
Abstract
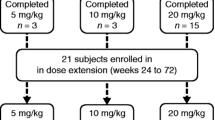
The objective of this study was to describe a large Italian cohort of patients with late-onset glycogen storage disease type 2 (GSDII) at various stages of disease progression and to evaluate the clinical effectiveness of alglucosidase alpha enzyme replacement therapy (ERT). Previous studies showed in late-onset patients ERT efficacy against placebo and variable response in uncontrolled studies. Seventy-four juvenile or adult GSDII patients were treated with ERT in a multicenter open label, non-randomized study, from 12 months up to 54 months. Recombinant human alpha glucosidase (rh-GAA) was injected by intravenous route at 20 mg/kg every second week. Patients were divided into three groups according to ERT duration: Group A received treatment for 12–23 months (n = 16), Group B for 24–35 months (n = 14), and Group C for more than 36 months (n = 44). Clinical assessment included a 6-min walk test (6MWT), forced vital capacity (FVC), the Walton and Gardner-Medwin score, the number of hours of ventilation, body mass index, echocardiography and blood creatine kinase (CK). Included in our cohort were 33 males and 41 females (M:F = 0.8:1), with a mean age at first symptoms of 28.3 years (range 2–55 years) and a mean age of 43 years at study entry (range 7–72 years). Seven wheelchair bound patients, as well as 27 patients requiring ventilation support, were included. After treatment we could observe an increase in distance walked on the 6MWT in the large majority of patients (48/58; 83%), with an overall mean increase of 63 m (from 320 ± 161 to 383 ± 178 m). After treatment in the majority of patients FVC was improved or unchanged (45/69; 65%). In ventilated patients we observed an improvement in average number of hours off the ventilator (from 15.6 to 12.1 h). Six patients stopped mechanical ventilation and two others started it. The effect of therapy was not related to ERT duration. Nine of 64 patients (13%) that underwent to echocardiography showed a variable degree of cardiac hypertrophy (left ventriculum or septum), and a positive effect was observed after 36 months of ERT in one adult case. Discontinuation of treatment occurred in four patients: one drop-off case, one patient died for a sepsis after 34 months of treatment and two patients stopped ERT for worsening of general clinical condition. Mild adverse effects were observed in four cases (5%). This study represents the largest cohort of late-onset GSDII patients treated with ERT, and confirm a positive effect of treatment. These results, obtained in a large case series on therapy, indicate a favourable effect of ERT therapy, even in more advanced stage of the disease.


Similar content being viewed by others


References
Hirschhorn R, Reuser AJ (2001) Glycogen storage disease type II: acid alphaglucosidase (acid maltase) deficiency. In: Scriver CR, Sly W, Valle D (eds) The metabolic and molecular bases of inherited disease, 8th edn edn. McGraw-Hill, New York, pp 3389–3420
Engel AG, Hirschhorn R (1994) Acid maltase deficiency. In: Engel A, Franzini-Armstrong C (eds) Myology: basic and clinical. McGraw-Hill, New York, pp 1533–1553
Laforet P, Nicolino M, Eymard PB et al (2000) Juvenile and adultonset acid maltase deficiency in France: genotype-phenotype correlation. Neurology 55:1122–1128
Hagemans ML, Winkel LP, Van Doorn PA, Hop WJ, Loonen MC, Reuser AJ, Van der Ploeg AT (2005) Clinical manifestation and naturai course of late-onset Pompe’s disease in 54 Dutch patients. Brain 128(Pt 3):671–677
Winkel LP, Hagemans ML, van Doorn PA, Loonen MC, Hop WJ, Reuser AJ, van der Ploeg AT (2005) The natural course of non-classic Pompe’s disease: a review of 225 published cases. J Neurol 252:875–884
Forsha D, Li JS, Smith PB, van der Ploeg AT, Kishnani P, Pasquali SK (2011) Late-onset treatment study investigators cardiovascular abnormalities in late-onset Pompe disease and response to enzyme replacement therapy. Genet Med 13:625–631
Slonim AE, Bulone L, Goldberg T, Minikes J, Slonim E, Galanko J, Martiniuk F (2007) Modification of the naturai history of adult-onset acid maltase deficiency by nutrition and exercise therapy. Muscle Nerve 35(l):70–77
van Capelle CI, Winkel LP, Hagemans ML, Shapira SK, Arts WF, van Doorn PA, Hop WC, Reuser AJ, van der Ploeg AT (2008) Eight years experience with enzyme replacement therapy in two children and one adult with Pompe disease. Neuromuscul Disord 18(6):447–452
Strothotte S, Strigi-Pill N, Grunert B, Kornblum C, Eger K, Wessig C, Deschauer M, Breunig F, Glocker FX, Vielhaber S, Brejova A, Hilz M, Reiners K, Muller-Felber W, Mengel E, Spranger M, Schoser B (2010) Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J Neurol 257:91–97
van der Ploeg AT, Clemens PR, Corzo D, Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS, Laforet P, Lake SL, Lange DJ, Leshner RT, Mayhew JE, Morgan C, Nozaki K, Park DJ, Pestronk A, Rosenbloom B, Skrinar A, van Capelle CI, van der Beek NA, Wasserstein M, Zivkovic SA (2010) A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J Med 362(15):1396–1406
Bembi B, Pisa FÉ, Gonfalonieri M, Ciana G, Fiumara A, Parini R, Rigoldi M, Moglia A, Costa A, Carlucci A, Danesino C, Pittis MG, Dardis A, Ravaglia S (2010) Long-term observational, non-randomized study of enzyme replacement therapy in late-onset glycogenosis type II. J Inherit Metab Dis 33:727–735
van Capelle CI, van der Beek NA, Hagemans ML, Arts WF, Hop WC, Lee P, Jaeken J, Frohn-Mulder IM, Merkus PJ, Corzo D, Puga AC, Reuser AJ, van der Ploeg AT (2010) Effect of enzyme therapy in juvenile patients with Pompe disease: a three-year open-label study. Neuromusc Disord 20:775–782
van der Ploeg AT (2010) Where do we stand in enzyme replacement therapy in Pompe’s disease? Neuromusc Disord 20:773–774
Conflict of interest
Drs Toscano and Angelini are members of the Pompe Global Advisory Board, sustained by Genzyme, and received reimbursement for the participation to the board meetings. Drs Angelini, Toscano, Bembi, Servidei, Filosto and Semplicini received support from Genzyme for organizing or participating to scientific meetings.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
C. Angelini, T. Mongini, A. Toscano: Coordinators of the Italian Group on GSDII.
The members of the Italian GSDII Group are listed in the Appendix.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Appendix: The Italian GSDII Group
Appendix: The Italian GSDII Group
L. Vercelli, Centre for Neuromuscular Diseases “P. Peirolo”, Department of Neurosciences, University of Torino, Torino, Italy; R. di Giacopo, Department of Neurology, Università Cattolica, Rome, Italy; V. Lucchini and G. P. Comi, Fondazione IRCCS Ca' Granda Osp. Policlinico, Dept. of Neurology, Centro Dino Ferrari, University of Milan, Italy; V. Tugnoli, Neurology Unit, S. Anna Hospital, Ferrara, Italy; M. Rigoldi, Rare Metabolic Diseases Unit, Department of Pediatrics, San Gerardo Hospital, Monza, Italy; M. B. Pasanisi, Immunology and Muscular Pathology Unit, National Neurological Institute Carlo Besta, Milan, Italy; R. Piras, Neuromuscular Unit, Department of Cardiovascular Science and Neurology, University of Cagliari, Italy; F. Giannini, Departments of Neurosciences, University of Siena, Italy; E. Barca, Department of Neurosciences, Psychiatry and Anaesthesiology, University of Messina, Messina, Italy; S. Gasperini, Clinic of Pediatric Neurology, Meyer Hospital, Firenze, Italy; G. Ricci, Neurological Institute, University of Pisa, Pisa, Italy; D. Diodato, Department of Neurological Sciences, Second University of Naples, Italy; A. Ariatti, Department of Medicine, Endocrinology, Metabolism and Geriatrics, University of Modena and Reggio Emilia, Modena, Italy.
Rights and permissions
About this article
Cite this article
Angelini, C., Semplicini, C., Ravaglia, S. et al. Observational clinical study in juvenile-adult glycogenosis type 2 patients undergoing enzyme replacement therapy for up to 4 years. J Neurol 259, 952–958 (2012). https://doi.org/10.1007/s00415-011-6293-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00415-011-6293-5