
Abstract
Background
T cell receptor-engineered T cell (TCR-T) therapy has shown promising efficacy in advanced solid tumours. Lymphodepleting (LD) chemotherapy improves TCR-T cell therapy efficacy but is associated with significant toxicities. Evidence is sparse regarding the optimum LD regimen for TCR-T cell therapy in solid tumours.
Methods
A systematic review was conducted of interventional, prospective clinical trials describing LD practices prior to TCR-T cell therapy in patients with advanced solid tumours. The objective was to define LD regimens administered prior to TCR-T cell therapy and their effects on specific safety and efficacy outcomes in this patient population.
Results
Searches returned 484 studies, 19 (231 patients) met the eligibility criteria. Cyclophosphamide (cyclo) 60 mg/kg daily (2 days), plus fludarabine (fludara) 25 mg/m2 daily (5 days) was the most common LD regimen (38% of studies). Higher dose LD regimens were associated with increased pooled incidence rates of febrile neutropaenia compared to low dose (0.64, [95% Confidence interval (CI): 0.50–0.78], vs. 0.39 [95% CI: 0.25–0.53], respectively) but were not significantly associated with higher objective responses (odds ratio: 1.05, 95%CI: 0.60–1.82, p = 0.86). A major shortfall in safety data reporting was identified; determination of LD regimen effects on many safety outcomes was not possible.
Conclusion
Standard consensus guidelines for the design and reporting of adoptive cell therapy (ACT) studies would facilitate accurate risk–benefit analysis for optimising LD regimens in patients with advanced solid tumours.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Patients with advanced solid tumours relapsed or refractory to standard of care therapies have a poor prognosis, with few further treatment options. Adoptive T cell therapy (ACT) has shown promising efficacy in various solid tumour types [1, 2]. Since early trials of tumour infiltrating lymphocytes (TILs), T cell therapies have evolved and include chimeric antigen receptor-T (CAR-T) cell and T cell receptor-engineered T (TCR-T) cell therapies [3]. TCR-T cells target a greater variety of antigens than CAR-T cells, which recognise only cell surface antigens, so may have greater efficacy in solid tumours, supported by response rates from recent clinical studies [4,5,6]. To date, no TCR-T cell therapies are licensed, albeit a positive risk benefit balance has been demonstrated in melanoma and other solid malignancies [6,7,8,9].
An essential requirement for ACT efficacy is the in vivo expansion and persistence of T cells once reinfused [1, 10, 11]. Clinical trial data show that lymphodepleting conditioning (LD) regimens consisting of cyclophosphamide (cyclo) and fludarabine (fludara) prior to cell reinfusion improves T cell in vivo expansion and persistence [12, 13].
ACT is associated with specific toxicities, including cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) [14]. Neurological adverse reactions are common to CAR-T cell therapy and cyclo/fludara dosing, so when given together, the lack of randomised controlled studies render causality assignment difficult for neurological toxicities [15].
Most safety and efficacy outcome data for LD regimens derive from CD19 CAR-T cell studies in haematological malignancies, which may not apply to TCR-T cell therapy in solid tumours. Differences include the direct access of CAR-T cells to tumour, high local concentrations of immune effector cells implicated in CRS, and neurological toxicity due to ‘on target, off tumour’ toxicity in haematological malignancies [16]. LD in haematological malignancies results in direct reduction of the malignant cell population. Bone marrow reserve is often lower than for patients with solid tumours, so rates of cytopaenias and bone marrow aplasia may differ for a given LD regimen [12]. Solid tumours inhabit an immunosuppressive microenvironment, the aim of LD being to reduce immunosuppressive cells.
Given the pivotal importance of LD in ACT, lack of randomised controlled clinical trials comparing LD regimens, and scarcity of TCR-T cell trials in solid tumours, leading to a lack of consensus regarding optimal regimens, a systematic review may help fill this knowledge gap. This review will summarise the lymphodepleting practices employed prior to TCR-T cell therapies and effects on safety and efficacy outcomes in patients with relapsed, refractory solid tumours, participating in interventional, prospective clinical studies.
Methods
Literature search and eligibility criteria
This review was written as part of a Master of Science (M.Sc.) thesis, planned prospectively with the supervisory committee and registered with the University of Manchester. It is reported per the Preferred Reporting Items for Systematic Reviews and Meta-analyses, ‘PRISMA-P’ guidelines [17, 18]. MEDLINE, Embase, ACP Journal Club, Cochrane Database of Systematic Reviews, Database of Abstracts of Reviews of Effects, Cochrane Central Register of Controlled Trials and Web of Science databases were searched, and literature retrieved, from 28 June to 4 August 2021. The search covered from the date of literature inception to 28 June through 4 August 2021. The search strategy used keywords (e.g. TCR, TCR-engineered T cells) and controlled vocabulary (e.g. receptors, antigen, T cell) related to study objectives, with no restrictions on the date of publication (full search strategy examples, Supplementary Tables 1a and 1b).
Studies were screened by a single reviewer, KO, using prespecified eligibility criteria, following the PICOTS (Population, Intervention, Comparison, Outcome, Timing, Study design) framework (Supplementary Table 2). References of related and systematic reviews were cross-referenced with the search strategy to include additional relevant studies. ClinicalTrials.gov was searched for updated results or missing methodological details not included in the original articles/abstracts.
Objectives
These were to define LD regimes administered prior to TCR-T cell infusion and effects on safety and efficacy outcomes in patients with solid tumours. LD was defined as a treatment to reduce the population of circulating lymphocytes, prior to infusion of TCR-T cells, including chemotherapy, radiotherapy and/or any other method specified. High-dose LD regimens included total doses of cyclo ≥ 120 mg/kg and fludara ≥ 100 mg/kg2. Specific safety outcomes included adverse events (AEs) of cytopaenias, bone marrow aplasia, infections, CRS, neurotoxicity, and Graft-versus-host disease (GvHD). Specific efficacy outcomes included objective response rate (ORR), defined by the Response Evaluation Criteria in Solid Tumours (RECIST) Guideline, v1.1.[19].
Risk of bias
No specific tool exists for risk of bias assessment of single-arm interventional studies; therefore, Grigor and colleagues’ risk of bias tool for interventional study designs was used to assess the quality of included studies [4, 20] (Supplementary Table 3). Studies that met all eligibility criteria but had a high risk of bias were included in the review but excluded from data analyses per the Cochrane Handbook recommended the best practice to reduce the risk of reporting bias [21].
Data synthesis and analysis
All analyses were conducted on an intention-to-treat population. Heterogeneity across subgroup analyses was tested by I-square (I2) and Chi-square tests. The Chi-square test measures the existence of a significant heterogeneity, whilst I2 quantifies the magnitude of heterogeneity in the effect size. If both tests showed low heterogeneity, the fixed effect model (Mantel–Haenszel method) was applied. If a high degree of heterogeneity was observed, a random-effect model was applied. A sensitivity analysis was conducted to identify sources of heterogeneity using “one-out” approach [22]. Single studies that lead to a decrease of I2 < 25% were reported if identified.
The analysis was conducted in STATA 16.0 [48]. The metaprop command was applied to calculate pooled incidence rates of AEs and ORR. To test the impact of the ordered values of the dose level categories of cyclo or fludara on the objective response status, nptrend command was used in STATA to run the Cuzick test.
Results
Characteristics of the included studies and participants
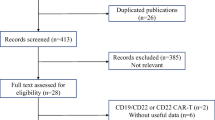
Systematic searches retrieved 484 potentially relevant citations; 433 were excluded based on abstract/title. 51 studies remaining were read in full, and 12 articles (185 patients) and 7 abstracts (46 patients) met the eligibility criteria (Supplementary Fig. 1). Two articles [23, 24] reported all outcome data for cohort 1, and efficacy data only for all 4 cohorts, respectively, for the same study. The four cohorts received different LD regimens. Data for all 4 cohorts were updated in a recent abstract [25]. Hence, both articles and abstract were included. Two abstracts [26, 27] reported data for two patient cohorts respectively, separately, receiving different LD regimens, within the same study; hence, both are included. All studies administered autologous T cells. No paediatric patients were included (Table 1).
Risk of bias and quality assessment of included studies
Risk of bias is summarised across all studies (Supplementary Fig. 2), with each risk of bias item assessment for individual studies presented (Supplementary Fig. 3). All studies were single-arm, non-randomised, and non-controlled, and therefore had a high risk of interpretation bias. Methods for measurement of safety and efficacy outcomes were not clearly stated in 40% of studies. Every study lacked an independent, blinded assessor for outcomes and showed a high risk of bias regarding patient recruitment. 40% of studies clearly reported AEs, with 95% of studies reporting ORR, demonstrating a strong reporting bias for favourable versus unfavourable outcomes. The follow-up period for data cut-off, reported in 53% of studies, was highly variable, potentially affecting data reporting (AEs resolution, delayed/chronic AEs, delayed responses, response duration, overall survival).
Lymphodepleting regimens prior to TCR-T cell therapy
Ten different dose levels were administered as LD chemotherapy across the 21 studies/cohorts. Cyclo 120 mg/kg (60 mg/kg daily, 2 days), plus fludara 125 mg/m2 (25 mg/m2 daily, 5 days), given in 75% of cases on days − 7 to − 6 (cyclo) and − 5 to − 1 (fludara) was the most commonly used in 8/21 (38%) studies/cohorts. Cyclo monotherapy without fludara was used in 14%. One study (5%) used no lymphodepletion [34].
TCR-T cell therapy targeted 7 different tumour antigens, with NY-ESO-1 the most frequent, doses ranging from 1 × 107 to 13 × 1010 cells. Four studies included separate cohorts receiving escalating doses of TCR-T cells. Interleukin-2 (IL-2) was used in 9/19 studies. Other adjunct therapies included colony stimulating factors, ipilimumab, and peptide antigen/primed dendritic cell vaccination (Supplementary Table 4).
The pooled incidence of adverse effects
Nine studies reported detailed adverse effects (AEs) data. Pooled incidence rates showed a trend for febrile neutropaenia and Grade ≥ 3 anaemia occurring more frequently among patients receiving high-dose LD regimens (Table 2). Pooled incidence rates of febrile neutropaenia among patients receiving high-dose LD regimens and low-dose LD regimens were 0.64 (95% CI 0.50–0.78) and 0.39 (95% CI 0.25–0.53), respectively (Fig. 1). Significant heterogeneity was observed in the high-dose subgroup and overall analyses of febrile neutropenia (I2 37.37% and 59.49%, respectively).

Pooled incidence rates of febrile neutropaenia according to lymphodepleting chemotherapy dose
Based on the lack of reported safety data, it was not possible to determine the effect of LD regimen on AEs of CRS, neurotoxicity, infection, or GvHD. (Supplementary Tables 5, 6).
Objective response rate (ORR)
The ORR ranged from 0 to 67% (Supplementary Table 7). Using the reported aggregate objective response rates, the pooled ORR for low-LD-dose and high-LD-dose cohorts were 0.31 (95% CI 0.15–0.46) and 0.36 (95% CI: 0.26–0.45), respectively (Supplementary Fig. 4). We were able to extract the individual cyclo and fludara doses with the corresponding response status for 231 patients. High LD dose was not associated with a higher likelihood of objective response (odds ratio: 1.05, 95%CI: 0.60–1.82, p = 0.86). We tested the impact of ordered dose levels of fludara and cyclo either alone or combined on the objective response (Fig. 2a). Only higher dose levels of fludara were associated with a significantly higher probability of objective responses (p = 0.03) (Fig. 2b). This was not significant with cyclo (p = 0.24) nor combined cyclo/fludara dose levels (p = 0.28) (Fig. 2c,d).

Impact of ordered dose levels of fludara and cyclo either alone or combined on the objective response rate. a Ten dose levels of cyclophosphamide and fludarabine were used across the included studies with seven different dose levels of cyclophosphamide and five for fludarabine. b Higher dose levels of fludara were associated significantly with a higher probability of objective responses, represented on the y-axis as the natural log of the sum of ranks of responses computed by Cuzick’s test. c Dose levels of cyclophosphamide were not significantly associated with a higher probability of objective responses. d Dose levels of cyclophosphamide and fludarabine were not significantly associated with a higher probability of objective responses
Discussion
This systematic review identified a major shortfall in reporting of data from TCR-T cell clinical trials in patients with solid tumours. Firstly, LD regimens were not sufficiently detailed in 9 of the potentially suitable 51 studies, increasing the potential for bias (missing data). Secondly, reporting of key, life-threatening AEs of CRS, neurological toxicity, infection, and GvHD was frequently inconsistent or missing.
For studies split into cohorts receiving different LD regimens, data were reported for the entire study population, not per cohort. Where reported, criteria used for CRS definition and severity were rarely cited. Some authors reported the three components of CRS (fever, hypotension, and hypoxia) as separate AEs [7]. Taken together, the lack of AE data reporting makes the correlation of LD regimens with safety outcome data, including the true rate of CRS for a given LD regimen, difficult. However, a trend for an association between high-dose LD regimens and the incidence of febrile neutropaenia and grade ≥ 3 anaemia was observed, albeit heterogeneity between high- and low-dose LD groups was significant. Dudley and colleagues first demonstrated that a LD regimen containing total doses of cyclo 120 mg/kg and fludara 100 mg/m2 significantly increased the efficacy of TIL therapy in metastatic melanoma, with manageable toxicity [13]. Adoption of this LD regimen in CAR-T cell therapy demonstrated that higher dose cyclo/fludara use was also associated with improved efficacy [38,39,40] but an increased toxicity burden in both haematological and solid tumours [12]. Consequently, a range of lower cyclo/fludara doses have been studied as LD in ACT therapy in solid tumours [12, 49,50,51]. For this review, we considered the original standard LD regimens containing total doses of cyclo ≥ 120 mg/kg and fludara ≥ 100 mg/m2 as ‘’high dose’’. Cyclophosphamide monotherapy used in 2 of the studies was considered a “low-dose” LD regimen. Higher dose fludara, but not higher dose cyclo nor cyclo/fludara, was associated with a significantly higher probability of objective responses (p = 0.03, p = 0.24, and p = 0.285, respectively) in this review, supported by data from two non-Hodgkin’s Lymphoma studies, where LD regimes containing fludarabine were associated with improved CAR-T cell expansion, persistence and efficacy, compared with non-fludarabine containing regimes [38, 39]. A multivariate analysis of factors affecting progression-free survival (PFS) in a study of CAR-T cells in B cell lymphoma, using low- or high-intensity LD, found that specific cytokine concentrations above the median were associated with improved PFS [40]. PFS was improved in patients receiving high-intensity LD and achieving a favourable cytokine profile, compared with those receiving the same high-intensity LD without achieving a favourable cytokine profile [40], suggesting that the effects of LD regimen on specific cytokine profiles are more important in terms of CAR-T cell efficacy than the intensity of LD. It remains to be seen whether this applies to TCR-T cell therapy in solid tumours, as data for cytokine profiles were not collected in this review.
Included studies employed a range of adjunct therapies. The rates of febrile neutropaenia were slightly lower (range 0–0.4) in the 2 studies reporting G-CSF use [23, 24] than for studies not reporting G-CSF use, but 2 of the cohorts used lower doses of cyclo with no fludara (100/0 and 50/0, [23]). As a potential routine component of TCR-T cell therapy, reporting of G-CSF use may have been omitted, making it difficult to determine its effect on AE incidence. The addition of IL-2 (9/19 studies) does not seem to have significantly influenced efficacy outcomes in this review, but data are confounded by variations in doses of IL-2 (72,000, 500,000, 720,000 units tds), doses of TCR-T cells and LD therapy, and use of other adjunct therapies (DC and peptide vaccinations). For these reasons, and due to inconsistent AE reporting (CRS, neurotoxicity, GvHD), the effect of adjunct therapies on safety and efficacy outcomes is therefore confounded and difficult to interpret.
Limitations of this review
Peer review of the search strategy may have reduced potential biases. The process of article and abstract selection from systematic searches for inclusion in the review, and quality assessments of the risk of bias for individual studies, were both intrinsically biased, relying on the interpretation and judgement of a single reviewer (KO), with no second reviewer or opportunity for discussion with peers (selection and reporter bias).
This review demonstrated a high level of clinical and methodological heterogeneity between and within studies of TCR-T cells in patients with solid tumours, in keeping with findings from similar systematic reviews/metanalyses of studies in CAR-T cells [4, 41]. Notable was the number of variables changing concurrently within a study and within small patient cohorts. Patients often had different tumour types, LD regimens, TCR-T cell doses or adjunct therapies, so it was impossible to decipher the effect of changing a single variable on an outcome. Therefore, the interpretation of the effect of LD regimen on safety and efficacy outcomes is heavily confounded by multiple factors, some largely outside study control, including patient characteristics (age, performance status, tumour burden at baseline, tumour and off-target expression of target antigen, number/nature of prior therapies, comorbidities, concomitant medications, cyclo/fludara PK), but also TCR-T cell variables (target antigen, cell dose, ‘fitness’[phenotype], persistence and expansion of cells, number of infusions of cells), adjunct therapies (IL-2, dendritic cell or peptide vaccination, immune checkpoint inhibitors) and follow-up periods.
Due to the rarity of TCR-T cell therapy, clinical trial designs should focus on key objectives and outcomes, fixing other variables so that confounding is minimised and the interpretation of an effect on an outcome is as robust as possible. Standardisation of clinical trial designs in TCR-T cell therapy and ACT in general, with a consensus for reporting LD regimen, TCR-T cell dose, adjunct therapies, defined safety/efficacy outcomes, and follow-up times, per the master protocols from the Pan American/World Health Organisations’ Master Protocol for clinical trials in COVID-19, may allow more valid data pooling and maximise available evidence [42]. Consensus should be reached on CRS definition and grading, neurological toxicities, and GvHD and adopted globally.
The risk of bias in the results was very high, i.e. the risk of over- or under-estimation of the true intervention effect (LD regimen) on safety and efficacy outcomes post TCR-T cell therapy. There are specific challenges involved in the production of robust, quality data for risk–benefit analyses of ACT using randomised controlled clinical trials [43]. TCR-T cell therapy is a bespoke product for patients with relapsed/refractory life-threatening disease, with no other suitable treatment options. Few patients are suitable for TCR-T cell therapy; their acquisition, manufacture, storage, shipping, clinical delivery, and patient care are highly complex, with a substantial production failure rate [1]. Together with high costs, these factors preclude larger clinical trials, and the lack of suitable alternative therapies prevents the ethical use of comparator arms.
The primary outcome for this review was to summarise the LD dosing regimens prior to TCR-T cell therapy in patients with solid tumours. A more accurate primary outcome would have been a summary of exposures of the chemotherapies used and safety and efficacy outcomes. Hepatic and renal function affects cyclo and fludara pharmacokinetics (PK), so for a given dose, there is high inter-patient variability in systemic exposure, despite adjustments for body weight or body surface area (BSA), potentially influencing outcomes [44,45,46,47]. Sparse PK sampling during LD may allow a more accurate correlation of cyclo/fludara exposure with safety and efficacy outcomes, with optimisation of future LD dosing, and improvement in LD-associated risks and benefits.
Conclusion
To our knowledge, this is the first systematic review of lymphodepleting practices employed prior to TCR-T cell therapy in patients with relapsed, refractory solid tumours, participating in interventional, prospective clinical studies. Such studies are infrequent, extremely heterogeneous, and the risk of bias for outcomes due to publication, methodological, reporting, and interpretation biases in this review is very high. The most commonly used LD regimen (38% of studies/cohorts) prior to TCR-T cell therapy was 60 mg/kg cyclo and 25 mg/m2 fludara, given daily for 2 and 5 days, (total doses 120 mg/kg, 125 mg/m2), respectively. Febrile neutropaenia and grade ≥ 3 anaemia tended to occur more frequently among patients receiving high-dose LD regimens. Higher-dose fludara (≥ 100 mg/m2) was associated with a significantly higher probability of objective responses, but this did not apply to cyclo nor combined cyclo/fludara at higher doses. Taken together, these data imply that lower doses of cyclophosphamide may be adopted without compromising TCR-T cell therapy efficacy outcomes.
Safety outcome reporting was inconsistent. Standardised TCR-T cell therapy clinical trial designs and data reporting may allow appropriate pooling of data and facilitate the development of robust evidence bases for future optimisation of LD regimens which may reduce morbidity mortality, shorten hospital stay, and reduce intensive care unit admissions, resulting in a longer and better quality of life for patients with advanced solid tumours with no other treatment options.
References
Tsimberidou A-M, Van Morris K, Vo HH, Eck S, Lin Y-F, Rivas JM, Andersson BS (2021) T-cell receptor-based therapy: an innovative therapeutic approach for solid tumors. J Hematol Oncol. https://doi.org/10.1186/s13045-021-01115-0
Garber K (2018) Driving T-cell immunotherapy to solid tumors. Nat Biotechnol 36:215–219. https://doi.org/10.1038/nbt.4090
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, Simon P, Lotze MT, Yang JC, Seipp CA et al (1988) Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. New England J Med 22(25):1676–1680
Grigor EJM, Fergusson D, Kekre N, Montroy J, Atkins H, Seftel MD, Daugaard M, Presseau J, Thavorn K, Hutton B, Holt RA, Lalu MM (2019) Risks and benefits of chimeric antigen receptor T-Cell (CAR-T) Therapy in Cancer: a systematic review and meta analysis. Transfus Med Rev 33(2):98–110
Newick K, O’Brien S, Moon E, Albelda SM (2017) CAR T cell therapy for solid tumors. Ann Rev Med 14(68):139–152. https://doi.org/10.1146/annurev-med-062315-120245 (Epub 2016 Nov 17 PMID: 27860544)
Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, Yang JC, Dudley ME, Wunderlich JR, Sherry RM, Kammula US, Hughes MS, Restifo NP, Raffeld M, Lee C-CR, Li YF, El-Gamil M, Rosenberg SA (2015) A pilot trial using lymphocytes genetically Engineered with an NY-ESO-1–reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res 21(5):1019–1027
Nagarsheth NB, Norberg SM, Sinkoe AL, Adhikary S, Meyer TJ, Lack JB, Warner AC, Schweitzer C, Doran SL, Korrapati S, Stevanović S, Trimble CL, Kanakry JA, Bagheri MH, Ferraro E, Astrow SH, Bot A, Faquin WC, Stroncek D, Gkitsas N, Highfill S, Hinrichs CS (2021) TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat Med 27(3):419–425
Doran SL, Stevanović S, Adhikary S, Gartner JJ, Jia L, Kwong MLM, Faquin WC, Hewitt SM, Sherry RM, Yang JC, Rosenberg SA, Hinrichs CS (2019) T-Cell receptor gene therapy for human papillomavirus-associated Epithelial Cancers: a first-in-Human, Phase I/II Study. J Clin Oncol 37(30):2759–2768
Lu Y-C, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, Yao X, Li YF, Robbins PF, Feldman SA, Van Der Bruggen P, Klebanoff CA, Goff SL, Sherry RM, Kammula US, Yang JC, Rosenberg SA (2017) Treatment of patients with metastatic cancer using a major histocompatibility complex class II–restricted T-Cell receptor targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol 35(29):3322–3329
Neelapu SS (2019) CAR-T efficacy: is conditioning the key? Blood Blood 133(17):1799–1800
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, Timmerman JM, Stiff PJ, Friedberg JW, Flinn IW, Goy A, Hill BT, Smith MR, Deol A, Farooq U, Mcsweeney P, Munoz J, Avivi I, Castro JE, Westin JR, Chavez JC, Ghobadi A, Komanduri KV, Levy R, Jacobsen ED, Witzig TE, Reagan P, Bot A, Rossi J, Navale L, Jiang Y, Aycock J, Elias M, Chang D, Wiezorek J, Go WY (2017) ‘Axicabtagene Ciloleucel CAR T-Cell therapy in refractory large B-Cell Lymphoma. New England J Med 377(26):2531–2544
Bechman N, Maher J (2021) Lymphodepletion strategies to potentiate adoptive T-cell immunotherapy – what are we doing; where are we going? Expert Opin Biol Ther 21:627–637. https://doi.org/10.1080/14712598.2021.1857361
Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL, Sherry RM, Marincola FM, Leitman SF, Seipp CA, Rogers-Freezer L, Morton KE, Nahvi A, Mavroukakis SA, White DE, Rosenberg SA (2002) A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother 25(3):243–251. https://doi.org/10.1097/01.CJI.0000016820.36510.89
Neelapu SS, Tummala S, Kebriaei P, Wierda W, Gutierrez C, Locke FL, Komanduri KV, Lin Y, Jain N, Daver N, Westin J, Gulbis AM, Loghin ME, De Groot JF, Adkins S, Davis SE, Rezvani K, Hwu P, Shpall EJ (2018) Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nat Rev Clin Oncol 15(1):47–62
Lowe KL, Mackall CL, Norry E, Amado R, Jakobsen BK, Binder G (2018) Fludarabine and neurotoxicity in engineered T-cell therapy. Gene Ther 25(3):176–191
Santomasso BD, Park JH, Salloum D, Riviere I, Flynn J, Mead E, Halton E, Wang X, Senechal B, Purdon T, Cross JR, Liu H, Vachha B, Chen X, Deangelis LM, Li D, Bernal Y, Gonen M, Wendel H-G, Sadelain M, Brentjens RJ (2018) Clinical and biological correlates of neurotoxicity associated with CAR T-cell Therapy in patients with B-cell acute lymphoblastic Leukemia. Cancer Discov 8:958–971. https://doi.org/10.1158/2159-8290.cd-17-1319
Shamseer L et al (2015) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ 349:g7647–g7647. https://doi.org/10.1136/bmj.g7647
Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M, Shekelle P, Stewart LA (2015) Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev 4(1):1. https://doi.org/10.1186/2046-4053-4-1
Eisenhauer EA, Therasse P, Bogaerts J, Schwatrz LH, Sargent D, Ford R et al (2009) New response evaluation criteria in solid tumors: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Guo B, Moga C, Harstall C, Schopflocher D (2016) A principal component analysis is conducted for a case series quality appraisal checklist. J Clin Epidemiol 69:199-207.e2
Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, (Eds.). Cochrane handbook for systematic reviews of interventions version 6.2 (updated February 2021). Cochrane, 2021. Available from www.training.cochrane.org/handbook
Patsopoulos NA, Evangelou E, Ioannidis JP (2008) Sensitivity of between-study heterogeneity in meta-analysis: proposed metrics and empirical evaluation. Int J Epidemiol 37(5):1148–1157. https://doi.org/10.1093/ije/dyn065
D’Angelo SP, Melchiori L, Merchant MS, Bernstein D, Glod J, Kaplan R, Grupp S, Tap WD, Chagin K, Binder GK, Basu S, Lowther DE, Wang R, Bath N, Tipping A, Betts G, Ramachandran I, Navenot J-M, Zhang H, Wells DK, Van Winkle E, Kari G, Trivedi T, Holdich T, Pandite L, Amado R, Mackall CL (2018) Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259T Cells in Synovial Sarcoma. Cancer Discov 8(8):944–957
Ramachandran I, Lowther DE, Dryer-Minnerly R, Wang R, Fayngerts S, Nunez D, Betts G, Bath N, Tipping AJ, Melchiori L, Navenot J-M, Glod J, Mackall CL, D’Angelo SP, Araujo DM, Chow WA, Demetri GD, Druta M, Van Tine BA, Grupp SA, Abdul Razak AR, Wilky B, Iyengar M, Trivedi T, Winkle EV, Chagin K, Amado R, Binder GK, Basu S (2019) Systemic and local immunity following adoptive transfer of NY-ESO-1 SPEAR T cells in synovial sarcoma. J ImmunoTherapy Cancer 7(1):1–4
D’Angelo S, Demetri G, Tine BV, Druta M, Glod J, Chow W, Pandya N, Hasan A, Chiou V, Tress J, Edwards J, Young T, Woessner M, Gyuerdieva A, Zajic S, Goodison S, Araujo D (2020) 298 Final analysis of the phase 1 trial of NY-ESO-1–specific T-cell receptor (TCR) T-cell therapy (letetresgene autoleucel; GSK3377794) in patients with advanced synovial sarcoma (SS). J Immunother Cancer 8(Suppl 3):A325–A325
Stadanlick J, Chagin K, Norry E, Trivedi T, D’Angelo S, Tap W, Druta M, Liebner D, Schuetzes S, Van Tine B, Britten C, Hasan A (2018) 33rd Annual meeting & pre-conference programs of the society for immunotherapy of Cancer (SITC 2018). J Immunother Cancer 6(S1):O18
D’Angelo S, Druta M, Van Tine BA, Liebner DA, Schuetze S, Hasan AN, Holmes AP, Huff A, Kapoor GS, Zajic S, Somaiah N (2021) Safety and efficacy of letetresgene autoleucel (lete-cel; GSK3377794) in advanced myxoid/round cell liposarcoma (MRCLS) following high lymphodepletion (Cohort 2): interim analysis. J Clin Oncol 39(15_suppl):11521–11521. https://doi.org/10.1200/JCO.2021.39.15_suppl.11521
Hong D, Clarke J, Johanns T, Kebriaei P, Heymach J, Galal A, Saibil S, Sacher A, Brophy F, Betts G, Bath N, William S, Tipping A, Tucci J, Luke R, Trivedi T, Lin Q, Navenot J-M, Fracasso P, Miller K, Norry E, Dudley M, Butler M (2020) 379 Initial safety, efficacy, and product attributes from the SURPASS trial with ADP-A2M4CD8, a SPEAR T-cell therapy incorporating an affinity optimized TCR targeting MAGE-A4 and a CD8α co-receptor. J Immunother Cancer 8(Suppl 3):A404–A404
Nowicki TS, Berent-Maoz B, Cheung-Lau G, Huang RR, Wang X, Tsoi J, Kaplan-Lefko P, Cabrera P, Tran J, Pang J, Macabali M, Garcilazo IP, Carretero IB, Kalbasi A, Cochran AJ, Grasso CS, Hu-Lieskovan S, Chmielowski B, Comin-Anduix B, Singh A, Ribas A (2019) A pilot trial of the combination of transgenic NY-ESO-1–reactive adoptive cellular therapy with dendritic cell vaccination with or without Ipilimumab. Clin Cancer Res 25(7):2096–2108
Hattori H, Ishihara M, Kitano S, Miyahara Y, Kato H, Mishima H, Yamamoto N, Funakoshi T, Kojima T, Sasada T, Sato E, Okamoto S et al (2019) A novel affinity-enhanced NY-ESO-1-targeting TCR-redirected T cell transfer exhibited early-onset cytokine release syndrome and subsequent tumour responses in synovial sarcoma patients. Ann Oncol 30(Suppl 5):1182PD. https://doi.org/10.1093/annonc/mdz253.008
Butler MO, Saibil S, Bonilla L, Sacher AG, Sotov V, Boross-Harmer S, Fyrsta M, Gray D, Nelles M, Le M, Lemiashkova D, Liu D et al (2019) ‘Adoptive T cell therapy with TBI-1301 results in gene-engineered T cell persistence and anti-tumour responses in patients with NY-ESO- 1 expressing solid tumours. Ann Oncol 30(Suppl 5):1183PD. https://doi.org/10.1093/annonc/mdz253.009
Moore T, Wagner CR, Scurti GM, Hutchens KA, Godellas C, Clark AL, Kolawole EM, Hellman LM, Singh NK, Huyke FA, Wang S-Y, Calabrese KM, Embree HD, Orentas R, Shirai K, Dellacecca E, Garrett-Mayer E, Li M, Eby JM, Stiff PJ, Evavold BD, Baker BM, Le Poole IC, Dropulic B, Clark JI, Nishimura MI (2018) Clinical and immunologic evaluation of three metastatic melanoma patients treated with autologous melanoma-reactive TCR-transduced T cells. Cancer Immunol Immunother 67(2):311–325
Hong DS, Butler MO, Johnson M, Olszanski AJ, Norry E, Van Winkle E, Chagin KD, Amado RG (2018) Initial safety assessment of MAGE-A4 SPEAR T-cells. Ann Oncol 29(8):1156. https://doi.org/10.1093/annonc/mdy288.029
Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, Sugino S, Ueda S, Ishikawa T, Kokura S, Naota H, Ohishi K, Shiraishi T, Inoue N, Tanabe M, Kidokoro T, Yoshioka H, Tomura D, Nukaya I, Mineno J, Takesako K, Katayama N, Shiku H (2015) Adoptive transfer of MAGE-A4 T-cell receptor Gene-Transduced Lymphocytes in patients with recurrent Esophageal Cancer. Clin Cancer Res 21(10):2268–2277
Chodon T, Comin-Anduix B, Chmielowski B, Koya RC, Wu Z, Auerbach M, Ng C, Avramis E, Seja E, Villanueva A, Mccannel TA, Ishiyama A, Czernin J, Radu CG, Wang X, Gjertson DW, Cochran AJ, Cornetta K, Wong DJL, Kaplan-Lefko P, Hamid O, Samlowski W, Cohen PA, Daniels GA, Mukherji B, Yang L, Zack JA, Kohn DB, Heath JR, Glaspy JA, Witte ON, Baltimore D, Economou JS, Ribas A (2014) Adoptive transfer of MART-1 T-cell receptor transgenic lymphocytes and dendritic cell vaccination in patients with metastatic Melanoma. Clin Cancer Res 20(9):2457–2465
Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, Phan GQ, Hughes MS, Kammula US, Miller AD, Hessman CJ, Stewart AA, Restifo NP, Quezado MM, Alimchandani M, Rosenberg AZ, Nath A, Wang T, Bielekova B, Wuest SC, Akula N, Mcmahon FJ, Wilde S, Mosetter B, Schendel DJ, Laurencot CM, Rosenberg SA (2013) Cancer regression and neurological toxicity following Anti-MAGE-A3 TCR gene therapy. J Immunother 36(2):133–151
Hong JJ, Rosenberg SA, Dudley M, Yang JC, White DE, Butman JA, Sherry RM (2010) Successful treatment of Melanoma Brain metastases with adoptive cell therapy. Clin Cancer Res 16(19):4892–4898
Kochenderfer JN, Somerville RPT, Lu T, Shi V, Bot A, Rossi J, Xue A, Goff SL, Yang JC, Sherry RM, Klebanoff CA, Kammula US, Sherman M, Perez A, Yuan CM, Feldman T, Friedberg JW, Roschewski MJ, Feldman SA, Mcintyre L, Toomey MA, Rosenberg SA (2017) Lymphoma remissions caused by Anti-CD19 chimeric antigen receptor T cells are associated with high serum Interleukin-15 levels. J Clin Oncol 35(16):1803–1813
Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, Hawkins R, Chaney C, Cherian S, Chen X, Soma L, Wood B, Li D, Heimfeld S, Riddell SR, Maloney DG (2016) Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+and CD4+CD19-specific chimeric antigen receptor–modified T cells. Sci Trans Med 8(355):355ra116-355ra116
Hirayama AV, Gauthier J, Hay KA, Voutsinas JM, Wu Q, Gooley T, Li D, Cherian S, Chen X, Pender BS, Hawkins RM, Vakil A, Steinmetz RN, Acharya UH, Cassaday RD, Chapuis AG, Dhawale TM, Hendrie PC, Kiem H-P, Lynch RC, Ramos J, Shadman M, Till BG, Riddell SR, Maloney DG, Turtle CJ (2019) The response to lymphodepletion impacts PFS in patients with aggressive non-Hodgkin lymphoma treated with CD19 CAR T cells. Blood 133(17):1876–1887
Anagnostou T, Riaz IB, Hashmi SK, Murad MH, Kenderian SS (2020) Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol 7(11):e816–e826. https://doi.org/10.1016/S2352-3026(20)30277-5
Pan American Health Organization and World Health Organisation (2020). A Multi-centre, Adaptive, Randomized, Double-Blind, Placebo controlled clinical trial of the safety and efficacy of investigational Therapeutics for the treatment of COVID-19 in Hospitalized patients. Available at: https://covid19-evidence.paho.org/handle/20.500.12663/386?show=full (Accessed 13 Sept 2021)
Abou-El-Enein M, Hey SP (2019) Cell and gene therapy trials: are we facing an ‘Evidence Crisis? EClinical Medicine 7:13–14. https://doi.org/10.1016/j.eclinm.2019.01.015
Helsby NA, Yong M, Kan M, Zoysa JR, Burns KE (2019) The importance of both CYP2C19 and CYP2B6 germline variations in cyclophosphamide pharmacokinetics and clinical outcomes. Br J Clin Pharmacol 85(9):1925–1934
Langenhorst JB, Dorlo TPC, Van Maarseveen EM, Nierkens S, Kuball J, Boelens JJ, Van Kesteren C, Huitema ADR (2019) Population Pharmacokinetics of Fludarabine in Children and adults during conditioning prior to Allogeneic Hematopoietic cell transplantation. Clin Pharmacokinet 58(5):627–637
De Jonge ME, Huitema ADR, Rodenhuis S, Beijnen JH (2005) Clinical Pharmacokinetics of Cyclophosphamide. Clin Pharmacokinet 44(11):1135–1164
Fludarabine Summary of Product Characteristics 02 April, 2020. Available at https://www.medicines.org.uk/emc/product/4530/smpc#gref
StataCorp (2019) Stata statistical software: Release 16 StataCorp LLC College Station, TX
Heczey A, Louis CU, Savoldo B, Dakhova O, Durett A, Grilley B, Liu H, Wu MF, Mei Z, Gee A, Mehta B, Zhang H, Mahmood N, Tashiro H, Heslop HE, Dotti G, Rooney CM, Brenner MK (2017) CAR T cells administered in combination with Lymphodepletion and PD-1 inhibition to patients with Neuroblastoma. Mol Ther 25(9):2214–2224
Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, Byatte AJ, Kirillova N, Valle JW, Sharma SK, Chester KA, Westwood NB, Halford SER, Nabarro S, Wan S, Austin E, Hawkins RE (2017) The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother 66(11):1425–1436
Nissani A, Lev-Ari S, Meirson T, Jacoby E, Asher N, Ben-Betzalel G, Itzhaki O, Shapira-Frommer R, Schachter J, Markel G, Besser MJ (2021) Comparison of non-myeloablative lymphodepleting preconditioning regimens in patients undergoing adoptive T cell therapy. J Immunother Cancer 9(5):e001743. https://doi.org/10.1016/j.ymthe.2017
Funding
This work was part of an application for the degree of Master of Science at Manchester University, funded by Owen Clinical Consulting Limited.
Author information
Authors and Affiliations
Contributions
FT proposed the study concept and objectives, KO designed the study, acquired the data and prepared all tables and figures except for table 2 and figures 2, 3b), 3c) and 3d) which were prepared by KS and RG. KS and RG performed all statistical analyses KO drafted the main manuscript text with input from RG and KS. All authors contributed to the analysis and interpretation of data. All authors critically reviewed the manuscript. KO obtained funding. Supervision: FT. KO had full access to all the data in the study and takes full responsibility for data integrity. KO and FT were involved in concept and design. All authors analysed or interpreted the data. KO, RG, and KS drafted the manuscript. All authors critically revised the manuscript. KO obtained funding. All authors were involved in administrative, technical, or material support. FT was involved in supervision.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no conflicts of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Owen, K., Ghaly, R., Shohdy, K.S. et al. Lymphodepleting chemotherapy practices and effect on safety and efficacy outcomes in patients with solid tumours undergoing T cell receptor-engineered T cell (TCR-T) Therapy: a systematic review and meta-analysis. Cancer Immunol Immunother 72, 805–814 (2023). https://doi.org/10.1007/s00262-022-03287-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00262-022-03287-1