
Abstract
An understanding of the natural history of beta cell responses is an essential prerequisite for interventional studies designed to prevent or treat type 1 diabetes. Here we review published data on changes in insulin responses in humans with type 1 diabetes. We also describe a new analysis of C-peptide responses in subjects who are at risk of type 1 diabetes and enrolled in the Diabetes Prevention Trial-1 (DPT-1). C-peptide responses to a mixed meal increase during childhood and through adolescence, but show no significant change during adult life; responses are lower in adults who progress to diabetes than in those who do not. The age-related increase in C-peptide responses may account for the higher levels of C-peptide observed in adults with newly diagnosed type 1 diabetes compared withhose in children and adolescents. Based on these findings, we propose a revised model of the natural history of the disease, in which an age-related increase in functional beta cell responses before the onset of autoimmune beta cell damage is an important determinant of the clinical features of the disease.
Similar content being viewed by others

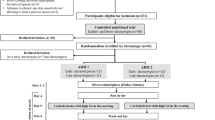

Introduction
Type 1 diabetes occurs when autoimmune destruction of beta cells has reached a stage at which metabolic stability is no longer maintained. At present it is not well understood when this autoimmune destruction begins in relation to the clinical presentation, or whether it follows a gradual or steep course. With the development of new immune modulatory therapies that may be able to arrest the autoimmune destructive process, understanding the details of this progression assumes great importance [1–4]. Knowledge of the state of beta cell function at each stage of the disease process allows the risks of therapy in terms of adverse events to be weighed against the benefit of recovering residual beta cell function. In this review, we discuss a new analysis of data from the Diabetes Prevention Trial-1 (DPT-1) on insulin secretion in individuals before and after the onset of type 1 diabetes. We also review existing information on insulin secretion in type 1 diabetes, and explore our understanding of the natural history of the disease and the merits of intervention at each stage [5, 6].
Beta cell mass in humans: technical considerations
There are few data concerning changes in beta cell mass in humans. One of the principal limitations of human studies is that it is not currently possible to measure beta cell mass in vivo. Instead, indirect methods based on secretory responses are used to approximate beta cell mass. These approaches have been validated in subjects undergoing syngeneic islet transplants, in whom measured insulin secretion was found to correlate well with the islet mass transplanted [7]. One limitation is that the mass that was viable after transplant was estimated rather than measured in these studies. Most investigations have used C-peptide responses to secretagogues as a measure of insulin secretion, but these measurements have experimental pitfalls that limit their precision under non-steady-state conditions [8]. There is, however, a method of calculating the insulin secretory rate (ISR) which takes the volume of distribution and half-life of C-peptide into account, and provides a better estimate of rapid changes in insulin secretion [9, 10]. This technique has been validated in normal individuals and studied in patients with type 2 diabetes [8, 11]. However, it has been little used to study insulin secretion in type 1 diabetes, and existing validation studies have generally involved young adults, who represent the majority of patients with type 1 diabetes [12], rather than young children. For this reason, evaluation of the C-peptide response to a secretagogue remains the most widely used surrogate measure of functional beta cell mass in children with type 1 diabetes. Since ISR is derived from serum C-peptide levels, there is a very close correlation between the two measures, with the caveat that during non-steady-state conditions, when secretory rates change rapidly, C-peptide levels may under- or overestimate insulin secretion [8].
Insulin secretion increases with growth and metabolic demands
Beta cell mass is not constant. In rodents, beta cell mass increases during normal growth, and is tightly controlled through replication of pre-existing beta cells, differentiation of precursor cells and cell death [13, 14]. In rodents, beta cell mass is stable from 2 to 20 days of life but increases six- to seven-fold between 1 and 7 months of age [15, 16]. After the neonatal period, beta cell replication and mass increase and apoptosis decreases in proportion to body weight and age [13]. Beta cell mass continues to increase well into adult life, eventually reaching a plateau [16].
The peak incidence of type 1 diabetes in humans is between the ages of 6 and 15 years, the time of rapid growth. An increase in insulin secretion during adolescence has been well documented, and is thought to occur in response to increased insulin resistance [17]. Insulin resistance in adolescents was first described by Amiel et al. in 1986 [18] and has since been confirmed by both the minimal model and glucose clamp techniques. An 18–35% reduction in insulin sensitivity occurs with adolescence [19–23]. In a large cross-sectional study of insulin action in normal adolescents between 10 and 14 years of age, insulin sensitivity decreased in early puberty (Tanner 2) with a nadir at mid-puberty (Tanner 3) and an almost complete return to prepubertal levels by the end of puberty (Tanner 5) [24]. Although the mechanism of insulin resistance is not well understood, it cannot be accounted for by adiposity, and changes in insulin secretion during puberty correlate with similar changes in components of the growth hormone axis [17, 18], but not with changes in gonadal sex steroids (oestradiol, testosterone and dihydrotestosterone); a correlation with changes in adrenal androgens (dehydroepiandrosterone sulphate and androstenedione) has also been reported [19–21, 25, 26].
A progressive increase in insulin secretion has been described in individuals at risk of type 1 diabetes. Schatz et al. reported results of stimulated insulin secretion in 26 subjects aged 3 to 57 years, all of whom had islet cell antibodies (ICA) and a decreased first-phase insulin response to intravenous glucose [27]. C-peptide increased with age, and an increase in insulin secretion was evident in both the peak and total area under the curve C-peptide response to an OGTT and a mixed meal tolerance test (MMTT). When subjects were divided into those who did and those who did not develop type 1 diabetes during 5.5 years of follow-up, the latter exhibited an increase in C-peptide with time, and the former did not.
Metabolic demand stimulates an increase in beta cell mass in animal models and in humans. Both beta cell hypertrophy and hyperplasia have been shown to account for increases in beta cell mass during conditions of increased demand, for example due to ablation of one insulin receptor allele or in Zucker Diabetic Fatty rats [28, 29]. Beta cell mass also expands with mild and transient hyperglycemia, and hyperglycemia may be the stimulus responsible for expansion of islets that are transplanted into rats following partial pancreatectomy [30, 31]. Replication, neogenesis, and transdifferentiation together with apoptosis have all been suggested to account for the development of new beta cells in these rodent models [14, 32]. Moreover, it has been suggested that glucagon-like peptide-1, which has been shown to stimulate beta cell replication and decrease beta cell apoptosis in rodents, plays a role in islet regeneration after partial pancreatectomy [33, 34]. In obese humans, insulin secretion is increased compared with lean individuals [10], and beta cell mass also increases during pregnancy. Women with longstanding type 1 diabetes, in whom C-peptide levels were not detectable before pregnancy, sometimes produces detectable levels of C-peptide during the third trimester [35, 36]. It has been postulated that an expansion of beta cell mass accounts for the hypoglycaemia that sometimes occurs during this period of gestation. After pregnancy, rodent studies have shown an involution of beta cells through apoptosis [13, 14].
Analysis of insulin secretion before and at diagnosis of type 1 diabetes: data from the DPT-1
In an attempt to improve understanding of changes in insulin production during the prediabetic period and at the time of diagnosis of type 1 diabetes, we analysed data from DPT-1 subjects who were at risk of developing type 1 diabetes. This trial recruited first- and second-degree relatives of patients with type 1 diabetes and tested whether treatment with oral or parenteral insulin would prevent the onset of diabetes [5, 6]. Here we report a secondary analysis of insulin secretion in this population. A total of 2,103 subjects were placed in four categories for risk of type 1 diabetes, based on levels of ICA, insulin autoantibodies, HLA type, and insulin and glucose responses to intravenous and oral glucose tolerance tests. The high-risk category consisted of 372 patients with at least a 50% probability of developing type 1 diabetes within 5 years. Subjects in this category received either subcutaneous insulin or no treatment. Another 338 patients, judged to have a 26 to 50% 5-year risk of developing diabetes, were defined as intermediate-risk, and received either oral insulin or placebo. For our analysis we included data from the untreated control groups (i.e. not treated with parenteral or oral insulin) in both the high-risk and intermediate-risk studies (n=340).
The enrolled subjects underwent a 2-h MMTT every 2 years (high-risk) or every 3 years (intermediate-risk). Of the 340 subjects assessed, 121 progressed to type 1 diabetes. The average age of onset of diabetes was 13.5±0.58 years. Progressors to type 1 diabetes were followed for an average of 2.54±0.14 years and underwent an average of 2.2 MMTTs (range 1 to 5). The non-progressors were followed for an average of 3.2±0.11 years and underwent an average of 2.2 MMTTs (range 1 to 5).
Insulin responses during the MMTTs were analysed both in cross-section and longitudinally, and differentiated according to whether the subjects progressed to diabetes or not over the follow-up periods. The AUC, reflective of the maximal C-peptide response to the mixed meal, was determined for each MMTT. In order to determine the relationship between age and the C-peptide response, a regression was performed separating children and adolescents (age 0–17 years) from adults (age >17 years). The data were also analysed by dividing the C-peptide AUC by the subjects’ BMIs to adjust for alterations in insulin secretion that occur in response to changes in insulin resistance secondary to changes in body composition [37].
In cross-sectional analysis, the rate of increase of the C-peptide response (AUC) in children and adolescents (0–17 years) who did not progress to diabetes was 15.2±1.79 (SEM) pmol ml−1 120 min−1 year−1 (r=0.324); in children and adolescents who did progress to diabetes, the rate of increase was 13.9±2.56 pmol ml−1 120 min−1 year−1 (r=0.397) [38]. The regression analysis that was performed (SigmaPlot↑; Systat, Richmond, CA, USA) used a linear mode. The p values for these regressions were both <0.0001. In adults (>17 years) who did not progress to diabetes there was no significant change in C-peptide response with age at the time of study, but in adults who did progress, the increase was 7.76±2.32 pmol ml−1 120 min−1 year−1 (r<0.589, p=0.0031). When C-peptide responses were corrected for BMI (C-peptide:BMI ratio), there was also a significant increase in the response with increasing age at the time of study, both in children and adolescents who did not progress to diabetes (0.403±0.108 pmol ml−1 120 min−1 kg−1 m−2 year−1 [r=0.207, p=0.0002]) and in those who did (0.362±0.126 pmol ml−1 120 min−1 kg−1 m−2 year−1 [r=0.222, p<0.0001]), but not in either group of adults (Fig. 1). The slopes describing these were not significantly different in children and adolescents who did and did not progress to diabetes, when the effect of the group (progressor vs non-progressor) on the change with age was analysed by ANOVA (Sigmaplot). However, using this approach, the effect of the group on the change in C-peptide AUC with age was significant (p=0.029), but not the effect of the group on the C-peptide AUC:BMI ratio. We found no evidence that a difference in BMI accounted for our findings, since BMI increased similarly in the progressor and non-progressor groups (Fig. 2).

C-peptide and insulin secretory responses to a mixed meal in subjects at risk of type 1 diabetes. MMTTs were performed in subjects at risk of type 1 diabetes. The C-peptide AUC:BMI ratios are plotted for each of the studies done on the 340 patients. Dark blue circles, subjects who developed diabetes; light blue circles, subjects who did not develop diabetes. Red regression line, p<0.0001 for relationships for progressors (age 0–17 years); green regression line, p=0.005 for relationships for non-progressors (age 0–17 years)

BMI in the DPT-1 study participants at the time of each MMTT. Graph shows BMI values from each of the studies done on the 340 patients. Dark blue circles, subjects who developed diabetes; light blue circles, subjects who did not develop diabetes. Regression lines: red, progressors (age 0–17 years), slope 0.58 kg m−2 year−1; purple, progressors (age >17 years), slope 0.25 kg m−2 year−1; green, non-progressors (age 0–17 years), slope 0.52 kg m−2 year−1; olive green, non-progressors (age >17 years), slope 0.1 kg m−2 year−1
To examine changes within individual subjects over time, we determined the slopes of the lines describing the C-peptide AUC:BMI ratio of all studies performed in a given individual before type 1 diabetes had been diagnosed. Of the 340 individuals studied in the cross-sectional analysis above, 223 (159 children and adolescents and 64 adults) underwent more than one MMTT and had data that could be used to calculate slopes. Each individual was assigned to an age group based on the average age at which the studies were done (Fig. 3). The increase in the average of the slopes in children and adolescents, but not in adults, was significant (0.938±0.358 pmol ml−1 120 min−1 kg−1 m−2 year−1 vs −0.011±0.242 pmol ml−1 120 min−1 kg−1 m−2 year−1, p<0.001 and NS). The slope was significantly greater in children and adolescents than in adults overall (p=0.028) (Fig. 3). In the adults who progressed to diabetes, the decrease in average slope was significant (−2.264±0.99 pmol ml−1 120 min−1 kg−1 m−2 year−1, p=0.05), the slope being significantly less in adults who progressed to diabetes than in those who did not progress (p<0.01).The average slope increased in children and adolescents who did not progress to diabetes (0.943±0.339 pmol ml−1 120 min−1 kg−1 m−2 year−1, p<0.01), but the difference between the slopes of the children and adolescents who did and did not progress to diabetes was not significant (p=0.41), due to the large variance in the slopes. A significantly greater proportion of adults who progressed to diabetes had negative slopes (8/9) compared with those who did not progress (22/55, Χ2 p<0.01), but there was no significant difference in the proportion of subjects with negative slopes among children and adolescents who did (21/48) or did not (35/111) progress to diabetes.

Slopes of C-peptide:BMI ratios in both children and adolescents, and in adults in the DPT-1. A slope describing the changes in the C-peptide AUC:BMI ratios over time was calculated for each subject who underwent more than one MMTT (64 adults, 159 children and adolescents). The mean±SEM of the slopes for progressors (n=57, red bars) and non-progressors (n=166, green bars) are shown. *p<0.05 or #p<0.01 for difference between mean values and 0 by paired t-test; **p<0.01 for child vs adult progressors; ***p=0.006 for adult progressor vs non-progressor by Mann–Whitney test
Is the increased insulin secretion during adolescence unique to this period?
Previous studies have reported increases in insulin resistance during adolescence that may account for changes in insulin response over that period of time [17]. Most of these studies were cross-sectional and were unable to distinguish between an effect of puberty and an effect of age [17, 39]. To determine whether the changes we found in children were confined to adolescence, we studied the changes over time in C-peptide responses (i.e. slopes) and stratified the subjects by age into three categories: prepubertal children (age <8 years, mean 6.32±0.215 years, n=27), adolescents (age 8–16 years, mean = 11.73 ± 0.212 years, n=132), and adults (age >17 years, mean 28.6±1.31 years, n=64). Both prepubertal and pubertal (p<0.05 for each) children who did not progress to diabetes showed significant increases in the C-peptide:BMI ratio over time. No significant increase was seen in the average slope in children who progressed to diabetes, nor was a significant difference seen between the average slopes of prepubertal and pubertal children who did and did not progress (Fig. 4). In an earlier longitudinal study of normal children, 29 pre-pubertal children and 31 children progressing into puberty were followed over time. The acute insulin response to an intravenous glucose tolerance test was found to increase by 30% in children progressing into puberty, while in those who remained prepubertal no increase in insulin secretion was seen [20]. In contrast, we found that the rate of increase in insulin responses over time was not significantly different in prepubertal and pubertal children, but was significantly different when adults were compared with children and adolescents (Fig. 3). This suggests that insulin secretion increases throughout this entire period of growth, consistent with an expansion of beta cell mass during this time period. A limitation of our analysis is that we were not able to separate individuals on the basis of Tanner stages, but rather did so by age. Future longitudinal studies in normal prepubertal and pubertal children will need to further evaluate insulin secretion during these periods of growth and development.

Slopes of C-peptide:BMI ratios in age subgroups in the DPT-1. The mean±SEM of the slopes for progressors and non-progressors were subdivided by age group (prepubertal, 0–8 years, n=27, yellow bars; pubertal, 8–17, n=132, orange bars; and adult, >17 years, n=64, red bars). The means were increased in the prepubertal and pubertal subjects who did not progress to diabetes (p<0.05 by paired t-test). There was a greater slope in pre-pubertal vs adult progressors and non-progressors (**p<0.01 by Mann–Whitney test)
Our analysis of the DPT-1 data shows that insulin secretion increases in childhood in individuals at risk of diabetes. This increase appears to occur before and during puberty, and then decreases during adulthood. The increase in the C-peptide response during childhood is similar in persons who do and do not progress to diabetes and, even in progressors, the slope in children and adolescents is increased in comparison with adults. We used a linear regression model to perform our analysis. Other models could be used to analyse the cross-sectional data, but the use of slopes describing the changes in each individual is the more accurate approach to determine changes over time because of the variability of the cross-sectional data on the C-peptide:BMI ratio at each time point.
It is not clear whether the changes in insulin secretion described by us would be similar in healthy individuals, but they are indicative of changes during progression to type 1 diabetes. It is possible that the islet inflammatory process affects beta cell mass and/or function in a positive or negative manner. Previous studies have shown an increase in beta cell proliferation as well as a loss of beta cell mass in NOD mice during the prediabetic period [40]. All the subjects we studied have evidence of active autoimmunity in the form of islet cell autoantibodies, but individual differences in the extent of the inflammatory process could affect the insulin secretory response as well as beta cell dynamics.
The approaches we used for analysis have some limitations. Our analysis involved measurement of the C-peptide response to an MMTT, which is an indirect reflection of beta cell mass. As noted above, C-peptide responses may not accurately reflect insulin secretion under non-steady-state conditions. Nonetheless, the responses are likely to reflect the peak response to the physiological stimulus over time. It is possible that a more robust stimulus for insulin secretion may uncover greater impairment in individuals who go on to develop type 1 diabetes. This is evident from comparison of insulin secretory responses at the time of diagnosis of type 1 diabetes in response to a mixed meal (52% of normal control subjects) with results for glucagon stimulation after a hyperglycaemic clamp (25% of normal control subjects) [1, 12]. We corrected the C-peptide AUC for the BMI in order to minimise the contribution of obesity and body size, which increase insulin resistance, to our analysis of insulin secretion [10, 41]. We made this correction since these changes during growth might have masked a trend in insulin secretion related to the development of type 1 diabetes, or might also have suggested that insulin secretion was increasing over a period of growth when it was constant in relation to body size. Instead, our studies indicate that, even when corrected for body mass, insulin secretion increases during growth in subjects at risk of type 1 diabetes. These findings are consistent with an expansion of beta cell mass during this period, but direct measurement would be needed to confirm this suggestion.
Can changes in insulin secretion over time in individuals at risk predict the development of type 1 diabetes?
It has been suggested that a relative decrease in insulin secretion occurs over time in high-risk individuals who progress to type 1 diabetes compared with those who do not progress, and that this may be a useful additional marker of risk [27]. We found such a correlation in adult patients, but not in young subjects. In children and adolescents who did and did not progress to type 1 diabetes there were no significant differences in patterns of insulin secretion over time, although the increase in C-peptide responses was slightly higher in non-progressors. The relatively small number of subjects in certain subgroups (e.g. prepubertal progressors, n=9) may account for our failure to detect a difference between progressors and non-progressors. It should also be appreciated that our division into progressors or non-progressors reflects a relatively brief observation period. Since the rate of progression to diabetes does not fall over time [5], some of the designated non-progressors will in time develop diabetes.
Insulin secretion at the time of diagnosis of type 1 diabetes
Historical data, based on autopsy studies and levels of stimulated C-peptide, have suggested a 67 to 90% loss of normal beta cell mass at the time of diagnosis of type 1 diabetes [42]. Recent studies using a hyperglycaemic clamp and glucagon stimulation have shown responses at diagnosis that are 75% lower than in age-matched healthy control subjects [1]. Nonetheless, the level of C-peptide secretion at diagnosis is of clinical importance. In the Diabetes Control and Complications Trial (DCCT), a stimulated C-peptide level of ≥0.2 pmol/ml was correlated with improved metabolic control and a decreased rate of complications when compared to patients with C-peptide levels below this threshold [43]. Stimulated C-peptide levels >0.2 pmol/ml were seen in 33% of subjects <18 years of age and in 48% of subjects >18 years of age with disease duration of <5 years. Our previous analysis of insulin secretion in recently diagnosed subjects with type 1 diabetes found that all 52 subjects had a C-peptide response of >0.2 pmol/ml in response to a mixed meal, and that the total insulin secretory response to this physiological stimulus was 52% of that in normal controls [12]. We found similar levels of residual insulin secretion in patients with new-onset type 1 diabetes in our current analysis. The percentage loss of ISR at the time of diagnosis in DPT-1 participants who progressed to diabetes was calculated by dividing the difference between the response before and after diagnosis by the response before diagnosis. The loss of C-peptide response to a mixed meal was remarkably small, and was similar in subjects below the age of 17 (mean −6.84±6.18%, range −96 to 136%) and those older than 17 (mean −16.1±12.1%, range −93 to 41%) (Fig. 5). Since clamp studies have shown that the impairment is greater than can be identified by an MMTT, these studies suggest that the level of insulin secretion that is commonly present at the time of diagnosis is nearly sufficient to meet daily needs, but is clearly reduced in comparison with normal in the face of stronger stimulation.

Loss of C-peptide at diagnosis of type 1 diabetes. The percentage loss of C-peptide:BMI ratios after diagnosis was calculated as: (C-peptide AUC/BMI AUC after diagnosis)−(before diagnosis) / (C-peptide AUC/BMI before diagnosis)×100. The average percentage loss was not significantly different in adults and children and adolescents. Red circles, ages 0 to 17 years (n= 58); purple circles, ages >17 years (n=12)
Age of onset is an important determinant of the absolute initial C-peptide response at diagnosis, but the loss of C-peptide response with further progression of type 1 diabetes is similar across ages. In a previous study, we found a significant relationship between age and the ISR to a 4-h MMTT in subjects with type 1 diabetes at the time of presentation. However, age was not a determinant of the decline in ISR over time after diagnosis [12]. A report from an ADA workshop panel on the use of C-peptide as an endpoint for studies of type 1 diabetes also indicated that levels of C-peptide were lower at diagnosis in younger individuals, but that the difference in the rates of decline after diagnosis in younger and older individuals was not clear [43]. Similar findings have been reported by others: Clarson et al. reported that stimulated C-peptide was associated with age of diagnosis in a group of 20 children with type 1 diabetes [44]. Ludvigsson et al. studied a group of subjects aged 6 to 17 years with a disease duration of 3 to 14 years, and also reported a positive correlation between age of diagnosis and residual C-peptide. Other studies reported that there was a similar decline in C-peptide in 204 consecutive cases followed for 7.4 years, but age at diagnosis and male sex were positively correlated with residual C-peptide at 5 years [45–47]. Another study reported that 25% of latent autoimmune diabetes for adults (LADA) subjects had a >50% decrease in fasting C-peptide 18 months after diagnosis, which was significantly greater than in subjects with type 2 diabetes, but children with type 1 diabetes were not included for comparison in the same analysis [48]. Thus, differences in the rate of progression (loss of beta cell responses) in younger vs older individuals with type 1 diabetes have not been found consistently.
Insulin secretion after prolonged duration of type 1 diabetes
Collectively, findings from a wide range of prospective studies of adults and children with autoimmune diabetes indicate that there is a persistent decline in insulin production over time to undetectable or near undetectable levels in subjects with type 1 diabetes [12, 42, 43, 49, 50]. Anatomical and functional studies have described the finding of insulin-containing cells in subjects with long disease duration, but the physiological significance of this finding is not known [51, 52]. In subjects in the DCCT with disease duration of between 5 and 15 years, 8% of adults and 3% of children had stimulated C-peptide levels >0.2 pmol/ml, and disease duration was less than 10 years in the majority of those who did [43]. Clinically useful insulin production persisting more than 5 years after diagnosis of type 1 diabetes is thus the exception rather than the rule. The fact that a higher percentage of older subjects have detectable C-peptide levels may be explained by a higher level of C-peptide at the time of diagnosis.
Given that age is an important determinant of C-peptide responses, we need to enquire whether ‘latent’ autoimmune diabetes in adults is different from autoimmune diabetes of children, or represents the same pathological process with greater insulin production at its outset. Schiffrin and coworkers found that lower age was associated with a more rapid rate of loss of C-peptide in a group of 151 patients with a mean age of 10.2±4.6 years, followed for 2 years [53]. Only few studies have prospectively measured insulin production in subjects with LADA. The UKPDS found that, among patients who were younger than 35 years old at diagnosis, 94% of those with ICA and 84% of those with anti-GAD65 antibodies required insulin therapy by 6 years, compared with 44% of subjects with ICA and 34% with anti-GAD65 antibodies when older than 55 years at diagnosis, clearly suggesting a more rapid progression of beta cell failure in younger versus older LADA patients [54]. In other studies, approximately 80% of ICA-positive subjects with adult onset type 2 diabetes developed complete beta cell failure after 12 years. Other data on the rates of loss of C-peptide suggest that it is similar throughout childhood and in ICA-positive adults compared with children [46, 55, 56]. However, one study suggested a more rapid rate of loss of C-peptide in adults with type 1 diabetes (mean age 45 years) than in LADA patients (mean age 59 years) after 10 years from diagnosis. However, the analysis of this study was limited to fasting C-peptide levels [57]. The available data therefore suggest that LADA may reflect the same pathological process as childhood type 1 diabetes, but starting with greater insulin production.
A revised model of the natural history of type 1 diabetes
In an early publication, Eisenbarth suggested a model of the natural history of type 1 diabetes, in which a relatively stable beta cell mass progressively declines after the onset of autoimmune destruction [58]. Our analysis indicates that in individuals at risk of type 1 diabetes, C-peptide responses increase progressively through early childhood and adolescence. Our findings suggest that an increase occurs during this time even in subjects who progress to diabetes. However, we were unable to determine whether response is impaired in comparison with individuals who are not at risk of diabetes. In adult life, the C-peptide response to a mixed meal test does not increase further. While we are unable to measure beta cell mass directly, these findings raise the possibility that an expansion of beta cell mass in humans occurs during growth, as has been shown previously in rodents [13, 59]. The changes in C-peptide responses during growth are substantial. The mean C-peptide AUC in children and adolescents who were non-progressors was 227±7.7 pmol/120 min, and therefore the slope of 15.2 represents an increase of approximately 6.6% per year. We suggest, therefore, that the model of the natural history of the disease should be modified to account for the increase in C-peptide responses that we found both during childhood and during adolescence (Fig. 6). It remains unclear whether the changes in C-peptide responses that we observed are directly related to changes in beta cell mass or function.

Revised model of changes in beta cell function in type 1 diabetes. This revised model, based on Eisenbarth [58], accounts for the increase in beta cell mass and function that occurs during normal growth and development in persons at risk of type 1 diabetes. The beta cell mass and function achieved prior to the initiation of autoimmunity affects the clinical course because the absolute production of insulin is a determinant of metabolic control. The rates of disease progression (absolute loss of beta cell function) are represented by the descending lines that begin with the trigger of autoimmune destruction (downward arrows). Rates are likely to be similar in younger and older individuals who develop type 1 diabetes. LLD, lower limit of detection. However, because of the differences in the absolute amount of insulin secretion at the time of disease discovery, the time taken to reach a critical C-peptide threshold of <0.2 pmol/ml or even the lower limit of C-peptide detection (0.03 pmol/ml) will be longer in older individuals (c and b) than in younger patients (a)
The loss of insulin secretion in a mixed meal response at the time of diagnosis is surprisingly small, but similar in younger and older individuals. With more intense provocation, such as a hyperglycaemic clamp followed by glucagon stimulation, a greater impairment can be shown. The limited published information on the rate of loss of C-peptide responses over time suggests that it may be similar at different ages. Because the absolute level of C-peptide is an important determinant of the quality and ease of metabolic control, the clinical differences between younger and older patients with type 1 diabetes may be related to the maturation of a full beta cell response in adults, which is cut short when children and adolescents develop diabetes before this maturation is complete. Because C-peptide response levels at the time patients develop diabetes are higher in older individuals than in children and adolescents, a longer period of time is needed before C-peptide levels become undetectable in adults.
Abbreviations
- DCCT:
-
Diabetes Control and Complications Trial
- DPT-1:
-
Diabetes Prevention Trial-1
- ICA:
-
islet cell antibodies
- ISR:
-
insulin secretory rate
- LADA:
-
latent autoimmune diabetes of adults
- MMTT:
-
mixed meal tolerance test
References
Keymeulen B, Vandemeulebroucke E, Ziegler et al (2005) Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med 352:2598–2608
Raz I, Elias D, Avron A, Tamir M, Metzger M, Cohen IR (2001) Beta-cell function in new-onset type 1 diabetes and immunomodulation with a heat-shock protein peptide (DiaPep277): a randomised, double-blind, phase II trial. Lancet 358:1749–1753
Herold KC, Hagopian W, Auger JA et al (2002) Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med 346:1692–1698
Herold KC, Gitelman SE, Masharani U et al (2005) A single course of anti-CD3 monoclonal antibody hOKT3{gamma}1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of Type 1 diabetes. Diabetes 54:1763–1769
Diabetes Prevention Trial—Type 1 Diabetes Study Group (2002) Effects of insulin in relatives of patients with type 1 diabetes mellitus. N Engl J Med 346:1685–1691
Skyler JS, Krischer JP, Wolfsdorf J et al (2005) Effects of oral insulin in relatives of patients with type 1 diabetes: the Diabetes Prevention Trial-Type 1. Diabetes Care 28:1068–1076
Teuscher AU, Kendall DM, Smets YF, Leone JP, Sutherland DE, Robertson RP (1998) Successful islet autotransplantation in humans: functional insulin secretory reserve as an estimate of surviving islet cell mass. Diabetes 47:324–330
Polonsky K, Frank B, Pugh W et al (1986) The limitations to and valid use of C-peptide as a marker of the secretion of insulin. Diabetes 35:379–386
Polonsky KS, Licinio-Paixao J, Given BD et al (1986) Use of biosynthetic human C-peptide in the measurement of insulin secretion rates in normal volunteers and type I diabetic patients. J Clin Invest 77:98–105
Polonsky KS, Given BD, Hirsch L et al (1988) Quantitative study of insulin secretion and clearance in normal and obese subjects. J Clin Invest 81:435–441
Polonsky KS, Given BD, Hirsch LJ et al (1988) Abnormal patterns of insulin secretion in non-insulin-dependent diabetes mellitus. N Engl J Med 318:1231–1239
Steele C, Hagopian WA, Gitelman S et al (2004) Insulin secretion in type 1 diabetes. Diabetes 53:426–433
Bonner-Weir S (2000) Islet growth and development in the adult. J Mol Endocrin 24:297–302
Scaglia L, Cahill CJ, Finegood DT, Bonner-Weir S (1997) Apoptosis participates in the remodeling of the endocrine pancreas in the neonatal rat. Endocrinology 138:1736–1741
Bouwens L, Rooman I (2005) Regulation of pancreatic beta cell mass. Physiol Rev 85:1255–1270
Finegood DT, Scaglia L, Bonner-Weir S (1995) (Perspective) Dynamics of β-cell mass in the growing rat pancreas: estimation with a simple mathematical model. Diabetes 44:249–256
Caprio S, Plewe G, Diamond MP et al (1989) Increased insulin secretion in puberty: a compensatory response to reductions in insulin sensitivity. J Pediatr 114:963–967
Amiel SA, Sherwin RS, Simonson DC, Lauritano AA, Tamborlane WV (1986) Impaired insulin action in puberty. A contributing factor to poor glycemic control in adolescents with diabetes. N Engl J Med 315:215–219
Saad RJ, Keenan BS, Danadian K, Lewy VD, Arslanian SA (2001) Dihydrotestosterone treatment in adolescents with delayed puberty: does it explain insulin resistance of puberty? J Clin Endocrinol Metab 86:4881–4886
Goran MI, Gower BA (2001) Longitudinal study on pubertal insulin resistance. Diabetes 50:2444–2450
Bloch CA, Clemons P, Sperling MA (1987) Puberty decreases insulin sensitivity. J Pediatr 110:481–487
Cook JS, Hoffman RP, Stene MA, Hansen JR (1993) Effects of maturational stage on insulin sensitivity during puberty. J Clin Endocrinol Metab 77:725–730
Brandou F, Brun JF, Mercier J (2005) Limited accuracy of surrogates of insulin resistance during puberty in obese and lean children at risk for altered glucoregulation. J Clin Endocrinol Metab 90:761–767
Moran A, Jacobs DR Jr, Steinberger J et al (1999) Insulin resistance during puberty: results from clamp studies in 357 children. Diabetes 48:2039–2044
Amiel SA, Caprio S, Sherwin RS, Plewe G, Haymond MW, Tamborlane WV (1991) Insulin resistance of puberty: a defect restricted to peripheral glucose metabolism. J Clin Endocrinol Metab 72:277–282
Arslanian S, Suprasongsin C, Janosky JE (1997) Insulin secretion and sensitivity in black versus white prepubertal healthy children. J Clin Endocrinol Metab 82:1923–1927
Schatz D, Cuthbertson D, Atkinson M et al (2004) Preservation of C-peptide secretion in subjects at high risk of developing type 1 diabetes mellitus—a new surrogate measure of non-progression? Pediatr Diabetes 5:72–79
Bruning JC, Winnay J, Bonner-Weir S, Taylor SI, Accili D, Kahn CR (1997) Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell 88:561–572
Pick A, Clark J, Kubstrup C, Pugh W, Bonner-Weir S, Polonsky K (1998) Role of apoptosis in failure of β cell mass compensation for insulin resistance and β cell defects in the male Zucker diabetic fatty (ZDF) rat. Diabetes 47:358–364
Montana E, Bonner-Weir S, Weir GC (1994) Transplanted beta cell response to increased metabolic demand. Changes in beta cell replication and mass. J Clin Invest 93:1577–1582
Bonner-Weir S, Deery D, Leahy JL, Weir GC (1989) Compensatory growth of pancreatic beta-cells in adult rats after short-term glucose infusion. Diabetes 38:49–53
Bonner-Weir S, Baxter LA, Schuppin GT, Smith FE (1993) A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 42:1715–1720
De Leon DD, Deng S, Madani R, Ahima RS, Drucker DJ, Stoffers DA (2003) Role of endogenous glucagon-like peptide-1 in islet regeneration after partial pancreatectomy. Diabetes 52:365–371
Xu G, Stoffers DA, Habener JF, Bonner-Weir S (1999) Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 48:2270–2276
Ilic S, Jovanovic L, Wollitzer AO (2000) Is the paradoxical first trimester drop in insulin requirement due to an increase in C-peptide concentration in pregnant Type I diabetic women? Diabetologia 43:1329–1330
Singer F, Horlick M, Poretsky L (1988) Recovery of beta-cell function postpartum in a patient with insulin-dependent diabetes mellitus. N Y State J Med 88:496–498
Sinaiko AR, Jacobs DR Jr, Steinberger J et al (2001) Insulin resistance syndrome in childhood: associations of the euglycemic insulin clamp and fasting insulin with fatness and other risk factors. J Pediatr 139:700–707
Sherry NA, Tsai EB, Herold KC (2005) Natural history of β-cell function in Type 1 diabetes. Diabetes 54(Suppl 2):S32–S39
Caprio S, Boulware D, Tamborlane V (1992) Growth hormone and insulin interactions. Horm Res 38 (Suppl 2):47–49
Sreenan S, Pick AJ, Levisetti M, Baldwin AC, Pugh W, Polonsky KS (1999) Increased beta-cell proliferation and reduced mass before diabetes onset in the nonobese diabetic mouse. Diabetes 48:989–996
Kim SH, Abbasi F, Reaven GM (2004) Impact of degree of obesity on surrogate estimates of insulin resistance. Diabetes Care 27:1998–2002
Faber OK, Binder C (1977) C-peptide response to glucagon. A test for the residual beta-cell function in diabetes mellitus. Diabetes 26:605–610
Palmer JP, Fleming GA, Greenbaum CJ et al (2004) C-peptide is the appropriate outcome measure for type 1 diabetes clinical trials to preserve beta-cell function: report of an ADA workshop, 21–22 October 2001. Diabetes 53:250–264
Clarson C, Daneman D, Drash AL, Becker DJ, Ehrlich RM (1987) Residual beta-cell function in children with IDDM: reproducibility of testing and factors influencing insulin secretory reserve. Diabetes Care 10:33–38
Ludvigsson J, Heding LG, Larsson Y, Leander E (1977) C-peptide in juvenile diabetics beyond the postinitial remission period. Relation to clinical manifestations at onset of diabetes, remission and diabetic control. Acta Paediatr Scand 66:177–184
Wallensteen M, Dahlquist G, Persson B et al (1988) Factors influencing the magnitude, duration, and rate of fall of B-cell function in type 1 (insulin-dependent) diabetic children followed for two years from their clinical diagnosis. Diabetologia 31:664–669
Snorgaard O, Lassen LH, Binder C (1992) Homogeneity in pattern of decline of beta-cell function in IDDM. Prospective study of 204 consecutive cases followed for 7.4 yr. Diabetes Care 15:1009–1013
Yang L, Zhou ZG, Huang G, Ouyang LL, Li X, Yan X (2005) Six-year follow-up of pancreatic beta cell function in adults with latent autoimmune diabetes. World J Gastroenterol 11:2900–2905
Faber OK, Binder C (1977) B-cell function and blood glucose control in insulin dependent diabetics within the first month of insulin treatment. Diabetologia 13:263–268
Madsbad S, McNair P, Faber OK, Binder C, Christiansen C, Transbol I (1980) Beta-cell function and metabolic control in insulin treated diabetics. Acta Endocrinol (Copenh) 93:196–200
Lohr M, Kloppel G (1987) Residual insulin positivity and pancreatic atrophy in relation to duration of chronic type 1 (insulin-dependent) diabetes mellitus and microangiopathy. Diabetologia 30:757–762
Daneman D, Clarson C (1987) Residual beta-cell function in children with type 1 diabetes: measurement and impact on glycemic control. Clin Invest Med 10:484–487
Schiffrin A, Suissa S, Weitzner G, Poussier P, Lalla D (1992) Factors predicting course of beta-cell function in IDDM. Diabetes Care 15:997–1001
Turner R, Stratton I, Horton V et al (1997) UKPDS 25: autoantibodies to islet-cell cytoplasm and glutamic acid decarboxylase for prediction of insulin requirement in type 2 diabetes. UK Prospective Diabetes Study Group. Lancet 350:1288–1293
Landin-Olsson M (2002) Latent autoimmune diabetes in adults. Ann N Y Acad Sci 958:112–116
Gottsater A, Landin-Olsson M, Lernmark A, Fernlund P, Sundkvist G (1994) Islet cell antibodies are associated with beta-cell failure also in obese adult onset diabetic patients. Acta Diabetol 31:226–231
Hosszufalusi N, Vatay A, Rajczy K et al (2003) Similar genetic features and different islet cell autoantibody pattern of latent autoimmune diabetes in adults (LADA) compared with adult-onset type 1 diabetes with rapid progression. Diabetes Care 26:452–457
Eisenbarth GS (1986) Type I diabetes mellitus. A chronic autoimmune disease. N Engl J Med 314:1360–1368
Bonner-Weir S (2000) Perspective: postnatal pancreatic beta cell growth. Endocrinology 141:1926–1929
Acknowledgements
This work was supported by grants R01DK57846, M01 RR00645, U19AI46132, M01RR01271 and P60DK20595 from the National Institutes of Health, and Special Grant 4-1999-711 and Center Grant 4-1999-841 from the Juvenile Diabetes Research Foundation. The DPT-1 was supported through cooperative agreements by the Division of Diabetes, Endocrinology, and Metabolic Diseases, NIDDK, NIH, and by NIAID, NICHD, and the National Center for Research Resources, NIH, as well as by the American Diabetes Association, the Juvenile Diabetes Foundation International, and various corporate sponsors.
Author information
Authors and Affiliations
Consortia
Corresponding author
Electronic supplementary materials
Rights and permissions
About this article
Cite this article
for the DPT-1 Study Group., Tsai, E.B., Sherry, N.A. et al. The rise and fall of insulin secretion in type 1 diabetes mellitus. Diabetologia 49, 261–270 (2006). https://doi.org/10.1007/s00125-005-0100-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0100-8