ijms18102048
Ontaneda D, Hyland M, Cohen JA (2012) Multiple sclerosis: new insights in pathogenesis and novel therapeutics. Annu Rev Med 63:389–404. https://doi.org/10.1146/annurev-med-042910-135833
Salzer J, Svenningsson R, Alping P, Novakova L, Bjorck A, Fink K, Islam-Jakobsson P, Malmestrom C, Axelsson M, Vagberg M, Sundstrom P, Lycke J, Piehl F, Svenningsson A (2016) Rituximab in multiple sclerosis: a retrospective observational study on safety and efficacy. Neurology 87(20):2074–2081. https://doi.org/10.1212/WNL.0000000000003331
Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, Hauser S, Waubant E, Vollmer T, Panitch H, Zhang J, Chin P, Smith CH, OLYMPUS trial group (2009) Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 66(4):460–471. https://doi.org/10.1002/ana.21867
D’Amico E, Zanghi A, Chisari CG, Fermo SL, Toscano S, Arena S, Patti F, Zappia M (2019) Effectiveness and safety of Rituximab in demyelinating diseases spectrum: an Italian experience. Mult Scler Relat Disord 27:324–326. https://doi.org/10.1016/j.msard.2018.09.041
Ternant D, Bejan-Angoulvant T, Passot C, Mulleman D, Paintaud G (2015) Clinical pharmacokinetics and pharmacodynamics of monoclonal antibodies approved to treat rheumatoid arthritis. Clin Pharmacokinet 54(11):1107–1123. https://doi.org/10.1007/s40262-015-0296-9
Ng CM, Bruno R, Combs D, Davies B (2005) Population pharmacokinetics of rituximab (anti-CD20 monoclonal antibody) in rheumatoid arthritis patients during a phase II clinical trial. J Clin Pharmacol 45(7):792–801. https://doi.org/10.1177/0091270005277075
Bar-Or A, Calabresi PA, Arnold D, Markowitz C, Shafer S, Kasper LH, Waubant E, Gazda S, Fox RJ, Panzara M, Sarkar N, Agarwal S, Smith CH (2008) Rituximab in relapsing-remitting multiple sclerosis: a 72-week, open-label, phase I trial. Ann Neurol 63(3):395–400. https://doi.org/10.1002/ana.21363
Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, Rocca MA (2018) Multiple sclerosis. Nat Rev Dis Primers 4(1):43. https://doi.org/10.1038/s41572-018-0041-4
Hagens MH, Killestein J, Yaqub MM, van Dongen GA, Lammertsma AA, Barkhof F, van Berckel BN (2018) Cerebral rituximab uptake in multiple sclerosis: a (89)Zr-immunoPET pilot study. Mult Scler 24(4):543–545. https://doi.org/10.1177/1352458517704507
Petereit HF, Rubbert-Roth A (2009) Rituximab levels in cerebrospinal fluid of patients with neurological autoimmune disorders. Mult Scler 15(2):189–192. https://doi.org/10.1177/1352458508098268
Komori M, Lin YC, Cortese I, Blake A, Ohayon J, Cherup J, Maric D, Kosa P, Wu T, Bielekova B (2016) Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann Clin Transl Neurol 3(3):166–179. https://doi.org/10.1002/acn3.293
Rubenstein JL, Fridlyand J, Abrey L, Shen A, Karch J, Wang E, Issa S, Damon L, Prados M, McDermott M, O’Brien J, Haqq C, Shuman M (2007) Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol 25(11):1350–1356. https://doi.org/10.1200/JCO.2006.09.7311
Schlachetzki F, Zhu C, Pardridge WM (2002) Expression of the neonatal Fc receptor (FcRn) at the blood-brain barrier. J Neurochem 81(1):203–206. https://doi.org/10.1046/j.1471-4159.2002.00840.x
Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, Ignoffo R, Aldape K, Shen A, Lee D, Grillo-Lopez A, Shuman MA (2003) Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood 101(2):466–468. https://doi.org/10.1182/blood-2002-06-1636
Topping J, Dobson R, Lapin S, Maslyanskiy A, Kropshofer H, Leppert D, Giovannoni G, Evdoshenko E (2016) The effects of intrathecal rituximab on biomarkers in multiple sclerosis. Mult Scler Relat Disord 6:49–53. https://doi.org/10.1016/j.msard.2016.01.001
Klein C, Lammens A, Schafer W, Georges G, Schwaiger M, Mossner E, Hopfner KP, Umana P, Niederfellner G (2013) Epitope interactions of monoclonal antibodies targeting CD20 and their relationship to functional properties. MAbs 5(1):22–33. https://doi.org/10.4161/mabs.22771
Morschhauser F, Marlton P, Vitolo U, Linden O, Seymour JF, Crump M, Coiffier B, Foa R, Wassner E, Burger HU, Brennan B, Mendila M (2010) Results of a phase I/II study of ocrelizumab, a fully humanized anti-CD20 mAb, in patients with relapsed/refractory follicular lymphoma. Ann Oncol 21(9):1870–1876. https://doi.org/10.1093/annonc/mdq027
Pawluczkowycz AW, Beurskens FJ, Beum PV, Lindorfer MA, van de Winkel JG, Parren PW, Taylor RP (2009) Binding of submaximal C1q promotes complement-dependent cytotoxicity (CDC) of B cells opsonized with anti-CD20 mAbs ofatumumab (OFA) or rituximab (RTX): considerably higher levels of CDC are induced by OFA than by RTX. J Immunol 183(1):749–758. https://doi.org/10.4049/jimmunol.0900632
Robak T, Robak E (2011) New anti-CD20 monoclonal antibodies for the treatment of B-cell lymphoid malignancies. BioDrugs 25(1):13–25. https://doi.org/10.2165/11539590-000000000-00000
Maloney DG (2007) Follicular NHL: from antibodies and vaccines to graft-versus-lymphoma effects. Hematology Am Soc Hematol Educ Program 2007:226–232. https://doi.org/10.1182/asheducation-2007.1.226
Maloney DG, Grillo-Lopez AJ, Bodkin DJ, White CA, Liles TM, Royston I, Varns C, Rosenberg J, Levy R (1997) IDEC-C2B8: results of a phase I multiple-dose trial in patients with relapsed non-Hodgkin’s lymphoma. J Clin Oncol 15(10):3266–3274. https://doi.org/10.1200/JCO.1997.15.10.3266
Piccio L, Naismith RT, Trinkaus K, Klein RS, Parks BJ, Lyons JA, Cross AH (2010) Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch Neurol 67(6):707–714. https://doi.org/10.1001/archneurol.2010.99
Bar-Or A, Fawaz L, Fan B, Darlington PJ, Rieger A, Ghorayeb C, Calabresi PA, Waubant E, Hauser SL, Zhang J, Smith CH (2010) Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann Neurol 67(4):452–461. https://doi.org/10.1002/ana.21939
Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, Moore CS, Michel L, Althekair F, Rajasekharan S, Gommerman JL, Prat A, Fillatreau S, Bar-Or A, Canadian BciMST (2015) Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med 7(310):310ra166. https://doi.org/10.1126/scitranslmed.aab4176
Graves J, Vinayagasundaram U, Mowry EM, Matthews IR, Marino JA, Cheng J, Waubant E (2014) Effects of rituximab on lymphocytes in multiple sclerosis and neuromyelitis optica. Mult Scler Relat Disord 3(2):244–252. https://doi.org/10.1016/j.msard.2013.10.003
Palanichamy A, Jahn S, Nickles D, Derstine M, Abounasr A, Hauser SL, Baranzini SE, Leppert D, von Budingen HC (2014) Rituximab efficiently depletes increased CD20-expressing T cells in multiple sclerosis patients. J Immunol 193(2):580–586. https://doi.org/10.4049/jimmunol.1400118
Olsson T, Barcellos LF, Alfredsson L (2017) Interactions between genetic, lifestyle and environmental risk factors for multiple sclerosis. Nat Rev Neurol 13(1):25–36. https://doi.org/10.1038/nrneurol.2016.187
Guan Y, Jakimovski D, Ramanathan M, Weinstock-Guttman B, Zivadinov R (2019) The role of Epstein-Barr virus in multiple sclerosis: from molecular pathophysiology to in vivo imaging. Neural Regen Res 14(3):373–386. https://doi.org/10.4103/1673-5374.245462
Naismith RT, Piccio L, Lyons JA, Lauber J, Tutlam NT, Parks BJ, Trinkaus K, Song SK, Cross AH (2010) Rituximab add-on therapy for breakthrough relapsing multiple sclerosis: a 52-week phase II trial. Neurology 74(23):1860–1867. https://doi.org/10.1212/WNL.0b013e3181e24373
Alping P, Frisell T, Novakova L, Islam-Jakobsson P, Salzer J, Bjorck A, Axelsson M, Malmestrom C, Fink K, Lycke J, Svenningsson A, Piehl F (2016) Rituximab versus fingolimod after natalizumab in multiple sclerosis patients. Ann Neurol 79(6):950–958. https://doi.org/10.1002/ana.24651
Scotti B, Disanto G, Sacco R, Guigli M, Zecca C, Gobbi C (2018) Effectiveness and safety of Rituximab in multiple sclerosis: an observational study from Southern Switzerland. PLoS ONE 13(5):e0197415. https://doi.org/10.1371/journal.pone.0197415
Granqvist M, Boremalm M, Poorghobad A, Svenningsson A, Salzer J, Frisell T, Piehl F (2018) Comparative effectiveness of rituximab and other initial treatment choices for multiple sclerosis. JAMA Neurol 75(3):320–327. https://doi.org/10.1001/jamaneurol.2017.4011
Honce JM, Nair KV, Sillau S, Valdez B, Miravalle A, Alvarez E, Schreiner T, Corboy JR, Vollmer TL (2019) Rituximab vs placebo induction prior to glatiramer acetate monotherapy in multiple sclerosis. Neurology 92(7):e723–e732. https://doi.org/10.1212/WNL.0000000000006916
Brown BA, Torabi M (2011) Incidence of infusion-associated reactions with rituximab for treating multiple sclerosis: a retrospective analysis of patients treated at a US centre. Drug Saf 34(2):117–123. https://doi.org/10.2165/11585960-000000000-00000
Dunn N, Juto A, Ryner M, Manouchehrinia A, Piccoli L, Fink K, Piehl F, Fogdell-Hahn A (2018) Rituximab in multiple sclerosis: frequency and clinical relevance of anti-drug antibodies. Mult Scler 24(9):1224–1233. https://doi.org/10.1177/1352458517720044
Hu Y, Nie H, Yu HH, Qin C, Wu LJ, Tang ZP, Tian DS (2019) Efficacy and safety of rituximab for relapsing-remitting multiple sclerosis: a systematic review and meta-analysis. Autoimmun Rev 18(5):542–548. https://doi.org/10.1016/j.autrev.2019.03.011
Salvarani C, Brown RD Jr, Muratore F, Christianson TJH, Galli E, Pipitone N, Cassone G, Huston J III, Giannini C, Warrington K, Hunder GG (2019) Rituximab therapy for primary central nervous system vasculitis: a 6 patient experience and review of the literature. Autoimmun Rev 18(4):399–405. https://doi.org/10.1016/j.autrev.2018.12.002
Alvarez KNIS E, Sillau S, Vollmer T (2019) Tolerability and safety of switching from rituximab to ocrelizumab: evaluating factors associated with infusion related reactions—Abstract: P1402. ECTRIMS Online Library. Alvarez E. 09/13/19; 278602
Joerger M (2012) Prevention and handling of acute allergic and infusion reactions in oncology. Ann Oncol 23(Suppl 10):x313-319. https://doi.org/10.1093/annonc/mds314
Heusele M, Clerson P, Guery B, Lambert M, Launay D, Lefevre G, Morell-Dubois S, Maillard H, Le Gouellec N, Hatron PY, Hachulla E (2014) Risk factors for severe bacterial infections in patients with systemic autoimmune diseases receiving rituximab. Clin Rheumatol 33(6):799–805. https://doi.org/10.1007/s10067-014-2509-2
Gottenberg JE, Ravaud P, Bardin T, Cacoub P, Cantagrel A, Combe B, Dougados M, Flipo RM, Godeau B, Guillevin L, Le Loet X, Hachulla E, Schaeverbeke T, Sibilia J, Baron G, Mariette X, AutoImmunity and Rituximab registry and French Society of Rheumatology (2010) Risk factors for severe infections in patients with rheumatoid arthritis treated with rituximab in the autoimmunity and rituximab registry. Arthritis Rheum 62(9):2625–2632. https://doi.org/10.1002/art.27555
Gea-Banacloche JC (2010) Rituximab-associated infections. Semin Hematol 47(2):187–198. https://doi.org/10.1053/j.seminhematol.2010.01.002
Luna G, Alping P, Burman J, Fink K, Fogdell-Hahn A, Gunnarsson M, Hillert J, Langer-Gould A, Lycke J, Nilsson P, Salzer J, Svenningsson A, Vrethem M, Olsson T, Piehl F, Frisell T (2019) Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol. https://doi.org/10.1001/jamaneurol.2019.3365
Keystone E, Fleischmann R, Emery P, Furst DE, van Vollenhoven R, Bathon J, Dougados M, Baldassare A, Ferraccioli G, Chubick A, Udell J, Cravets MW, Agarwal S, Cooper S, Magrini F (2007) Safety and efficacy of additional courses of rituximab in patients with active rheumatoid arthritis: an open-label extension analysis. Arthritis Rheum 56(12):3896–3908. https://doi.org/10.1002/art.23059
Salzer J, Lycke J, Wickstrom R, Naver H, Piehl F, Svenningsson A (2016) Rituximab in paediatric onset multiple sclerosis: a case series. J Neurol 263(2):322–326. https://doi.org/10.1007/s00415-015-7979-x
Durozard P, Maarouf A, Boutiere C, Ruet A, Brochet B, Vukusic S, Carra-Dalliere C, Labauge P, Mathey G, Debouverie M, Papeix C, Maillart E, Lubetzki C, Bensa C, Gout O, Giannesini C, Stankoff B, Ciron J, Brassat D, Pelletier J, Rico Lamy A, Audoin B (2019) Efficacy of rituximab in refractory RRMS. Mult Scler 25(6):828–836. https://doi.org/10.1177/1352458518772748
de Flon P, Gunnarsson M, Laurell K, Soderstrom L, Birgander R, Lindqvist T, Krauss W, Dring A, Bergman J, Sundstrom P, Svenningsson A (2016) Reduced inflammation in relapsing-remitting multiple sclerosis after therapy switch to rituximab. Neurology 87(2):141–147. https://doi.org/10.1212/WNL.0000000000002832
Rommer PS, Dorner T, Freivogel K, Haas J, Kieseier BC, Kumpfel T, Paul F, Proft F, Schulze-Koops H, Schmidt E, Wiendl H, Ziemann U, Zettl UK, GRAID Investigators (2016) Safety and clinical outcomes of rituximab treatment in patients with multiple sclerosis and neuromyelitis optica: experience from a National Online Registry (GRAID). J Neuroimmune Pharmacol 11(1):1–8. https://doi.org/10.1007/s11481-015-9646-5
Alldredge B, Jordan A, Imitola J, Racke MK (2018) Safety and efficacy of rituximab: experience of a single multiple sclerosis center. Clin Neuropharmacol 41(2):56–59. https://doi.org/10.1097/WNF.0000000000000268
Tony HP, Burmester G, Schulze-Koops H, Grunke M, Henes J, Kotter I, Haas J, Unger L, Lovric S, Haubitz M, Fischer-Betz R, Chehab G, Rubbert-Roth A, Specker C, Weinerth J, Holle J, Muller-Ladner U, Konig R, Fiehn C, Burgwinkel P, Budde K, Sorensen H, Meurer M, Aringer M, Kieseier B, Erfurt-Berge C, Sticherling M, Veelken R, Ziemann U, Strutz F, von Wussow P, Meier FM, Hunzelmann N, Schmidt E, Bergner R, Schwarting A, Eming R, Hertl M, Stadler R, Schwarz-Eywill M, Wassenberg S, Fleck M, Metzler C, Zettl U, Westphal J, Heitmann S, Herzog AL, Wiendl H, Jakob W, Schmidt E, Freivogel K, Dorner T, GRAID Investigators (2011) Safety and clinical outcomes of rituximab therapy in patients with different autoimmune diseases: experience from a national registry (GRAID). Arthritis Res Ther 13(3):R75. https://doi.org/10.1186/ar3337
Hellgren J, Risedal A, Kallen K (2020) Rituximab in multiple sclerosis at general hospital level. Acta Neurol Scand 141(6):491–499. https://doi.org/10.1111/ane.13225
Mok CC (2013) Rituximab for the treatment of rheumatoid arthritis: an update. Drug Des Devel Ther 8:87–100. https://doi.org/10.2147/DDDT.S41645
Pyrpasopoulou A, Douma S, Vassiliadis T, Chatzimichailidou S, Triantafyllou A, Aslanidis S (2011) Reactivation of chronic hepatitis B virus infection following rituximab administration for rheumatoid arthritis. Rheumatol Int 31(3):403–404. https://doi.org/10.1007/s00296-009-1202-2
Chen KL, Chen J, Rao HL, Guo Y, Huang HQ, Zhang L, Shao JY, Lin TY, Jiang WQ, Zou DH, Hu LY, Wirian ML, Cai QQ (2015) Hepatitis B virus reactivation and hepatitis in diffuse large B-cell lymphoma patients with resolved hepatitis B receiving rituximab-containing chemotherapy: risk factors and survival. Chin J Cancer 34(5):225–234. https://doi.org/10.1186/s40880-015-0015-9
Seto WK, Chan TS, Hwang YY, Wong DK, Fung J, Liu KS, Gill H, Lam YF, Lie AK, Lai CL, Kwong YL, Yuen MF (2014) Hepatitis B reactivation in patients with previous hepatitis B virus exposure undergoing rituximab-containing chemotherapy for lymphoma: a prospective study. J Clin Oncol 32(33):3736–3743. https://doi.org/10.1200/JCO.2014.56.7081
Di Bisceglie AM, Lok AS, Martin P, Terrault N, Perrillo RP, Hoofnagle JH (2015) Recent US Food and Drug Administration warnings on hepatitis B reactivation with immune-suppressing and anticancer drugs: just the tip of the iceberg? Hepatology 61(2):703–711. https://doi.org/10.1002/hep.27609
(FDA) FaDA (2020) FDA Drug safety communication: boxed warning and new recommendations to decrease risk of hepatitis B reactivation with the immune-suppressing and anti-cancer drugs Arzerra (ofatumumab) and Rituxan (rituximab). https://www.fda.gov/drugs/drug-safety-and-availability/fda-drug-safety-communication-boxed-warning-and-new-recommendations-decrease-risk-hepatitis-b. Accessed Nov 2020
Goldberg SL, Pecora AL, Alter RS, Kroll MS, Rowley SD, Waintraub SE, Imrit K, Preti RA (2002) Unusual viral infections (progressive multifocal leukoencephalopathy and cytomegalovirus disease) after high-dose chemotherapy with autologous blood stem cell rescue and peritransplantation rituximab. Blood 99(4):1486–1488. https://doi.org/10.1182/blood.v99.4.1486
Ennishi D, Terui Y, Yokoyama M, Mishima Y, Takahashi S, Takeuchi K, Ikeda K, Tanimoto M, Hatake K (2008) Increased incidence of interstitial pneumonia by CHOP combined with rituximab. Int J Hematol 87(4):393–397. https://doi.org/10.1007/s12185-008-0066-7
Sikkema T, Schuiling WJ, Hoogendoorn M (2013) Progressive multifocal leukoencephalopathy during treatment with rituximab and CHOP chemotherapy in a patient with a diffuse large B-cell lymphoma. BMJ Case Rep. https://doi.org/10.1136/bcr-2012-008142
Oshima Y, Tanimoto T, Yuji K, Tojo A (2019) Drug-associated progressive multifocal leukoencephalopathy in multiple sclerosis patients. Mult Scler 25(8):1141–1149. https://doi.org/10.1177/1352458518786075
Narula S, LaRosa DF, Kamoun M, Dalmau J, Levinson AI (2007) Progressive multifocal leukoencephalopathy in a patient with common variable immunodeficiency and abnormal CD8+ T-cell subset distribution. Ann Allergy Asthma Immunol 98(5):483–489. https://doi.org/10.1016/S1081-1206(10)60764-8
Stasi R, Del Poeta G, Stipa E, Evangelista ML, Trawinska MM, Cooper N, Amadori S (2007) Response to B-cell depleting therapy with rituximab reverts the abnormalities of T-cell subsets in patients with idiopathic thrombocytopenic purpura. Blood 110(8):2924–2930. https://doi.org/10.1182/blood-2007-02-068999
Iacobaeus E, Burkill S, Bahmanyar S, Hakim R, Bystrom C, Fored M, Olsson T, Brundin L, Montgomery S (2018) The national incidence of PML in Sweden, 1988–2013. Neurology 90(6):e498–e506. https://doi.org/10.1212/WNL.0000000000004926
Classifying PML risk with disease modifying therapies—FormSus. (2020). http://formsus.datasus.gov.br/novoimgarq/58543/13614899_312364.pdf
Prosperini L, Scarpazza C, Imberti L, Cordioli C, De Rossi N, Capra R (2017) Age as a risk factor for early onset of natalizumab-related progressive multifocal leukoencephalopathy. J Neurovirol 23(5):742–749. https://doi.org/10.1007/s13365-017-0561-9
Kartau M, Sipila JO, Auvinen E, Palomaki M, Verkkoniemi-Ahola A (2019) Progressive Multifocal Leukoencephalopathy: current Insights. Degener Neurol Neuromuscul Dis 9:109–121. https://doi.org/10.2147/DNND.S203405
Kridin K, Ahmed AR (2020) Post-rituximab immunoglobulin M (IgM) hypogammaglobulinemia. Autoimmun Rev 19(3):102466. https://doi.org/10.1016/j.autrev.2020.102466
Schneider P, MacKay F, Steiner V, Hofmann K, Bodmer JL, Holler N, Ambrose C, Lawton P, Bixler S, Acha-Orbea H, Valmori D, Romero P, Werner-Favre C, Zubler RH, Browning JL, Tschopp J (1999) BAFF, a novel ligand of the tumor necrosis factor family, stimulates B cell growth. J Exp Med 189(11):1747–1756. https://doi.org/10.1084/jem.189.11.1747
Ireland S, Monson N (2011) Potential impact of B cells on T cell function in multiple sclerosis. Mult Scler Int 2011:423971. https://doi.org/10.1155/2011/423971
Marcinno A, Marnetto F, Valentino P, Martire S, Balbo A, Drago A, Leto M, Capobianco M, Panzica G, Bertolotto A (2018) Rituximab-induced hypogammaglobulinemia in patients with neuromyelitis optica spectrum disorders. Neurol Neuroimmunol Neuroinflamm 5(6):e498. https://doi.org/10.1212/NXI.0000000000000498
Etemadifar M, Salari M, Mirmosayyeb O, Serati M, Nikkhah R, Askari M, Fayyazi E (2017) Efficacy and safety of rituximab in neuromyelitis optica: review of evidence. J Res Med Sci 22:18. https://doi.org/10.4103/1735-1995.200275
Boleto G, Avouac J, Wipff J, Forien M, Dougados M, Roux C, Kahan A, Dieude P, Allanore Y (2018) Predictors of hypogammaglobulinemia during rituximab maintenance therapy in rheumatoid arthritis: a 12-year longitudinal multi-center study. Semin Arthritis Rheum 48(2):149–154. https://doi.org/10.1016/j.semarthrit.2018.02.010
Isvy A, Meunier M, Gobeaux-Chenevier C, Maury E, Wipff J, Job-Deslandre C, Kahan A, Allanore Y (2012) Safety of rituximab in rheumatoid arthritis: a long-term prospective single-center study of gammaglobulin concentrations and infections. Joint Bone Spine 79(4):365–369. https://doi.org/10.1016/j.jbspin.2011.12.004
Barmettler S, Ong MS, Farmer JR, Choi H, Walter J (2018) Association of immunoglobulin levels, infectious risk, and mortality with rituximab and hypogammaglobulinemia. JAMA Netw Open 1(7):e184169. https://doi.org/10.1001/jamanetworkopen.2018.4169
Rozman S, Sonc M, Novakovic BJ (2012) Late-onset neutropenia following primary treatment of diffuse large B-cell lymphoma with rituximab-containing therapy. Leuk Lymphoma 53(10):1945–1948. https://doi.org/10.3109/10428194.2012.679266
Tesfa D, Palmblad J (2011) Late-onset neutropenia following rituximab therapy: incidence, clinical features and possible mechanisms. Expert Rev Hematol 4(6):619–625. https://doi.org/10.1586/EHM.11.62
Rigal J, Ciron J, Lepine Z, Biotti D (2020) Late-onset neutropenia after RITUXIMAB therapy for multiple sclerosis, neuromyelitis optica spectrum disorders and MOG-antibody-associated diseases. Mult Scler Relat Disord 41:102019. https://doi.org/10.1016/j.msard.2020.102019
Alcala C, Gascon F, Perez-Miralles F, Gil-Perotin S, Navarre A, Bosca I, Coret F, Casanova B (2018) Efficacy and safety of rituximab in relapsing and progressive multiple sclerosis: a hospital-based study. J Neurol 265(7):1690–1697. https://doi.org/10.1007/s00415-018-8899-3
Yamout BI, El-Ayoubi NK, Nicolas J, El Kouzi Y, Khoury SJ, Zeineddine MM (2018) Safety and efficacy of rituximab in multiple sclerosis: a retrospective observational study. J Immunol Res 2018:9084759. https://doi.org/10.1155/2018/9084759
Wadstrom H, Frisell T, Askling J, Anti-Rheumatic Therapy in Sweden (ARTIS) Study Group (2017) Malignant neoplasms in patients with rheumatoid arthritis treated with tumor necrosis factor inhibitors, tocilizumab, abatacept, or rituximab in clinical practice: a nationwide cohort study from Sweden. JAMA Intern Med 177(11):1605–1612. https://doi.org/10.1001/jamainternmed.2017.4332
Alping P, Askling J, Burman J, Fink K, Fogdell-Hahn A, Gunnarsson M, Hillert J, Langer-Gould A, Lycke J, Nilsson P, Salzer J, Svenningsson A, Vrethem M, Olsson T, Piehl F, Frisell T (2020) Cancer risk for fingolimod, natalizumab, and rituximab in multiple sclerosis patients. Ann Neurol 87(5):688–699. https://doi.org/10.1002/ana.25701
Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, Kallenberg CG, St Clair EW, Turkiewicz A, Tchao NK, Webber L, Ding L, Sejismundo LP, Mieras K, Weitzenkamp D, Ikle D, Seyfert-Margolis V, Mueller M, Brunetta P, Allen NB, Fervenza FC, Geetha D, Keogh KA, Kissin EY, Monach PA, Peikert T, Stegeman C, Ytterberg SR, Specks U, RAVE-ITN Research Group (2010) Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 363(3):221–232. https://doi.org/10.1056/NEJMoa0909905
Oddis CV, Reed AM, Aggarwal R, Rider LG, Ascherman DP, Levesque MC, Barohn RJ, Feldman BM, Harris-Love MO, Koontz DC, Fertig N, Kelley SS, Pryber SL, Miller FW, Rockette HE, RIM Study Group (2013) Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial. Arthritis Rheum 65(2):314–324. https://doi.org/10.1002/art.37754
Fervenza FC, Appel GB, Barbour SJ, Rovin BH, Lafayette RA, Aslam N, Jefferson JA, Gipson PE, Rizk DV, Sedor JR, Simon JF, McCarthy ET, Brenchley P, Sethi S, Avila-Casado C, Beanlands H, Lieske JC, Philibert D, Li T, Thomas LF, Green DF, Juncos LA, Beara-Lasic L, Blumenthal SS, Sussman AN, Erickson SB, Hladunewich M, Canetta PA, Hebert LA, Leung N, Radhakrishnan J, Reich HN, Parikh SV, Gipson DS, Lee DK, da Costa BR, Juni P, Cattran DC, Investigators M (2019) Rituximab or cyclosporine in the treatment of membranous nephropathy. N Engl J Med 381(1):36–46. https://doi.org/10.1056/NEJMoa1814427
Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, Savage CO, Segelmark M, Tesar V, van Paassen P, Walsh D, Walsh M, Westman K, Jayne DR, European Vasculitis Study Group (2010) Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 363(3):211–220. https://doi.org/10.1056/NEJMoa0909169
Stan MN, Garrity JA, Carranza Leon BG, Prabin T, Bradley EA, Bahn RS (2015) Randomized controlled trial of rituximab in patients with Graves’ orbitopathy. J Clin Endocrinol Metab 100(2):432–441. https://doi.org/10.1210/jc.2014-2572
Diaz-Manera J, Martinez-Hernandez E, Querol L, Klooster R, Rojas-Garcia R, Suarez-Calvet X, Munoz-Blanco JL, Mazia C, Straasheijm KR, Gallardo E, Juarez C, Verschuuren JJ, Illa I (2012) Long-lasting treatment effect of rituximab in MuSK myasthenia. Neurology 78(3):189–193. https://doi.org/10.1212/WNL.0b013e3182407982
Owczarczyk K, Hellmann M, Fliedner G, Rohrs T, Maizus K, Passon D, Hallek M, Rubbert A (2008) Clinical outcome and B cell depletion in patients with rheumatoid arthritis receiving rituximab monotherapy in comparison with patients receiving concomitant methotrexate. Ann Rheum Dis 67(11):1648–1649. https://doi.org/10.1136/ard.2007.087023
Mease PJ, Cohen S, Gaylis NB, Chubick A, Kaell AT, Greenwald M, Agarwal S, Yin M, Kelman A (2010) Efficacy and safety of retreatment in patients with rheumatoid arthritis with previous inadequate response to tumor necrosis factor inhibitors: results from the SUNRISE trial. J Rheumatol 37(5):917–927. https://doi.org/10.3899/jrheum.090442
Kaegi C, Wuest B, Schreiner J, Steiner UC, Vultaggio A, Matucci A, Crowley C, Boyman O (2019) Systematic review of safety and efficacy of rituximab in treating immune-mediated disorders. Front Immunol 10:1990. https://doi.org/10.3389/fimmu.2019.01990
Kimby E (2005) Tolerability and safety of rituximab (MabThera). Cancer Treat Rev 31(6):456–473. https://doi.org/10.1016/j.ctrv.2005.05.007
Yri OE, Torfoss D, Hungnes O, Tierens A, Waalen K, Nordoy T, Dudman S, Kilander A, Wader KF, Ostenstad B, Ekanger R, Meyer P, Kolstad A (2011) Rituximab blocks protective serologic response to influenza A (H1N1) 2009 vaccination in lymphoma patients during or within 6 months after treatment. Blood 118(26):6769–6771. https://doi.org/10.1182/blood-2011-08-372649
Nazi I, Kelton JG, Larche M, Snider DP, Heddle NM, Crowther MA, Cook RJ, Tinmouth AT, Mangel J, Arnold DM (2013) The effect of rituximab on vaccine responses in patients with immune thrombocytopenia. Blood 122(11):1946–1953. https://doi.org/10.1182/blood-2013-04-494096
Cho A, Bradley B, Kauffman R, Priyamvada L, Kovalenkov Y, Feldman R, Wrammert J (2017) Robust memory responses against influenza vaccination in pemphigus patients previously treated with rituximab. JCI Insight 2(12):e93222. https://doi.org/10.1172/jci.insight.93222
Hoyer BF, Manz RA, Radbruch A, Hiepe F (2005) Long-lived plasma cells and their contribution to autoimmunity. Ann N Y Acad Sci 1050:124–133. https://doi.org/10.1196/annals.1313.014
Sutter JA, Kwan-Morley J, Dunham J, Du YZ, Kamoun M, Albert D, Eisenberg RA, Luning Prak ET (2008) A longitudinal analysis of SLE patients treated with rituximab (anti-CD20): factors associated with B lymphocyte recovery. Clin Immunol 126(3):282–290. https://doi.org/10.1016/j.clim.2007.11.012
Zrzavy T, Kollaritsch H, Rommer PS, Boxberger N, Loebermann M, Wimmer I, Winkelmann A, Zettl UK (2019) Vaccination in multiple sclerosis: friend or foe? Front Immunol 10:1883. https://doi.org/10.3389/fimmu.2019.01883
(MabThera) EAoM-I-R (2020) https://www.ema.europa.eu/en/documents/product-information/mabthera-epar-product-information_en.pdf
Administration FaD (2020) https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/103705s5367s5388lbl.pdf
Gelfand JM, Cree BAC, Hauser SL (2017) Ocrelizumab and other CD20(+) B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics 14(4):835–841. https://doi.org/10.1007/s13311-017-0557-4
JR (2019) Rituximab for treatment of aggressive multiple sclerosis during pregnancy: a case study. Academy of American Neurology Annual Meeting, May 4–10, 2019; Philadelphia, PA Abstract P42–103
LaHue S KK, Rutatangwa A, et al (2019) Minimal concentrations of rituximab in the breastmilk of women treated for multiple sclerosis. Academy of American Neurology Annual Meeting, May 4–10, 2019; Philadelphia, PA Abstract P42–097
Matias-Guiu J, Gomez-Pinedo U, Montero-Escribano P, Gomez-Iglesias P, Porta-Etessam J, Matias-Guiu JA (2020) Should we expect neurological symptoms in the SARS-CoV-2 epidemic? Neurologia 35(3):170–175. https://doi.org/10.1016/j.nrl.2020.03.001
Salama S, Ahmed SF, Ibrahim Ismail I, Alroughani R (2020) Impact of coronavirus disease (COVID-19) pandemic on multiple sclerosis care. Clin Neurol Neurosurg 197:106203. https://doi.org/10.1016/j.clineuro.2020.106203
Sokolowska M, Lukasik Z, Agache I, Akdis CA, Akdis D, Akdis M, Barcik W, Brough H, Eiwegger T, Eliaszewicz A, Eyerich S, Feleszko W, Gomez Casado C, Hoffmann-Sommergruber K, Janda J, Jimenez-Saiz R, Jutel M, Knol E, Kortekaas Krohn I, Kothari A, Makowska J, Moniuszko M, Morita H, O’Mahony L, Nadeau K, Ozdemir C, Pali-Scholl I, Palomares O, Papaleo F, Prunicki M, Schmidt-Weber CB, Sediva A, Schwarze J, Shamji MH, Tramper-Stranders G, van de Veen W, Untersmayr E (2020) Immunology of COVID-19: mechanisms, clinical outcome, diagnostics and perspectives—a report of the European Academy of Allergy and Clinical Immunology (EAACI). Allergy. https://doi.org/10.1111/all.14462
Novi G, Mikulska M, Briano F, Toscanini F, Tazza F, Uccelli A, Inglese M (2020) COVID-19 in a MS patient treated with ocrelizumab: does immunosuppression have a protective role? Mult Scler Relat Disord 42:102120. https://doi.org/10.1016/j.msard.2020.102120
Tay MZ, Poh CM, Renia L, MacAry PA, Ng LFP (2020) The trinity of COVID-19: immunity, inflammation and intervention. Nat Rev Immunol 20(6):363–374. https://doi.org/10.1038/s41577-020-0311-8
Safavi F, Nourbakhsh B, Azimi AR (2020) B-cell depleting therapies may affect susceptibility to acute respiratory illness among patients with multiple sclerosis during the early COVID-19 epidemic in Iran. Mult Scler Relat Disord 43:102195. https://doi.org/10.1016/j.msard.2020.102195
Maria Pia Sormani NDR, Irene Schiavetti Luca, Carmisciano CC, Lucia Moiola, Marta Radaelli, Paolo, Immovilli Marco Capobianco MT, Paola Zaratin, Gioacchino Tedeschi GC, Mario Alberto Battaglia, Francesco Patti MS, and the Musc-19 study group (2020) Disease modifying therapies and COVID-19 severity in multiple sclerosis. Lancet Neurol. Available at SSRN: https://doi.org/10.2139/ssrn.3631244
Coles ALM, Giovannoni G, Anderson P, Dorsey-Campbell R, Qualie M, Vieira JJ (2020) ABN guidance on the use of disease-modifying therapies in multiple sclerosis in response to the threat of a coronavirus epidemic. https://cdn.ymaws.com/www.theabn.org/resource/collection/65C334C7-30FA-45DB-93AA-74B3A3A20293/02.04.20_ABN_Guidance_on_DMTs_for_MS_and_COVID19_VERSION_4_April_2nd.pdf. Accessed Nov 2020
Tepasse PR, Hafezi W, Lutz M, Kuhn J, Wilms C, Wiewrodt R, Sackarnd J, Keller M, Schmidt HH, Vollenberg R (2020) Persisting SARS-CoV-2 viraemia after rituximab therapy: two cases with fatal outcome and a review of the literature. Br J Haematol 190(2):185–188. https://doi.org/10.1111/bjh.16896
Wurm H, Attfield K, Iversen AK, Gold R, Fugger L, Haghikia A (2020) Recovery from COVID-19 in a B-cell-depleted multiple sclerosis patient. Mult Scler 26(10):1261–1264. https://doi.org/10.1177/1352458520943791
Baker D, Roberts CAK, Pryce G, Kang AS, Marta M, Reyes S, Schmierer K, Giovannoni G, Amor S (2020) COVID-19 vaccine-readiness for anti-CD20-depleting therapy in autoimmune diseases. Clin Exp Immunol 202(2):149–161. https://doi.org/10.1111/cei.13495
ClinicalTrials.gov (2020) A study to evaluate the effects of ocrelizumab on immune responses in participants with relapsing forms of multiple sclerosis. Accessed 22 Nov 2020
Montero-Escribano P, Matias-Guiu J, Gomez-Iglesias P, Porta-Etessam J, Pytel V, Matias-Guiu JA (2020) Anti-CD20 and COVID-19 in multiple sclerosis and related disorders: A case series of 60 patients from Madrid. Spain Mult Scler Relat Disord 42:102185. https://doi.org/10.1016/j.msard.2020.102185
D’Amico E, Chisari CG, Gitto L, Zanghi A, Toscano S, Patti F (2019) Pharmacoeconomics of synthetic therapies for multiple sclerosis. Expert Opin Pharmacother 20(11):1331–1340. https://doi.org/10.1080/14656566.2019.1615880
Du FH, Mills EA, Mao-Draayer Y (2017) Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun Highlights 8(1):12. https://doi.org/10.1007/s13317-017-0100-y
Rognoni C, Bertolani A, Jommi C (2018) Budget impact analysis of rituximab biosimilar in Italy from the hospital and payer perspectives. Glob Reg Health Technol Assess 5(1):1–11. https://doi.org/10.1177/228424031878428
Hartung DM (2017) Economics and cost-effectiveness of multiple sclerosis therapies in the USA. Neurotherapeutics 14(4):1018–1026. https://doi.org/10.1007/s13311-017-0566-3
Genenentech (2020) OCREVUS pricing and financial support https://www.ocrevus.com/patient/cost.html Accessed 20 Nov 2020
Mullard A (2017) Bracing for the biosimilar wave. Nat Rev Drug Discov 16(3):152–154. https://doi.org/10.1038/nrd.2017.36
Simoens S (2011) Biosimilar medicines and cost-effectiveness. Clinicoecon Outcomes Res 3:29–36. https://doi.org/10.2147/CEOR.S12494
Montserrat E (2003) Rituximab in chronic lymphocytic leukemia. Semin Oncol 30(1S2):34–39. https://doi.org/10.1053/sonc.2003.50033
Boremalm M, Juto A, Axelsson M, Novakova L, Frisell T, Svenningsson A, Lycke J, Piehl F, Salzer J (2019) Natalizumab, rituximab and fingolimod as escalation therapy in multiple sclerosis. Eur J Neurol 26(8):1060–1067. https://doi.org/10.1111/ene.13936
Roll P, Palanichamy A, Kneitz C, Dorner T, Tony HP (2006) Regeneration of B cell subsets after transient B cell depletion using anti-CD20 antibodies in rheumatoid arthritis. Arthritis Rheum 54(8):2377–2386. https://doi.org/10.1002/art.22019
Carson KR, Evens AM, Richey EA, Habermann TM, Focosi D, Seymour JF, Laubach J, Bawn SD, Gordon LI, Winter JN, Furman RR, Vose JM, Zelenetz AD, Mamtani R, Raisch DW, Dorshimer GW, Rosen ST, Muro K, Gottardi-Littell NR, Talley RL, Sartor O, Green D, Major EO, Bennett CL (2009) Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 113(20):4834–4840. https://doi.org/10.1182/blood-2008-10-186999
Clifford DB, Ances B, Costello C, Rosen-Schmidt S, Andersson M, Parks D, Perry A, Yerra R, Schmidt R, Alvarez E, Tyler KL (2011) Rituximab-associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol 68(9):1156–1164. https://doi.org/10.1001/archneurol.2011.103
Nielsen AS, Miravalle A, Langer-Gould A, Cooper J, Edwards KR, Kinkel RP (2012) Maximally tolerated versus minimally effective dose: the case of rituximab in multiple sclerosis. Mult Scler 18(3):377–378. https://doi.org/10.1177/1352458511418631
Shirani A, Okuda DT, Stuve O (2016) Therapeutic advances and future prospects in progressive forms of multiple sclerosis. Neurotherapeutics 13(1):58–69. https://doi.org/10.1007/s13311-015-0409-z
Studer V, Rossi S, Motta C, Buttari F, Centonze D (2014) Peripheral B cell depletion and central proinflammatory cytokine reduction following repeated intrathecal administration of rituximab in progressive Multiple Sclerosis. J Neuroimmunol 276(1–2):229–231. https://doi.org/10.1016/j.jneuroim.2014.08.617
Svenningsson A, Bergman J, Dring A, Vagberg M, Birgander R, Lindqvist T, Gilthorpe J, Bergenheim T (2015) Rapid depletion of B lymphocytes by ultra-low-dose rituximab delivered intrathecally. Neurol Neuroimmunol Neuroinflamm 2(2):e79. https://doi.org/10.1212/NXI.0000000000000079
Ellrichmann G, Bolz J, Peschke M, Duscha A, Hellwig K, Lee DH, Linker RA, Gold R, Haghikia A (2019) Peripheral CD19(+) B-cell counts and infusion intervals as a surrogate for long-term B-cell depleting therapy in multiple sclerosis and neuromyelitis optica/neuromyelitis optica spectrum disorders. J Neurol 266(1):57–67. https://doi.org/10.1007/s00415-018-9092-4
Yi JS, Decroos EC, Sanders DB, Weinhold KJ, Guptill JT (2013) Prolonged B-cell depletion in MuSK myasthenia gravis following rituximab treatment. Muscle Nerve 48(6):992–993. https://doi.org/10.1002/mus.24063
Memon AB, Javed A, Caon C, Srivastawa S, Bao F, Bernitsas E, Chorostecki J, Tselis A, Seraji-Bozorgzad N, Khan O (2018) Long-term safety of rituximab induced peripheral B-cell depletion in autoimmune neurological diseases. PLoS ONE 13(1):e0190425. https://doi.org/10.1371/journal.pone.0190425
Kim SH, Kim W, Li XF, Jung IJ, Kim HJ (2011) Repeated treatment with rituximab based on the assessment of peripheral circulating memory B cells in patients with relapsing neuromyelitis optica over 2 years. Arch Neurol 68(11):1412–1420. https://doi.org/10.1001/archneurol.2011.154
Jurczak W, Dlugosz-Danecka M (2020) Rituximab biosimilars in clinical practice. Leuk Lymphoma 61(7):1523–1524. https://doi.org/10.1080/10428194.2020.1788016
Initiative GaB (2020) EMA approval for rituximab biosimilar Truxima
(EMA) EMA (2020) Rixathon https://www.ema.europa.eu/en/medicines/human/EPAR/rixathon. Accessed 10 Nov 2020
Yoo DH, Suh CH, Shim SC, Jeka S, Cons-Molina FF, Hrycaj P, Wiland P, Lee EY, Medina-Rodriguez FG, Shesternya P, Radominski S, Stanislav M, Kovalenko V, Sheen DH, Myasoutova L, Lim MJ, Choe JY, Lee SJ, Lee SY, Kwon TS, Park W (2017) A multicentre randomised controlled trial to compare the pharmacokinetics, efficacy and safety of CT-P10 and innovator rituximab in patients with rheumatoid arthritis. Ann Rheum Dis 76(3):566–570. https://doi.org/10.1136/annrheumdis-2016-209540
Visser J, Feuerstein I, Stangler T, Schmiederer T, Fritsch C, Schiestl M (2013) Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 27(5):495–507. https://doi.org/10.1007/s40259-013-0036-3
Jurczak W, Moreira I, Kanakasetty GB, Munhoz E, Echeveste MA, Giri P, Castro N, Pereira J, Akria L, Alexeev S, Osmanov E, Zhu P, Alexandrova S, Zubel A, Harlin O, Amersdorffer J (2017) Rituximab biosimilar and reference rituximab in patients with previously untreated advanced follicular lymphoma (ASSIST-FL): primary results from a confirmatory phase 3, double-blind, randomised, controlled study. Lancet Haematol 4(8):e350–e361. https://doi.org/10.1016/S2352-3026(17)30106-0
Smolen JS, Cohen SB, Tony HP, Scheinberg M, Kivitz A, Balanescu A, Gomez-Reino J, Cen L, Zhu P, Shisha T (2017) A randomised, double-blind trial to demonstrate bioequivalence of GP2013 and reference rituximab combined with methotrexate in patients with active rheumatoid arthritis. Ann Rheum Dis 76(9):1598–1602. https://doi.org/10.1136/annrheumdis-2017-211281
Perez T, Rico A, Boutiere C, Maarouf A, Roudot M, Honore S, Pelletier J, Bertault-Peres P, Audoin B (2020) Comparison of rituximab originator (MabThera((R))) to biosimilar (Truxima((R))) in patients with multiple sclerosis. Mult Scler. https://doi.org/10.1177/1352458520912170
Whittam DH, Tallantyre EC, Jolles S, Huda S, Moots RJ, Kim HJ, Robertson NP, Cree BAC, Jacob A (2019) Rituximab in neurological disease: principles, evidence and practice. Pract Neurol 19(1):5–20. https://doi.org/10.1136/practneurol-2018-001899
Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, Honig LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N, Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F, Dalmau J (2013) Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 12(2):157–165. https://doi.org/10.1016/S1474-4422(12)70310-1
Benedetti L, Briani C, Franciotta D, Fazio R, Paolasso I, Comi C, Luigetti M, Sabatelli M, Giannini F, Mancardi GL, Schenone A, Nobile-Orazio E, Cocito D (2011) Rituximab in patients with chronic inflammatory demyelinating polyradiculoneuropathy: a report of 13 cases and review of the literature. J Neurol Neurosurg Psychiatry 82(3):306–308. https://doi.org/10.1136/jnnp.2009.188912
Salvarani C, Brown RD Jr, Huston J 3rd, Morris JM, Hunder GG (2014) Treatment of primary CNS vasculitis with rituximab: case report. Neurology 82(14):1287–1288. https://doi.org/10.1212/WNL.0000000000000293
Damato V, Evoli A, Iorio R (2016) Efficacy and safety of rituximab therapy in neuromyelitis optica spectrum disorders: a systematic review and meta-analysis. JAMA Neurol 73(11):1342–1348. https://doi.org/10.1001/jamaneurol.2016.1637
Palace J, Leite MI, Jacob A (2012) A practical guide to the treatment of neuromyelitis optica. Pract Neurol 12(4):209–214. https://doi.org/10.1136/practneurol-2012-000237
Yang CS, Yang L, Li T, Zhang DQ, Jin WN, Li MS, Su N, Zhangning N, Liu Q, Shao ZH, Yu C, Shi FD (2013) Responsiveness to reduced dosage of rituximab in Chinese patients with neuromyelitis optica. Neurology 81(8):710–713. https://doi.org/10.1212/WNL.0b013e3182a1aac7
Zhang M, Zhang C, Bai P, Xue H, Wang G (2017) Effectiveness of low dose of rituximab compared with azathioprine in Chinese patients with neuromyelitis optica: an over 2-year follow-up study. Acta Neurol Belg 117(3):695–702. https://doi.org/10.1007/s13760-017-0795-6
Jones RB, Furuta S, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, Savage CO, Segelmark M, Tesar V, van Paassen P, Walsh M, Westman K, Jayne DR, European Vasculitis S (2015) Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis: 2-year results of a randomised trial. Ann Rheum Dis 74(6):1178–1182. https://doi.org/10.1136/annrheumdis-2014-206404
National Institute for Health and Care Excellence technology appraisal guidance TA308 Rituximab in combination with glucocorticoids for treating anti-neutrophil cytoplasmic antibody-associated vasculitis. https://www.nice.org.uk/guidance/ta308/chapter/1-Guidance. Accessed 16 Jun 2020
Iorio R, Damato V, Alboini PE, Evoli A (2015) Efficacy and safety of rituximab for myasthenia gravis: a systematic review and meta-analysis. J Neurol 262(5):1115–1119. https://doi.org/10.1007/s00415-014-7532-3
Tandan R, Hehir MK 2nd, Waheed W, Howard DB (2017) Rituximab treatment of myasthenia gravis: a systematic review. Muscle Nerve 56(2):185–196. https://doi.org/10.1002/mus.25597
Amor S, Baker D, Khoury SJ, Schmierer K, Giovanonni G (2020) SARS-CoV-2 and multiple sclerosis: not all immune depleting DMTs are equal or bad. Ann Neurol 87(6):794–797. https://doi.org/10.1002/ana.25770
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
Clara Grazia Chisari received grant for congress participation from Almiral, Biogen, Merck Serono, Novartis, Roche, Sanofi and TEVA. Eleonora Sgarlata declares no conflict of interest. Sebastiano Arena declares no conflict of interest. Simona Toscano declares no conflict of interest. Maria Luca declares no conflict of interest. Francesco Patti has received honoraria for speaking activities by Bayer Schering, Biogen, Merck Serono, Novartis and Sanofi Aventis; he also served as advisory board member the following companies: Bayer Schering, Biogen Idec, Merck Serono, Novartis; he was also funded by Pfizer and FISM for epidemiological studies; finally he received grant for congress participation from Bayer Schering, Biogen Idec, Merck Serono, Novartis, Roche, Sanofi Aventis and TEVA.
Rights and permissions
About this article

Cite this article
Chisari, C.G., Sgarlata, E., Arena, S. et al. Rituximab for the treatment of multiple sclerosis: a review. J Neurol 269, 159–183 (2022). https://doi.org/10.1007/s00415-020-10362-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-020-10362-z