
Abstract
A two-step strategy combining assisted benchmark testing (entry controls) and External Quality Assessments (EQAs) with blinded simulated clinical specimens to enhance and maintain the quality of nucleic acid amplification testing was developed. This strategy was successfully applied to 71 diagnostic laboratories in The Netherlands when upscaling the national diagnostic capacity during the SARS-CoV-2 pandemic. The availability of benchmark testing in combination with advice for improvement substantially enhanced the quality of the laboratory testing procedures for SARS-CoV-2 detection. The three subsequent EQA rounds demonstrated high quality testing with regard to specificity (99.6% correctly identified) and sensitivity (93.3% correctly identified). Even with the implementation of novel assays, changing workflows using diverse equipment and a high degree of assay heterogeneity, the overall high quality was maintained using this two-step strategy. We show that in contrast to the limited value of Cq value for absolute proxies of viral load, these Cq values can, in combination with metadata on strategies and techniques, provide valuable information for laboratories to improve their procedures. In conclusion, our two-step strategy (preparation phase followed by a series of EQAs) is a rapid and flexible system capable of scaling, improving, and maintaining high quality diagnostics even in a rapidly evolving (e.g. pandemic) situation.
Similar content being viewed by others


Introduction
High quality pathogen detection systems, with both high sensitivity and specificity, are of paramount importance for public health and individual patient diagnostics1,2,3. In The Netherlands, diagnostic laboratories have the option to choose their own experimental workflows in contrast to many other countries where one or only a few central testing facilities for the whole country are used (e.g. Denmark4) or a single workflow type is implemented in multiple decentralized laboratories (e.g. USA5). At the start of the SARS-CoV-2 pandemic, no laboratory diagnostic tests for specific SARS-CoV-2 detection were available. Various initiatives were taken to develop specific SARS-CoV-2 tests, including ours at the national reference laboratories for public health action in emerging situations (Dutch National Institute for Public Health and the Environment (RIVM) and Erasmus Medical Centre (Erasmus MC))1,6,7. We were involved in the validation of real-time reverse transcription PCR (rRT-PCR) assays for the detection of the novel SARS-CoV-2 virus1. This initial assay was based on limited genomic information and developed by Corman et al.1 and implemented for Dutch national SARS-CoV-2 testing. In an emerging pathogen situation, like SARS-CoV-2, reference and clinical materials of confirmed positive and negative specimens are largely lacking and procedures for at least verification of the assays with standardized controls is needed. A complicating element was the evolution of the virus resulting in potential mismatched primers leading to false-negative results8,9,10,11,12. A widely applied method to evaluate the quality of the complete workflows in diagnostic laboratories (from extraction of nucleic acid to specific virus target detection) is through an External Quality Assessment (EQA)13,14,15,16. If the test results are unsatisfactory, additional in-depth analyses of the individual components of the workflow can be carried out. In addition, sharing detailed (anonymised) information about workflows and procedures from other laboratories might suggest alternatives and possible solutions.
Here, we describe the application of the combination of an initial benchmark testing (entry-control) procedure using simulated clinical specimens, provision of positive control material and confirmatory testing of patient clinical specimens at the reference laboratory, in which feedback and assistance are offered, followed by periodic EQAs for SARS-CoV-2 molecular diagnostic testing using Nucleic Acid Amplification Tests (NAAT) in 71 diagnostic laboratories in The Netherlands in 2020 and 2021. Passing benchmark testing was necessary for a laboratory to be able to start diagnostic testing or high throughput testing for the general population. We demonstrate that the introduction of the benchmark testing phase before an EQA was highly effective and efficient, and resulted in high quality diagnostic testing. An important aspect of this study is the exploration of additional analysis methods of some steps of/in the workflows. We applied Bayesian statistical modelling to estimate the contribution of the choice of target gene on the Cq values and composed a model that incorporates the effect of individual laboratories. These strategies can identify sensitive steps in the workflows and be helpful to uncover valuable information for the laboratories to improve their performance. Furthermore, the abundant information on Cq values resulting from a high number of different workflows at different laboratories for the same viral concentration specimens, in combination with metadata on strategies and techniques, provided valuable information in the use of Cq values as absolute proxy for viral load. We suggest applying the two-stage strategy and the associated analysis strategy as components of diagnostic preparedness plans for a much wider range of (re-) emerging pathogens of public health concern.
Materials and methods
Benchmark testing
Blinded simulated clinical specimen panels (benchmark panel) for sensitivity and specificity analyses were prepared and distributed by the RIVM in collaboration with Erasmus MC. Preparation was performed as previously described17,18. Briefly, specimens were prepared in Minimal Essential Medium with Hanks’ salts and Hep2 cells to simulate clinical specimens. The panels contained a randomized dilution series of cultured SARS-CoV-2 and specimens with other related or different viruses were included as analytical specificity controls. A detailed description of the composition of the specimens is given in Supplementary Table 1. Initially, SARS-CoV-1 and also SARS-CoV-2 were included as RNA. As soon as they were available, inactivated Dutch SARS-CoV-2 isolates were included to assess the extraction component in the workflows. Laboratories were asked to report test panel results, as well as information about specimen input volume, extraction volume, elution volume, PCR/NAAT-reaction volume, devices and kits/reagents implemented, and target gene (sequences) for their assays. Alongside the benchmark panels, a positive control specimen initially containing SARS-CoV-1, rapidly replaced by SARS-CoV-2 when available, and validated primers and probes and/or their nucleotide sequence1 were supplied for implementing laboratory developed tests in the phase when no commercial detection kits were available. Laboratories implementing solely sample-to-result assays were given the option to test a reduced benchmark panel of four specimens to reduce costly and scarce testing cartridges. In addition, the participating laboratories were requested to supply a minimum of five SARS-CoV-2 positive and 10 SARS-CoV-2 negative tested clinical specimens derived from their own COVID-19 diagnostic pipeline for confirmatory testing at the reference laboratories. Together, these procedures were considered an entry benchmark test. In the event the results returned by a laboratory were unsatisfactory, the laboratory could request another benchmark panel after taking corrective actions. In exceptional cases multiple rounds of benchmark testing were performed. Furthermore, advice was offered by the reference laboratories to improve the technical procedures including the handling of the specimen, the execution of the testing methods, and data analysis. A laboratory’s performance was considered satisfactory during the benchmark testing phase when it was able to test the full panel and the confirmation specimens without false-positive or false-negative results. After a laboratory’s performance was satisfactory it had the freedom to implement other SARS-CoV-2 diagnostic assays, so the new test would be cross-referenced to their primary verified workflow. Laboratories were encouraged to request additional benchmark panels and apply for additional confirmatory testing of clinical specimens to verify novel SARS-CoV-2 diagnostic workflows.
External Quality Assessment
Three rounds of EQA were performed, in November 2020, February 2021, and May 2021. The EQA panels, consisting of 10 specimens each, were produced in similar fashion as the benchmark panels and their components are described in Table 1. Copies of SARS-CoV-2 E-gene RNA per mL were determined by digital droplet PCR as described previously17,18. For sensitivity analyses of SARS-CoV-2, the specimens containing 1.28 × 103 and 1.28 × 105 copies of SARS-CoV-2/mL (referred to as SARS2_L and SARS2_H, respectively) are fundamental as these mimic clinical samples most realistically. The specimen with the lowest virus concentration (1.28 × 102 copies; indicated as SARS2_Edu) was included to get insight into the detection limits of the various workflows. The SARS-CoV-1 containing specimen was included to get an insight into both assay specificity and target gene specificity for pathogens highly similar to SARS-CoV-2, especially as primers and probes specific for SARS-Betacoronaviruses (Sarbecoviruses) are being used1. This educational specimen and the SARS-CoV-1 containing specimen were not included in the judgment of the performance of a specific assay regarding applicability for diagnostic testing. Before shipping, the prepared panel was validated at the reference laboratories to confirm expected results. Laboratories were also asked to submit the same metadata as for the benchmark testing phase. The performance per workflow was divided over three performance categories based on the number of false-negative results for SARS-CoV-2, false-positive results for SARS-CoV-2 or inconclusive test results for the non-educational specimens: ‘Excellent’ (100% correct), ‘Mediocre’ (maximally one false positive or negative, or up to two inconclusive) and ‘Unsatisfactory’ (more than one false positive or negative and/or more than two inconclusive). An inconclusive or incorrect result can occur from inadequate specimen preparation or processing or not optimal limit of detection of the NAAT. Specifically, an inconclusive result can be the consequence of differences in individual target results of multi-target tests leading to no clear conclusion concerning the pathogen presence in the tested specimen; not negative and not positive.
Statistical analyses
Statistical analyses were based on a Bayesian model using R (version 4.2.2) and Rstan (R package version 2.21.7) where the measured Cq-value \(Cq_{j}\) was assumed to be linearly dependent on the true Cq-value \(\mu\). Errors were assumed normally distributed with standard deviation \(\sigma\), so that for data point \(j\) we have:
The Cq-value \(\mu_{j}\) is modelled as a sum of components:
The component \(\mu_{0}\) is the baseline Cq-value in the specimen labelled as ‘SARS2_H’ containing 1.28 × 105 copies of SARS-CoV-2 per mL, with prior value set to \(\mu_{0} \sim N\left( {30,3} \right)\). The component \(\mu_{d\left[ j \right]}^{{{\text{dilution}}}}\) is the contribution of the dilution factor at dilution \(d\left[ j \right]\) of data point \(j\) (which takes values 0 = ’ SARS2_H’, 1 = ’ SARS2_L’, and 2 = ’ SARS2_Edu’). The dilution labelled ‘SARS2_H’ (lowest dilution factor; containing 1.28 × 105 copies of SARS-CoV-2 per mL), is defined as the baseline Cq-value contribution to the dilution-specific term of the model, hence we set \(\mu_{0}^{{{\text{dilution}}}} = 0\). For the other dilutions labelled with ‘SARS2_L’ (medium dilution factor; containing 1.28 × 103 copies of SARS-CoV-2 per mL) and ‘SARS2_Edu’ (highest dilution factor; containing 1.28 × 102 copies of SARS-CoV-2 per mL) we expect a correction of respectively \(2 \times \log \left( {10} \right)/{\text{log}}\left( 2 \right)\) and 3\(\times \log \left( {10} \right)/{\text{log}}\left( 2 \right)\), since we have 2 and 3 log10 decreases, and theoretically each halving of the number of genomic copies increases the Cq-value by one. Hence we set priors \(\mu_{1}^{{{\text{dilution}}}} \sim N\left( {2\frac{{\log \left( {10} \right)}}{\log \left( 2 \right)}, 0.5} \right)\) and \(\mu_{2}^{{{\text{dilution}}}} \sim N\left( {3\frac{{\log \left( {10} \right)}}{\log \left( 2 \right)}, 0.5} \right)\).
The components \(\mu_{t\left[ j \right]}^{{{\text{target}}}}\) and \(\mu_{l\left[ j \right]}^{{{\text{laboratory}}}}\) are the gene-target and laboratory-specific contributions to the Cq-value. We model those as random effects, i.e. the values they take are assumed to stem from a common distribution:
The parameters \(\sigma^{{{\text{target}}}}\) and \(\sigma^{{{\text{laboratory}}}}\) measure how similar gene-target and laboratory specific Cq-value contributions are. Those are also estimated from the data. We set priors that encode our belief that more than two log10 units difference is unlikely:
Additionally, we enforce sum-to-zero constraints to \(\mu_{t\left[ j \right]}^{{{\text{target}}}}\) and \(\mu_{l\left[ j \right]}^{{{\text{laboratory}}}}\).
Results that were marked ‘no detection’ were treated differently. For those values the Cq-value was not reported, because no Cq value was generated at all or it is above some Cq threshold. This assessment by the laboratory is unknown to us and could vary between gene-target and dilution. As a substitute we recorded the highest Cq-value found for each combination of ‘dilution’ and ‘gene target’, and used this value (denoted \(c\left[ j \right]\)) as the censoring level of sample \(j\). The censoring is then implemented by using not the probability density function (PDF) for modelling Eq. (1), but the cumulative complementary PDF. This models that Cq-value of non-detects in SARS-CoV-2 containing specimens is somewhere above the censoring level \(c\left[ j \right]\), in the tail of the normal distribution Eq. (1).
Results
Supporting laboratories to validate and improve their SARS-CoV-2 testing, the benchmark test
As part of the response to the spread of the novel SARS-CoV-2 virus, the reference laboratories assessed and helped to improve the quality of newly introduced workflows for the testing of SARS-CoV-2 in diagnostic laboratories during the early stages of the pandemic. The procedure consisted of two stages, a benchmark testing phase, consisting of the combination of a benchmark panel and a series of confirmation samples, and three rounds of confirmatory EQAs (Fig. 1).

Preparation of diagnostic laboratories for molecular testing of/on SARS-CoV-2 infections in The Netherlands. Already before the first confirmed Dutch SARS-CoV-2 case (February 27th, 2020), the RIVM and Erasmus MC validated and implemented a SARS-CoV-2 RT-PCR test designed by German colleagues1. (A) During the benchmark testing phase an increasing number of commercial assays became available and were applied in an expanding range of diagnostic laboratories. The benchmark phase was followed by three External Quality Assessment rounds (EQA1-EQA3). (B) National data on number of persons tested for SARS-Cov-2 and on number of notified cases were obtained from RIVM data36.
An important aspect of this arrangement was the support offered by the reference laboratories, to assist in the introduction of the workflows and subsequent evaluation thereof. In the benchmark phase multiple technical issues were encountered by some of the laboratories based on the results of the benchmark panel and the confirmation samples that were sent to RIVM. Sensitivity issues were experienced by 15/71 laboratories (21.1%). Also, specificity issues were identified, as 2/71 laboratories (2.8%) were unable to differentiate between SARS-CoV-2 and other (seasonal) coronaviruses. In both cases, RNA isolation and/or amplification techniques were adjusted or substituted which solved the issues. One manufacturer was contacted to improve the performance of three of their kits since laboratories using these kits were experiencing both specificity and sensitivity issues. Contamination issues either during inter-facility specimen transport within the testing laboratory or during testing were experienced in 9/71 laboratories (12.7%). Overall, 56/71 laboratories (78.9%) immediately reached the ‘Excellent’ score whereas 15/71 laboratories (21.1%) needed to implement several adaptations to reach the desired quality level confirmed by testing and passing with another panel. The type of adjustments ranged from fine-tuning their workflow by changing the volumes used during RNA amplification to changing the RNA isolation and/or RNA amplification technique entirely before performance became ‘Excellent’ and passing the benchmark phase.
Performance of diagnostic laboratories over three EQA rounds
After successfully passing the benchmark test, laboratories took part in up to three EQA rounds which were performed over the course of 7 months. Some laboratories were added to the SARS-CoV-2 testing laboratory network and started and finished the benchmark test after already one or two EQA rounds were completed and therefore could not partake in all three EQA rounds. Other laboratories did not submit data for all EQA rounds despite finishing the benchmark test. In total 53 laboratories participated in EQA1, 60 in EQA2 and 68 in EQA3. The composition of the EQA panels was adapted each round to reflect the occurrence of novel SARS-CoV-2 variants of concern. A schematic overview of the performance of all individual 277 workflows submitted by the 71 laboratories spread over the three EQA rounds is given in Fig. 2.

Performance of SARS-CoV-2 diagnostic workflows over three External Quality Assessment (EQA) rounds. Identical workflows used in subsequent EQA rounds are displayed as bars at the same height. Unique workflows are displayed as a bar with no bar to their left and/or right. In total, cumulated over all three EQA rounds 413 excellent performances (127 in EQA round 1, 136 in EQA round 2 and 150 in EQA round 3), 54 mediocre performances (17 in EQA round 1, 17 in EQA round 2 and 20 in EQA round 3), and 22 unsatisfactory performances (7 in EQA round 1, 7 in EQA round 2 and 8 in EQA round 3) were reported. Full details about the workflows used in each EQA round can be found in Supplemental Figs. 4, 5 and 6.
Many laboratories submitted datasets of multiple workflows which culminated to a total of 489 data sets. The composition of the various workflows was subject to considerable change over time (Supplementary Figs. 4, 5 and 6). An overview of the various target genes applied by the laboratories is given in Supplementary Fig. 1A. Some workflows were deployed in all three EQA rounds while others were used only in one or two rounds (Fig. 2). Remarkably, the overall performance of the workflows did not improve in subsequent rounds. The quality of assays was consistent over the three EQA rounds with approximately 85% of the implemented assays having a 100% score (performance category ‘Excellent’) (Fig. 2). A cumulative overview of the performance on all tested specimens is given in Table 2.
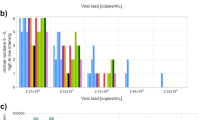
As expected, a virus concentration as low as 1.28 × 102 digital copies of E-gene/mL (the educational specimen SARS2_Edu) is a challenge for multiple workflows and resulted in a high proportion (40.1%) of false-negative test results. The various SARS-CoV-2 variants were detected with high accuracy (specificity 99.7%). The specificity of the testing procedures was high, 99.6% of the non-SARS-CoV-2 containing specimens were not mistaken for SARS-CoV-2 except for SARS-CoV-1 which was included in the panels as an educational specimen. Most workflows (53.3%) failed to distinguish SARS-CoV-2 from the closely related SARS-CoV-1 resulting in false-positive results because some workflows solely implemented the E-gene based primers as described by Corman et al.1, which cannot discriminate between the two pathogens. However, due to the absence of circulating SARS-CoV-1 since its elimination in 2003, this was not considered a problem. Remarkably, newly developed and implemented assays showed the same high level of quality as pre-existing ones (Fig. 2). Overall, the quality of the implemented workflows was high and stable over time during the study period in which new Variants of Concern emerged. The spread of the reported Cq values by the laboratories over the three sensitivity SARS-CoV-2 specimens is visualized in Fig. 3, in which a subdivision over the target genes is given. Whereas the data for most target genes are produced from multiple assays, the Cq values from the multiplex E-gene/N2-gene are all derived from a single type of cartridge-based sample-to-result assay (Cepheid, Xpert® Xpress SARS-CoV-2/Flu/RSV assay). We observed the least spread of Cq values with this last assay (24.2–30.5), whereas the spread overall for the other assays was 18.11–39.02 for specimen SARS2_H. For each of the targets a lower concentration resulted in a higher Cq value (connectors between specimens for individual workflows not shown in Fig. 3).

Reported Cq values and the fraction of specimens reported negative for the three SARS-CoV-2 virus concentrations: SARS2_H (1.28 × 105 copies of E-gene/mL); SARS2_L (1.28 × 103 copies of E-gene/mL); SARS2_Edu (1.28 × 102 copies of E-gene/mL). The results are further subdivided per target gene. Target genes utilized less than 10 times are not included. Boxplot hinges represent the 25th and 75th percentile, the middle vertical line is the median, and vertical lines extend to the smallest and largest values no further than 1.5 times the interquartile range from the hinges (values beyond this point can be considered outliers).
The reported Cq values are systematically higher than theoretically expected based on the dilutions (Supplementary Fig. 2). Supplementary Fig. 3 shows the predicted versus the reported Cq values of the individual target genes when plotted against each other. These data demonstrate that there is no strict linear correlation between Cq value and viral concentration in the studied concentration range and that this is independent of the choice of target gene.
Quantification of the contribution of some parameters to workflow performance
To infer the quantitative effect of the choice of target genes on the assay read-out parameter Cq, we applied Bayesian statistic modelling. This method estimates the likelihood of a Cq value as a distribution while correcting for confounding factors (for details see “Methods” section). To determine what the effect of the chosen target genes on the reported Cq values is, we modelled this effect for the SARS-CoV-2 specimens SARS2_H, SARS2_L and SARS2_Edu. Figure 4 shows the (mean) effect of target gene selection on derived Cq values for an assay. The predicted range is a parameter of the number of data points and that over all individual target genes, the mean values are distributed over a range of about 3.5 Cq values. Such data could be useful for selecting a new target gene for an assay if necessary. Similarly as for the target genes, we modelled the contribution of ‘Laboratory’ on the reported Cq values (for details see Methods section). This ‘Laboratory’ effect on the predicted Cq values for all types of assays ranged from -2.3 to 3.2 from the mean (Supplementary Fig. 1, panel B). The laboratories CS and CT occupy a relatively separate position which can possibly be attributed to the relative high number of specimens incorrectly reported as SARS-CoV-2 negative due to sensitivity issues. Thus, even when adjusting for the ‘Laboratory effect’ the difference in Cq values between laboratories for same concentration specimens remained considerable, further illustrating that taking Cq values as absolute proxy for viral load between laboratories and assays has limited value.

Visual representation of the effect of ‘Target gene’ on the reported Cq values as probability distributions. The X-axis depicts the spread in predicted Cq value per target gene (Y-axis) relative to the mean Cq value of all target genes combined. The median (black dot) and 50% and 95% distribution intervals (black lines) are indicated per target gene. The total number of datapoints for each target gene is shown in brackets. The different sizes of datasets are displayed using light blue (range < 20), blue (range 20–99) and dark blue (100+).
Discussion
This study describes a successful strategy for assessment, increasing and maintaining the quality of molecular diagnostics for SARS-CoV-2 in a very heterogeneous laboratory landscape by combining a benchmark testing phase and an EQA phase. These establishment and evaluation procedures were of great importance for setting up diagnostic testing facilities throughout The Netherlands in an early phase of the pandemic.
The Netherlands chose to implement decentralised testing with a wide variety of SARS-CoV-2 assays, the same approach that was also chosen for The Netherlands during the 2009 influenza pandemic16. This strategy has challenges as it is potentially difficult to maintain a homogenous high-quality level in a heterogenous testing landscape. This issue can be resolved by a well-designed test-implementation system with regular EQA and inter-laboratory comparisons as shown in this study. Importantly, a laboratory network implementing a multitude of assays essentially reduces the risk of collapse of the complete testing landscape (don’t put all of your eggs in one basket)19. During the COVID-19 pandemic, multiple issues were encountered including manufacturing problems, contamination of primers/probes20 and drop-outs because of genomic mutations in target genes8,9,10,11,12,21. In contrast to The Netherlands, the USA took the approach of decentralised testing with one assay type, similar to what they did during the 2009 influenza pandemic22,23. Although this method generally allows for quick and relatively simple upscaling of diagnostic capacity, when this strategy was implemented for SARS-CoV-2 in 2020 in the USA it had its challenges, namely contamination of primers/probes with synthetic template and improper primer/probe design20,24,25,26 which impaired the testing system. The CDC had a similar experience when implementing an mpox assay in their laboratory network in 202227,28. While we acknowledge that this topic is too complex to be discussed thoroughly in our paper, we feel it is worth briefly mentioning in this discussion as a way of starting or adding to pandemic preparedness systems discourse.
An important characteristic of the strategy of implementing heterogenous assays in The Netherlands was the presence of a ‘preparation phase’ (benchmark testing). In this phase laboratories could already make use of readily available blinded panels of simulated clinical specimens containing SARS-CoV-2 and other viruses during the early stage of the pandemic, and in addition receive advice and support from the reference laboratories. We observed that during this preparation phase the performance of several laboratories improved considerably, resulting in high quality testing in these laboratories and meeting set requirements for inclusion in the list of qualified SARS-CoV-2 diagnostics laboratories29. Most issues were found in high volume laboratories that previously did not perform diagnostics on human-derived specimens, which included veterinary laboratories and newly set up laboratories specific for SARS-CoV-2 testing, among others.
After finishing the benchmark phase, 84.5% of all submitted workflows performed up to the desired level in the subsequent individual EQA rounds. This is remarkable, as according to the benchmark inclusion criteria, all workflows were expected to perform ‘Excellent’ in the EQA rounds. It is possible that not all new workflows implemented in laboratories were pre-tested in the benchmark phase. However, our data does not provide a clear explanation for this observation.
The first published SARS-CoV-2 EQA was performed, primarily focused on frontline diagnostic laboratories, in April/May of 202030. In this first EQA, 365 of 406 laboratories from 36 countries submitted 521 datasets. All core samples from the EQA were correctly reported by 86.3% of participating laboratories and 83.1% of the datasets30, similar to our study. In another early SARS-CoV-2 EQA (which focused more on “expert” and reference laboratories, rather than frontline diagnostic laboratories) among 68 diagnostic laboratories spread over 35 European countries14, the test performances were of significantly lower quality than in our study (39.7% versus 84.5% of workflows scored all core specimens correct). The percentage of false positives or negatives in our study were 3.2% false negative, 0.1% false positive, whereas Fischer and colleagues found 8.6% false negative and 1.1% false positive results in their European study14. The Fischer et al. EQA was performed in June and July 2020 while our EQAs started in December that same year14. As laboratories had more time to set up their assays before the start of our study compared to the laboratories partaking in the Fischer et al. study14, the difference in diagnostic quality between the two studies might be partly due to more experience in COVID-19 diagnostics at the partaking laboratories. A major difference with our study is that the Fischer et al. or Matheeussen et al. study did not involve a benchmark testing procedure in advance of the EQAs14,30. The benchmark testing phase in our study started as early as March 2020 and could be considered an individual EQA with strict targets to be met by the laboratories. Nevertheless, this actually shows the benefit of our systematically applied entry benchmark testing approach that was (largely) lacking in other approaches. Based on our results, we expect that the availability of blinded testing panels to validate assays during implementation and compare performance with that of other laboratories, in combination with technical support, could improve the quality of the diagnostic testing performance in laboratories elsewhere. It is of note that this strategy is widely applicable and can cover other (novel) pathogens as well.
Other national SARS-CoV-2 diagnostic testing EQAs were performed and documented in Japan (94.1% correct reporting)31, South Korea (93.2% correct reporting)15, and Austria (93% correct reporting)32, with mostly similar results. Comparing these EQAs, or the original EQA from Matheeussen et al.30, with our EQA program is challenging, as sample quantification and preparation were done differently (in our study, using Minimal Essential Medium with Hanks’ salts and Hep2 cells instead of transport medium for sample preparation, varying methods for virus concentration determination, and using inactivated SARS-CoV-2 virus instead of RNA or pseudovirus constructs). It is of additional value when, in addition to the test results of the panel specimens, detailed information about the technical and procedural aspects, the so-called metadata, are shared with the organizer of an EQA. Communicating an overview of these anonymized and aggregated data, which cannot be collected within individual laboratories, among all EQA participants might be informative for an individual laboratory to compare its own quality level with its peers and especially, for getting suggestions for alternatives in case of suboptimal performance. In this report, we have taken this analysis a step further and demonstrate the possibility to get insights into specific aspects of the workflows. Such information can hint at steps in the procedures that are critical and it provides a quantitative estimate of its impact. Here we show a comparison of the consequences of the various target genes used by the laboratories on the workflows. This provided a direct comparison of target genes and suggests validated alternatives in case a gene target is no longer available because of mutations. Such analyses can also be performed for other elements of the workflows or even as a comparison between laboratories as we demonstrate.
A much debated topic is the use of Cq values as a measure for the absolute amount of virus in a clinical specimen. Differences in the amount of mRNA for protein production between the various targeted genes besides the presence of viral genomic and subgenomic RNA and differences in stability of the various RNAs will influence the amount of substrate for the RT-PCR reaction33,34. This is supposedly also in part reflected in the results of our analyses of the influence of target genes on Cq values. These factors limit a direct use of Cq values for virus concentration, apart from the difficulty to collect a specimen from a host in a reproducible way. Even standardised sample-to-result assays, which excludes some technical variation by making use of cartridges, showed substantial spread in Cq values in our study. Therefore, Cq values without calibration to international standards cannot be used to determine the amount of virus reliably and can at most provide a rough estimate, as for all workflows analyzed a decrease in concentration resulted in an increase in Cq value. The N2 target region seemed to be most sensitive target with the highest percentage of workflows with a positive result for the SARS2_Edu specimen, although Cq values were the highest compared to the other targets for this specimen. Differential generation of subgenomic mRNA’s35 and differences in reaction efficacy at low target concentration in a specimen—we show amplification is not exponential anymore (Supplementary Fig. 2 and 3)—could explain this phenomenon.
We consider the system of initial provision of validated primers and probes and protocols for laboratory developed tests and subsequent entry benchmark testing as an excellent way to develop and improve molecular diagnostic testing of pathogens in emerging situations requiring rapid availability of validated assays and high testing capacity. Technical and logistic assistance from a public health institute and/or expert laboratory is an important component. As this report demonstrates, the program is highly flexible and fast, allowing laboratories to design or purchase their own preferred assays and workflows while verifying and maintaining high quality testing. The addition of timely followup EQA rounds are necessary to maintain the overall quality of the diagnostic network. The collection and exchange of metadata is a valuable component and sophisticated statistical analyses can provide informative insight to the laboratories into components of their workflows. Importantly, the strategies here described are applicable to other pathogens as well and can be of great value in improving preparedness for novel pathogen detection and contribute to the advance of public health in a continuously developing and changing diagnostic field.
Data availability
Raw data is available as supplemental file.
References
Corman, V. M. et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 25, 2000045. https://doi.org/10.2807/1560-7917.ES.2020.25.3.2000045 (2020).
Corman, V. M. et al. Specific detection by real-time reverse-transcription PCR assays of a novel avian influenza A(H7N9) strain associated with human spillover infections in China. Eurosurveillance 18, 20461. https://doi.org/10.2807/ese.18.16.20461-en (2013).
Cameron, A. R., Meyer, A., Faverjon, C. & Mackenzie, C. Quantification of the sensitivity of early detection surveillance. Transbound. Emerg. Dis. 67, 2532–2543. https://doi.org/10.1111/tbed.13598 (2020).
Danmark, T. About TestCenter Denmark. https://tcdk.ssi.dk/english (2023).
CDC. Overview of Testing for SARS-CoV-2, the virus that causes COVID-19. https://www.cdc.gov/coronavirus/2019-ncov/lab/multiplex-faq.html (2023).
CDC. Research Use Only 2019-Novel Coronavirus (2019-nCoV) Real-time RT-PCR Primers and Probes. https://www.cdc.gov/coronavirus/2019-ncov/lab/rt-pcr-panel-primer-probes.html (2020).
Li, D., Zhang, J. & Li, J. Primer design for quantitative real-time PCR for the emerging Coronavirus SARS-CoV-2. Theranostics 10, 7150–7162. https://doi.org/10.7150/thno.47649 (2020).
Wang, H. et al. A deletion in the N gene of SARS-CoV-2 may reduce test sensitivity for detection of SARS-CoV-2. Diagn. Microbiol. Infect. Dis. 102, 115631–115631. https://doi.org/10.1016/j.diagmicrobio.2021.115631 (2022).
Borges, V. et al. Tracking SARS-CoV-2 lineage B.1.1.7 dissemination: Insights from nationwide spike gene target failure (SGTF) and spike gene late detection (SGTL) data, Portugal, week 49 2020 to week 3 2021. Euro Surveill. 26, 2100131. https://doi.org/10.2807/1560-7917.ES.2021.26.10.2100130 (2021).
Popping, S. et al. Diminished amplification of SARS-CoV-2 ORF1ab in a commercial dual-target qRT-PCR diagnostic assay. J. Virol. Methods 300, 114397. https://doi.org/10.1016/j.jviromet.2021.114397 (2022).
Artesi, M. et al. A recurrent mutation at position 26340 of SARS-CoV-2 is associated with failure of the E gene quantitative reverse transcription-PCR utilized in a commercial dual-target diagnostic assay. J. Clin. Microbiol. https://doi.org/10.1128/jcm.01598-20 (2020).
Li, Z. et al. A confirmed case of SARS-CoV-2 pneumonia with negative routine reverse transcriptase-polymerase chain reaction and virus variation in Guangzhou, China. Clin. Infect. Dis. 73, e426–e433. https://doi.org/10.1093/cid/ciaa941 (2021).
Görzer, I. et al. First results of a national external quality assessment scheme for the detection of SARS-CoV-2 genome sequences. J. Clin. Virol. 129, 104537. https://doi.org/10.1016/j.jcv.2020.104537 (2020).
Fischer, C. et al. Variable sensitivity of SARS-CoV-2 molecular detection in European expert laboratories: External quality assessment, June and July 2020. J. Clin. Microbiol. https://doi.org/10.1128/jcm.02676-20 (2021).
Sung, H. et al. Nationwide external quality assessment of SARS-CoV-2 molecular testing, South Korea. Emerg. Infect. Dis. J. 26, 2353. https://doi.org/10.3201/eid2610.202551 (2020).
Meijer, A. et al. Preparing the outbreak assistance laboratory network in the Netherlands for the detection of the influenza virus A(H1N1) variant. J. Clin. Virol. 45, 179–184. https://doi.org/10.1016/j.jcv.2009.06.003 (2009).
Wolters, F. et al. Multi-center evaluation of cepheid xpert® xpress SARS-CoV-2 point-of-care test during the SARS-CoV-2 pandemic. J. Clin. Virol. 128, 104426. https://doi.org/10.1016/j.jcv.2020.104426 (2020).
Sluimer, J. et al. Multi-center evaluation of Cepheid Xpert(R) Xpress SARS-CoV-2/Flu/RSV molecular point-of-care test. J. Clin. Virol. Plus 1, 100042. https://doi.org/10.1016/j.jcvp.2021.100042 (2021).
Saavedra, M. D. C. Don Quixote. (Penguin Classics, 2003).
Mogling, R. et al. Delayed laboratory response to COVID-19 caused by molecular diagnostic contamination. Emerg. Infect. Dis. 26, 1944–1946. https://doi.org/10.3201/eid2608.201843 (2020).
Wang, C. Y. T., Buckley, C., Bletchly, C., Harris, P. & Whiley, D. Contamination of SARS-CoV-2 RT-PCR probes at the oligonucleotide manufacturer. Pathology 52, 814–816. https://doi.org/10.1016/j.pathol.2020.08.002 (2020).
Jernigan, D. B. et al. Detecting 2009 pandemic influenza A (H1N1) virus infection: Availability of diagnostic testing led to rapid pandemic response. Clin. Infect. Dis. 52(Suppl 1), S36-43. https://doi.org/10.1093/cid/ciq020 (2011).
Dawood, F. S. et al. Emergence of a novel swine-origin influenza A (H1N1) virus in humans. N. Engl. J. Med. 360, 2605–2615. https://doi.org/10.1056/NEJMoa0903810 (2009).
Lee, J. S. et al. Analysis of the initial lot of the CDC 2019-Novel Coronavirus (2019-nCoV) real-time RT-PCR diagnostic panel. PLoS One 16, e0260487. https://doi.org/10.1371/journal.pone.0260487 (2021).
Vergano, D. The Government Asked Us Not To Release Records From The CDC’s First Failed COVID Test. Here They Are. https://www.buzzfeednews.com/article/danvergano/covid-test-false-positive-failure-cdc-documents (2021).
SERVICES, D. O. H. H. Summary of the Findings of the Immediate Office of the General Counsel’s Investigation Regarding CDC’s Production of COVID-19 Test Kits. https://www.washingtonpost.com/context/summary-of-the-findings-of-the-immediate-office-of-the-general-counsel-s-investigation-regarding-cdc-s-production-of-covid-19-test-kits/a750fbf7-9a4f-4062-8fcc-c6cf42600578/ (2020).
Wu, F., Oghuan, J., Gitter, A., Mena, K. D. & Brown, E. L. Wide mismatches in the sequences of primers and probes for Monkeypox virus diagnostic assays. medRxiv https://doi.org/10.1101/2022.08.10.22278644 (2022).
CDC. Lab Alert: MPXV TNF Receptor Gene Deletion May Lead to False Negative Results with Some MPXV Specific LDTs. https://www.cdc.gov/locs/2022/09-02-2022-lab-alert-MPXV_TNF_Receptor_Gene_Deletion_May_Lead_False_Negative_Results_Some_MPXV_Specific_LDTs.html (2022).
RIVM. Aanvullende informatie diagnostiek COVID-19. https://lci.rivm.nl/covid-19/bijlage/aanvullend (2020).
Matheeussen, V. et al. International external quality assessment for SARS-CoV-2 molecular detection and survey on clinical laboratory preparedness during the COVID-19 pandemic, April/May 2020. Euro Surveill. https://doi.org/10.2807/1560-7917.ES.2020.25.27.2001223 (2020).
Asai, S. et al. Nationwide external quality assessment of SARS-CoV-2 nucleic acid amplification tests in Japan. Int. J. Infect. Dis. 115, 86–92. https://doi.org/10.1016/j.ijid.2021.11.022 (2022).
Buchta, C. et al. The versatility of external quality assessment for the surveillance of laboratory and in vitro diagnostic performance: SARS-CoV-2 viral genome detection in Austria. Clin Chem Lab Med 59, 1735–1744. https://doi.org/10.1515/cclm-2021-0604 (2021).
Dimcheff, D. E. et al. Severe acute respiratory syndrome coronavirus 2 total and subgenomic RNA viral load in hospitalized patients. J. Infect. Dis. 224, 1287–1293. https://doi.org/10.1093/infdis/jiab215 (2021).
Toppings, N. B., Oberding, L. K., Lin, Y.-C., Evans, D. & Pillai, D. R. The role of subgenomic RNA in discordant results from reverse transcription-polymerase chain reaction tests for COVID-19. Archiv. Pathol. Lab. Med. 146, 805–813. https://doi.org/10.5858/arpa.2021-0630-SA (2022).
Valadan, R. et al. Differential gene expression analysis of common target genes for the detection of SARS-CoV-2 using real time-PCR. AMB Express 12, 112. https://doi.org/10.1186/s13568-022-01454-2 (2022).
RIVM. Covid-19 afgenomen testen per datum van de afspraak en veiligheidsregio. https://data.rivm.nl/meta/srv/dut/catalog.search#/metadata/0f3336f5-0f16-462c-9031-bb60adde4af1 (2023).
Acknowledgements
The technicians of the RIVM (Lisa Wijsman, Sharon van den Brink, Barbara Favié, Annemarie van den Brandt) and Erasmus MC (Daphne Mulders) are thanked for skilfully evaluating the testing material. We would also like to thank Bart Haagmans (Erasmus MC) and Lia van der Hoek (Amsterdam University Medical Hospital) for supplying us with material with which our panels were made. We would also like to thank Pleunie van Alphen (PAMM), Greet Boland, Anne Wensing, Martin Rakke, Edwin Fries (UMCU), Roy Masius, Anne Vogel, Anco Molijn (Eurofins NMDL-LCPL), Roland Doorn (Tergooi MC, CBSL), Martijn Mostert (Diakonessenhuis Utrecht), Patricia Glasius (Noordwest Ziekenhuisgroep Alkmaar), Caroline M.A. Swanink, Angelino T. Tromp (Rijnstate Velp), Kelly Mertens, Nico van Lisdonk, Cindy Houwen (Star-SHL) and Stefan Küchler (SYNLAB Jena ONCOSCREEN) for their contributions to the datasets sent by the Working Group SARS-CoV-2 Diagnostics The Netherlands. This investigation was performed by order, and for the account, of the Ministry of Health, Welfare and Sport, project number V/190028/22/PR.
Author information
Authors and Affiliations
Consortia
Contributions
J.S.: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, visualization, editing, project administration. W.M.R. van den A.: formal analysis, writing—original draft, visualization. G.G.: conceptualization, methodology, investigation, resources. A.S.: methodology, formal analysis, data curation, visualization. B. van der V., J.C., N.H.C., J.V., J.G.: investigation. R.M.: writing—review and editing. Working Group SARS-CoV-2 Diagnostics The Netherlands: Investigation. D.E.: writing—review and editing. L.D.P.: writing—review and editing. A.M.: conceptualization, validation, writing—review and editing, supervision, project administration, funding acquisition.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sluimer, J., van den Akker, W.M.R., Goderski, G. et al. High quality of SARS-CoV-2 molecular diagnostics in a diverse laboratory landscape through supported benchmark testing and External Quality Assessment. Sci Rep 14, 1378 (2024). https://doi.org/10.1038/s41598-023-50912-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-023-50912-9
This article is cited by
-
External quality assessments for SARS-CoV-2 genome detection in Austria
Wiener klinische Wochenschrift (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
