
Abstract
The Poxviridae family members vaccinia and variola virus enter mammalian cells, replicate outside the nucleus and produce virions that travel to the cell surface along microtubules, fuse with the plasma membrane and egress from infected cells toward apposing cells on actin-filled membranous protrusions. We show that cell-associated enveloped virions (CEV) use Abl- and Src-family tyrosine kinases for actin motility, and that these kinases act in a redundant fashion, perhaps permitting motility in a greater range of cell types. Additionally, release of CEV from the cell requires Abl- but not Src-family tyrosine kinases, and is blocked by STI-571 (Gleevec), an Abl-family kinase inhibitor used to treat chronic myelogenous leukemia in humans. Finally, we show that STI-571 reduces viral dissemination by five orders of magnitude and promotes survival in infected mice, suggesting possible use for this drug in treating smallpox or complications associated with vaccination. This therapeutic approach may prove generally efficacious in treating microbial infections that rely on host tyrosine kinases, and, because the drug targets host but not viral molecules, this strategy is much less likely to engender resistance compared to conventional antimicrobial therapies.
Similar content being viewed by others


Main
Vaccinia virus and variola major are members of the Poxviridae family1,2. Vaccinia serves as the vaccinating agent for variola major, the cause of smallpox. In 1980, the World Health Organization declared smallpox eradicated. Routine vaccinations for smallpox ceased in 1972, and though immunity in vaccinated individuals does not seem to have abated3,4, a large proportion of the general population is still considered extremely susceptible to variola and other poxviruses in the event of an intentional or unintentional release4,5. Moreover, vaccination itself is known to produce serious side effects in immunosuppressed individuals6,7,8, raising the possibility that vaccination may no longer prove completely effective as a means to control an outbreak6.
Upon entry into the cell, pox virions move to juxtanuclear locations where they replicate up to 104 concatameric genomes2. The concatamers resolve into unit genomes and are packaged in individual enveloped particles called intracellular mature virions9 (IMV), some of which are wrapped in additional membranes to form intracellular enveloped virions10 (IEV). Cytolysis releases IMV from the cell. Before cytolysis, some IEV travel toward the host cell periphery through a kinesin-microtubule transport system11,12,13,14. To exit the cell, the IEV particle fuses with the plasma membrane of the host cell to form a cell-associated enveloped virus (CEV), leaving behind one of its two outer membranes10,15. CEV either detach directly, or initiate actin polymerization to propel the particle on an actin-filled membrane protuberance toward an apposing cell and then detach10. Once detached, CEV form extracellular enveloped virus10 (EEV). It has been proposed that IMV are readily detected and inactivated by the host immune system in vivo, but CEV and EEV evade immune detection and mediate spread of the infection15.
The vaccinia virus protein A36R is located in the membrane surrounding the IEV and is required for actin polymerization16 and virulence17. Previous reports suggest that the mammalian tyrosine kinase c-Src localizes to virions and phosphorylates A36R18. Once phosphorylated, A36R facilitates detachment of kinesin19, and, following virion fusion with the plasma membrane, recruitment and activation of host cell proteins, including Nck, Grb2, N-WASP and Arp2/3, which initiate actin polymerization beneath the particle20,21,22. The mechanism by which CEV detach from the cell or from the actin protuberance (thereby generating EEV) and enter apposing cells is less understood, and no role for tyrosine kinases in this process has been established.
Investigations of the mechanism of actin polymerization by the bacterial pathogen enteropathogenic E. coli23 have shown that phosphorylation of the virulence factor Tir24 and subsequent recruitment of actin-polymerizing factors25 are mediated by several redundant tyrosine kinases26. The similarity of the phosphorylation sites in the bacterial Tir protein and vaccinia virus A36R (Supplementary Fig. 1 online)20 led us to consider whether the role of tyrosine kinases in vaccinia virus actin polymerization might be similarly complex.
Here we show that redundant Src- and Abl-family kinases mediate actin motility, that Abl-family kinases mediate efficient release of infectious EEV, and that inhibitors of Abl-family kinases such as the 2-phenylpyrimidine STI-571 (also called imatinib mesylate or Gleevec)27 block infectious EEV release, and limit spread of infection in vivo and promote survival. STI-571 has proven clinically useful in treating chronic myelogenous leukemia (CML) in humans, a disease resulting from dysregulation of c-Abl28,29. Our results raise the possibility of using STI-571 to treat variola infections or complications associated with vaccination.
Results
Abl- and Src-family proteins localize in vaccinia actin tails
To test whether Src- and Abl-family tyrosine kinases participate in vaccinia actin motility, we first determined whether endogenous proteins resembling kinases in these families localized on the virion at the tip of the actin tail. 3T3 cells were exposed to vaccinia virus, and then stained with antibodies against c-Src, c-Fyn, c-Yes, c-Abl (encoded by Abl1) and the Abl-related kinase c-Arg (encoded by Abl2). Infected cells were identified by staining with 4,6-diamidino-2-phenylindole (DAPI) to label extranuclear replication centers (Supplementary Fig. 2 online), or by staining with α-TW2.3, an antibody that recognizes a vaccinia protein expressed early in infection30 (Supplementary Fig. 2 online). Virions on actin tails were recognized by DAPI staining or by fluorescence of a vaccinia virus with green fluorescent protein (GFP)-B5R fusion protein (GFP-VV)31 localized in the membrane of the virion (Fig. 1a and Supplementary Fig. 2 online). Actin tails are seen as intense phalloidin staining directly apposed to the virion (Fig. 1a).

(a) GFP-VV–infected 3T3 cell stained with Cy3-phalloidin to visualize actin and α-Arg-Cy5 polyclonal antibody. Blue, GFP-VV; red, Arg; green, actin. The virion and c-Arg colocalize. (b) Merged images of 3T3 cells exposed to vaccinia virus and stained with α-phosphotyrosine polyclonal antibody (α-R-pY) to recognize extranuclear replication centers (blue), FITC-phalloidin to recognize actin (green) and α-Abl 8E9 (red). The white boxes in b are shown in expanded scale to the right. (c) Merged image of 3T3 cell exposed to vaccinia virus and stained with FITC-phalloidin to recognize actin (green), together with α-Src (left), α-Fyn (middle) or α-Yes (right; all red). (d) Merged images of 3T3 cell exposed to vaccinia virus and stained with FITC-phalloidin to recognize actin (green), together with α-Abl-Cy3 (blue) and α-Arg-Cy5 (red). Images are from different locations within the same cell. c-Abl but not c-Arg is evident at the tip of the actin tail in the left panel, c-Arg but not c-Abl in the middle panel, and both kinases in the right panel. (e) Quantification of distribution of Src- and Abl-family kinases in vaccinia actin tails on infected 3T3 cells. For Abl-family kinases, tails contain one or the other kinase, but few contain both. (f) Merged image of 3T3 cell exposed to vaccinia and stained with FITC-phalloidin to recognize actin (green), together with α-phosphotyrosine (pY)-412 polyclonal antibody (blue, arrow) which recognizes activated Abl or activated Arg. pY-412 staining is evident in one tail in this image but not in others, consistent with the lack of Abl or Arg staining in all tails. Scale bars, 5 μm.
An endogenous protein recognized by α-Arg polyclonal antibody was enriched at the tips of the actin tails relative to the cytoplasm (Fig. 1a). Proteins recognized by the α-Abl monoclonal antibody 8E9 (Fig. 1b) or AB3 (data not shown), α-Src polyclonal antibody (Fig. 1c), α-Fyn monoclonal antibody (Fig. 1c), and α-Yes monoclonal antibody (Fig. 1c) were also enriched at the tips of the actin tails relative to the cytoplasm. Each kinase was detectable in only a fraction of actin tails. For example, c-Abl was detectable in some tails, but not in others within the same cell (Fig. 1b). Staining with combinations of antibodies (e.g., α-Abl together with α-Arg) indicated that tails containing one kinase did not generally contain detectable levels of another kinase type (Fig. 1d), though some tails did contain both kinase types (Fig. 1d,e). Similar results were obtained with combinations of other kinase-specific antibodies (data not shown), though because many were of similar isotype, testing all combinations was not feasible. Of the five Src- and Abl-family kinases, proteins resembling c-Fyn were the most frequently observed in actin tails in 3T3 cells (Fig. 1e). PDGFR, FGFR, Lck, FAK, Ntk, Lyn, Jak1, Csk, Tyk2 and Pyk2 did not appear localized (data not shown), suggesting that localization is specific for Src- and Abl-family kinases. Finally, staining with antibody recognizing phosphorylated Y412 (α-pY412) in the activation domain of Abl and Arg32 indicated that these kinases were active in tails (Fig. 1f). Staining with α-pY412 was specific for c-Abl or c-Arg, and was not evident in tails formed in cells lacking c-Abl and c-Arg26. All antibodies were specific and did not recognize epitopes in cells lacking these kinases (Supplementary Fig. 2 online), and showed no cross-reactivity with other family members as judged by transfection experiments in cells lacking the kinases26.
Localization and distribution of exogenously expressed and endogenous kinases were similar. For example, c-Arg bound to yellow fluorescent protein (YFP–c-Arg) was present in only a fraction of actin tails in transfected cells (Supplementary Fig. 2 online), and even in cells expressing high levels of YFP–c-Arg, some tails contained no YFP–c-Arg, suggesting that localization of overexpressed kinase was specific (Supplementary Fig. 2 online). Colocalization with virions was not observed for other overexpressed proteins including GFP, YFP or the kinase Hck (data not shown). The possibility exists that kinases sequentially associate with tails, or that one kinase triggers recruitment of another, or that different virion types recruit different kinases. Together, these data suggest that Abl- and Src-family kinases localize in vaccinia actin tails, though other kinases may localize as well.
Redundant roles of Abl- and Src-family kinases in actin motility
To determine whether individual Src- and/or Abl-family tyrosine kinases were necessary for actin tail formation, we infected 3T3 cells derived from mice lacking lacking c-Src (Src−/− ; Fig. 2a), c-Src and Yes (Src−/−Yes1−/− ; data not shown), c-Fyn and c-Yes (Fyn−/−Yes1−/− ; data not shown) or c-Src, c-Fyn and c-Yes (Src−/−Fyn−/−Yes1−/− ; Fig. 2b), or from mice lacking c-Abl alone (Abl1−/− ; data not shown), c-Arg alone (Abl2−/− ; data not shown), or both c-Abl and c-Arg (Abl1−/−Abl2−/− ; Fig. 2c). No differences were apparent in the capacity to form actin tails in these cell lines compared to wild-type cells (Fig. 2a–c), though the proportion of tails occupied by particular kinases differed depending on the cell line and kinase (Supplementary Fig. 2 online). Despite the differences in distribution of kinases, neither c-Abl, c-Arg, c-Src, c-Fyn nor c-Yes alone seemed necessary for vaccinia actin motility.

(a–c) Cell lines derived from Src−/− (a), Src−/−Fyn−/−Yes1−/− (b), or Abl1−/−Abl2−/− mice (c), were exposed to vaccinia (a) or GFP-VV (b,c) and stained with Cy3-phalloidin. (d) Images of 3T3 cells infected with vaccinia for 8 h and treated with 10 μM PD-166326 either for the duration of the infection (upper panels) or for 10 min before fixation (lower panels). Cells were stained with DAPI and α-pY polyclonal antibody to recognize infected cells, and FITC-phalloidin to recognize actin. Blue, DAPI; green, actin; red, pY. p.i. indicates post-infection. (e) Quantification of cells with actin tails after 8 h of infection with vaccinia. 3T3 cells were exposed to 0.1% DMSO, 25 μm PP2, 10 μm PD-166326. (f) Measurement of viral yield by single-step growth curves in BSC-40 cells infected with vaccinia virus at an MOI of 5, and treated with 10 μM PD-166326, or DMSO, or 10 μM STI-571. PD-166326 has no effect on viral yield except at the initial time point. STI-571 does not reduce viral yield. (g) Merged images of cells exposed to DMSO (upper) or 10 μM PD-166236 (lower), infected with GFP-VV (green) for 8 h and stained with α-pY monoclonal antibody 4G10 (blue) and Alexa 546-phalloidin (red). The α-pY 4G10 monoclonal antibody recognizes residues in the tip of the actin tail in DMSO-treated cells. But α-pY 4G10 monoclonal antibody staining is not evident upon PD-166326 treatment (Supplementary Fig. 2 online). The effect of PD-166326 was selective because targets in the replication centers recognized by α-R-pY were unaffected (d). Scale bars, 5 μm.
To determine whether Abl- or Src-family kinases are sufficient for actin motility, we first identified inhibitors of tyrosine kinases that block actin motility, and then determined whether mutant kinases resistant to such inhibitors could support actin motility with the inhibitor present. To identify inhibitors of actin motility, we assessed the effects of pyrido[2,3-d]-pyrimidine (PD) compounds (Supplementary Fig. 3 online), which competitively inhibit binding of ATP to Abl- and Src-family kinases33,34,35. Treatment with 10 μM PD-166326 reduced the percentage of infected cells with actin tails from 96% to 0% (Fig. 2d,e). Concentrations of PD-166326 less than 1 μM were without effect (data not shown). Staining with α-TW2.3 was evident in cells treated with 10 μM PD-166326, suggesting that the drug did not block viral entry (data not shown). Staining with DAPI or α-phosphotyrosine polyclonal antibody (Fig. 2d) showed the presence of extranuclear replication centers in the presence of 10 μM PD-166326. In a one-step growth curve (multiplicity of infection (MOI) of 5), viral yield was unaffected by PD-166326 except at the initial time point, at which it was reduced by 10-fold from 106 PFU to 105 PFU (Fig. 2f) compared to carrier control. Consistent with an effect on cell-to-cell spread, PD-166326 reduced plaque diameter (Supplementary Fig. 3 online), and viral yield in a one-step growth curve at low MOI (0.1) indicated a 1,000-fold reduction in viral yield from 108 PFU to 105 PFU at all time points (Supplementary Fig. 3 online). Addition of 10 μM PD-166326 8 h after infection for as little as 20 min also resulted in loss of actin motility (Fig. 2d). Compounds structurally related to PD-166326 (e.g., SKI-DV-1-10, 10 μM) were as effective as PD-166326 in blocking actin tails (data not shown), and PP1 and PP2, which similarly inhibit activity of Src- and Abl-family kinases36,37, also blocked actin tails at concentrations of 25 μM or greater, as reported18 (Fig. 2e). The effects of PD-166326 did not result from nonspecific block of actin polymerization, as PD-166326 had no effect on actin tails induced by Listeria monocytogenes or Shigella flexneri26. PD-166326 also blocked localization on the virion of factors crucial for actin polymerization. Colocalization with the virion of phosphotyrosine, Nck, N-WASP, Grb-2 and Arp2/3 (refs. 21,22) was not evident in GFP-VV–infected cells treated with 10 μM PD-166326 (Fig. 2g, Supplementary Fig. 3 online and data not shown). Together, these results suggest that PD-166326 blocks tyrosine kinase activity essential for actin motility.
To determine whether Abl, Arg or Yes was sufficient for vaccinia actin motility, we assessed whether each kinase could support vaccinia actin motility in the absence of activity from other Src- or Abl-family kinases. Specifically, we tested whether expression of PD-resistant alleles of c-Abl or c-Arg allowed actin motility to persist in the presence of PD-166326. Mutations within the ATP binding pocket disrupt van der Waals interaction between PD-166326 and the kinases (Abl-T315I and Arg-T314I), and increase the Ki of PD-166326 from 10 nM to 1 μM as measured by in vitro kinase assay26. As seen in Fig. 3a, actin tails were evident in PD-treated cells expressing YFP–c-Arg-T314I (Fig. 3a), but not in cells expressing endogenous c-Arg (Fig. 3a), or in cells overexpressing wild-type c-Arg (Supplementary Fig. 3 online) or in cells expressing c-Abl-T315I (Fig. 3b). Tails were also evident in PD-treated cells expressing c-Yes-T348I (Fig. 3c), but not c-Yes (Supplementary Fig. 3 online). These data indicate that c-Arg and c-Yes, but not c-Abl, are sufficient among tyrosine kinases for vaccinia actin motility.

Pseudocolors in parentheses refer to merged images. (a) 3T3 cells transfected with YFP–c-Arg-T314I (green), treated with 10 μM PD-166326 and exposed to vaccinia virus for 8 h, and then stained with 594-phalloidin (red) to recognize actin, and DAPI (blue). Actin tails are present in the cell expressing YFP–c-Arg-T314I (upper), but not in the nonexpressing cell (lower). (b) 3T3 cells transfected with c-Abl-T315I, treated with 10 μM PD-166326 and exposed to vaccinia for 8 h, and then stained with FITC-phalloidin (green), DAPI (blue) and α-Abl monoclonal antibody 8E9 (red). Actin tails are absent in the cell expressing c-Abl-T315I. (c) Src−/−Fyn−/−Yes1−/− cells transfected with c-Yes-T348I, treated with 10 μM PD-166326 and exposed to vaccinia for 8 h, and then stained with FITC-phalloidin (green), DAPI (blue) and α-Yes monoclonal antibody (red). Actin tails are present in the cell. (d) 3T3 cells treated with 10 μM STI-571 and exposed to vaccinia for 8 h. Cells were stained with DAPI and α-pY polyclonal antibody (red) to recognize infected cells, and FITC-phalloidin (green). Actin tails are present. (e) Src−/−Fyn−/−Yes1−/− cells treated with 10 μM STI-571 and exposed to GFP-VV (green) for 8 h, and stained with 546-phalloidin (red). Actin tails are absent despite the presence of GFP-VV at the cell periphery. (f) Src−/−Fyn−/−Yes1−/− cells treated with DMSO (0.1%) and exposed to GFP-VV for 8 h, and stained as in e. In the merged image, GFP-VV is present at the tip of the actin tails. (g,h) Src−/−Yes1−/− cells (g) or Fyn−/−Yes1−/− cells (h) treated with 10 μM STI-571, exposed to vaccinia for 8 h, and stained with FITC-phalloidin (green), and DAPI (blue). Actin tails are present in these cells. Scale bars, 5 μm.
To determine whether other Src-family kinases were sufficient for vaccinia actin motility, we treated Src−/−Fyn−/−Yes1−/− cells or cell lines lacking subsets of Src-family kinases with 10 μM STI-571 (Supplementary Fig. 4 online), a 2-phenylpyrimidine compound that inhibits Abl- but not Src-family kinases27. STI-571 did not block actin motility in wild-type 3T3 cells (Fig. 3d), even at concentrations as high as 25 μM, and did not reduce viral yield (Fig. 2f) or the appearance of viral replication centers (Fig. 3d). Addition of 10 μM STI-571 severely limited vaccinia actin motility in Src−/−Fyn−/−Yes1−/− cells, reducing the average number of actin tails per cell by 16-fold from 48 to ∼3 per cell (Supplementary Fig. 4 online) with 30% of cells having none (Fig. 3e), but did not affect transit of GFP-labeled virions to the cell periphery (Fig. 3e). The carrier for STI-571, DMSO, did not exert an effect (Fig. 3f). To determine whether c-Fyn or c-Src were sufficient for vaccinia actin motility, we tested the effects of 10 μM STI-571 on vaccinia actin motility in Src−/−Yes1−/− cells (Fig. 3g and Supplementary Fig. 4 online) and Fyn−/−Yes1−/− cells (Fig. 3h). Treatment with STI-571 also had no detectable effects on the number of actin tails per cell (Supplementary Fig. 4 online). Together, these results suggest that c-Arg, c-Yes, c-Src and c-Fyn are all sufficient for vaccinia actin motility.
To determine whether redundant tyrosine kinases mediate plaque formation, monolayers of 3T3, cells lacking various kinases or cells treated with PD-166326 or STI-571 were infected with vaccinia virus (Fig. 4). In accordance with a requirement of actin tails for cell-to-cell spread15,38, plaques formed on wild-type 3T3 cells or cells lacking various kinases with equal efficacy (Fig. 4d–f). Treatment of 3T3 or BSC-40 cells with 10 μM PD-166326 reduced plaque size to 'pinpoints' (Fig. 4a,b and Supplementary Fig. 3 online). Treatment with 10 μM STI-571 did not produce significant changes in plaque number (Fig. 4a,c) in BSC-40 cells, though this compound did reduce plaque size (Fig. 4a,c and Supplementary Fig. 4 online). Single-step growth assays indicated that 10 μM STI-571 had little effect on viral yield in BSC-40 cells (Fig. 2f and Supplementary Fig. 3 online) or Src−/−Fyn−/−Yes1−/− cells (MOI of 5; Supplementary Fig. 4 online). But treatment with 10 μM STI-571 reduced plaque size to pinpoints in Src−/−Fyn−/−Yes1−/− (Fig. 4f,g). Together, these data suggest that redundant Abl- and Src-family kinases mediate actin motility and cell-to-cell spread, but have little effect on viral replication.

Plaque assays of vaccinia virus strain WR on BSC-40 cells (a–c) or 3T3 cells derived from wild-type mice (d) or from Abl1−/−Abl2−/− mice (e), or Src−/−Fyn−/−Yes1−/− mice (f,g). Cells were fixed 3 (a–c) or 4 (d–g) d after infection. Vaccinia virus was added to wells depicted in the three right columns at the dilutions indicated. The titer of undiluted vaccinia added to all wells was 104 PFU/ml, as measured on BSC-40 cells. We added 10 μM PD-166326 1 h after infection to the wells depicted in b, and 10 μM STI571 to the wells depicted in c and g. PD-166326 blocked plaque formation in BSC-40 cells and 3T3 cells (Supplementary Fig. 3 online), whereas STI-571 only blocked plaque formation in the absence of c-Src, c-Fyn and c-Yes (g).
Abl-family kinases mediate release of infectious EEV
We next determined whether tyrosine kinases mediated long-range spread of vaccinia virus by facilitating release of infectious EEV from the plasma membrane. To do this, wild-type 3T3 cells or 3T3 cells lacking various tyrosine kinases were infected with vaccinia virus. Supernatants containing EEV were collected from cells 24 h after infection. At this time point, the supernatant contains plaque-forming units (PFU) composed of EEV, and to a lesser extent IMV released from lysed cells39. The supernatant was then used to assess plaque formation in BSC-40 cells (Fig. 5). Analysis of plaques indicated that supernatants from wild-type 3T3 cells (Fig. 5a), Src−/−Fyn−/−Yes1−/− cells (Fig. 5b), Abl1−/− cells (Fig. 5c), and Abl2−/− cells (Fig. 5d) all contained approximately the same number of PFU, but supernatants from Abl1−/−Abl2−/− cells (Fig. 5e,h) contained ∼70% less PFU. Such a decrease could not be accounted for by lower infectivity of Abl1−/−Abl2−/− cells compared to wild-type cells because in both cell types, the same number of plaques formed (Fig. 4d,e) and viral yields were equivalent (Supplementary Fig. 5 online). In accordance with these data, treatment of wild-type cells (BSC-40 or 3T3) with 10 μM STI-571 caused a 60–70% reduction in EEV (Fig. 5f–h), without significantly affecting viral yield (Fig. 2f and Supplementary Figs. 3 and 5 online). Treatment with 10 μM STI-571 also blocked the formation of 'comets' apposed to plaques in BSC-40 cells (Supplementary Fig. 4 online), a phenomenon associated with EEV40. The decrease in EEV observed with STI-571 may also account for the reduction in plaque size in BSC-40 cells treated with this compound (Fig. 4a,c and Supplementary Fig. 4 online). The effect of STI-571 on EEV release was not restricted to vaccinia virus strain WR. Release of EEV by vaccinia virus strain IHD-J, which releases 40-fold more EEV than strain WR41, was similarly inhibited (Fig. 5h). In some experiments, supernatants were incubated with 2D5 monoclonal antibody to neutralize contaminating IMV39,42 (Fig. 5h and Supplementary Fig. 6 online).

(a–e) Plaque assays of BSC-40 cells infected with supernatants (SN) derived from uninfected cells or vaccinia virus strain WR–infected wild-type 3T3 cells (a) or 3T3 cells derived from Src−/−Fyn−/−Yes1−/− mice (b), Abl1−/− (c), Abl2−/− (d), or Abl1−/−Abl2−/− mice (e). (f,g) Plaque assays of BSC-40 cells infected with supernatants derived from uninfected BSC-40 cells (first column) or vaccinia-infected BSC-40 cells. For a–g, supernatant was added neat or diluted 1:10. For f and g, initial infections, but not plaque assays, were carried out in the absence of drug (f), or in the presence of 10 μM STI-571 (g). Plaques are present except when both c-Abl and c-Arg are absent (e), or when their activity is blocked with drug (g). (h) Left, quantification of EEV from supernatants of vaccinia-infected BSC-40 cells or BSC-40 cells treated with 10 μM STI-571. Supernatants were treated with 2D5 monoclonal antibody to reduce contamination from IMV. Middle, quantification of EEV from supernatants of vaccinia virus strain WR–infected 3T3 cells, or 3T3 cells treated with 10 μM STI-571, or Abl1−/−Abl2−/− cells. Supernatants were treated with 2D5 monoclonal antibody to reduce contamination from IMV. Right, effects of STI-571 on VV-IHD-J EEV. Quantification of EEV from supernatants of vaccinia virus strain IHD-J–infected 3T3 cells or 3T3 cells treated with 5 μM STI-571. Supernatants were treated with 2D5 monoclonal antibody to reduce contamination from IMV. As with vaccinia virus strain WR EEV, STI-571 reduced vaccinia virus strain IHD-J EEV production.
Treatment of 3T3 cells with 10 μM PD-166326 likewise reduced the number of EEV (data not shown). But because PD-166326 blocks actin tails (Fig. 2d,e), we could not distinguish whether the drug caused a decrease in the number of virions reaching the cell surface or blocked EEV release or both. Taken together, these observations suggest that c-Abl and c-Arg, but not Src-family kinases, are each sufficient for release of infectious EEV in vitro, and together they are necessary.
STI-571 promotes survival in vaccinia-infected mice
EEV are less immunogenic than other forms of the virus43, and have been proposed to mediate spread of infection and virulence in vivo17,44,45. The involvement of Abl-family kinases in release of infectious EEV in vitro suggested that STI-571 might control vaccinia infection in vivo. STI-571 at 100 mg/kg/d or the saline carrier was delivered to mice through Alzet osmotic pumps placed subcutaneously. This drug concentration has been used in mouse leukemia models for a period of months without considerable side effects, and was equivalent to doses used in humans with CML46. We measured by quantitative PCR47 the number of viral genomes in ovaries 4 d after infection. In untreated animals or animals with pumps containing carrier, we detected ∼107 copies/250 ng DNA (Fig. 6a). The detection limit of the assay, determined by serial dilution, was 10 viral genomes. Treatment with STI-571 reduced viral genome copies in ovaries by five orders of magnitude from ∼107 to less than 102 copies (Fig. 6a; Fisher exact test, P < 10−6). In accordance with an inhibition of EEV release (Fig. 5h), these results suggest that STI-571 inhibits spread of vaccinia virus in vivo.

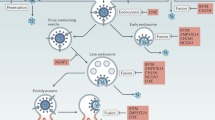
(a) STI-571 reduces vaccinia virus dissemination. We intraperitoneally infected or left uninfected 6-week-old C57/B6 mice with 104 PFU vaccinia virus strain WR. Infected mice were left untreated or treated with PBS (carrier) or STI-571 (100 mg/kg/d). We quantified the number of viral genomes in ovaries 4 d after infection. Combined data from three separate experiments are shown (n = 18 mice per condition). The line in each data set represents the median number of viral genomes. (b) STI-571 promotes survival in vaccinia-infected mice. We intranasally infected 6-week-old C57/B6 mice with 2 × 104 PFU vaccinia virus strain IHD-J. In some mice, we implanted osmotic pumps containing PBS or STI-571 (100 mg/kg/d) 1 d before infection. The percentage of mice surviving after infection is plotted as a function of time after infection (n = 12 mice for each condition). All STI-571–treated mice survive after 15 d compared to 30% of carrier or untreated mice. (c) Summary of effects of tyrosine kinases on vaccinia virus motility and EEV formation. (1) Vaccinia virus is produced in juxtanuclear replication centers, or 'factories,' forming intracellular mature virions (IMV; blue). (2) Some virions are then wrapped in additional membranes within a trans-golgi network (TGN; IEV, red). (3) IEV then travel toward the cell surface on microtubules where they fuse with the plasma membrane (4) and emerge outside the cell (CEV). (5) Some CEV initiate actin polymerization using c-Src, c-Fyn, c-Yes or c-Arg in a redundant fashion, and egress toward an adjacent cell on an actin-filled membranous protrusion or 'tail.' Actin tails are blocked by PD-166326, but not by STI-571. Using c-Abl or c-Arg, CEV associated with the plasma membrane or at the tip of actin tails detach to form EEV, which mediate virulence in vivo (6). Formation of infectious EEV is inhibited by STI-571 and PD-166326.
We next assessed whether STI-571 promotes survival in response to a lethal challenge. Mice were intranasally inoculated with 104 PFU vaccinia virus strain IHD-J, and killed upon loss of 30% of their body weight. With this inoculum, 30–40% of control or carrier-treated mice survived, compared to 100% of STI-571–treated mice (Fig. 6b; P < 10−6, Fisher exact test). These data suggest that STI-571 prevents lethality probably by preventing dissemination of virus without interfering with immune clearance.
Discussion
Previous reports concluded that Src or Src-family kinases mediate release of virions from microtubules19 and actin motility18. But these studies were based on localization of c-Src to the virion, and block of virion detachment from microtubules or actin motility with kinase-dead alleles of Src and with PP1, an inhibitor of Src-family kinases36. Our data showing actin motility in Src−/−Fyn−/−Yes1−/− cells suggest that kinase requirements are more complex. In this regard, PP1 and PP2 have been recently found to additionally inhibit Abl-family kinases37, and kinase-dead Src may have competed with not only c-Src but also other Src- or Abl-family kinases. Results presented here suggest that c-Arg, c-Src, c-Fyn and c-Yes are all likely to mediate release of virions from microtubules and actin motility, though the possibility exists that additional functionally redundant tyrosine kinases, defined by their sensitivity to STI-571 and PD-166326, also suffice. This possibility is supported by our observation that only ∼80% of the tails in Src−/−Fyn−/−Yes1−/− cells contain both c-Abl and c-Arg. Our results additionally indicate a role for Abl and Arg, but not Src-family kinases, in release of infectious EEV. A model summarizing the dependence of actin tails and EEV release on tyrosine kinases, and the effects of the various inhibitors, is presented in Fig. 6c.
Why might vaccinia virus use multiple kinases in infected cells? One possibility is that multiple kinases ensure phosphorylation of targets crucial for pathogenesis and broaden the range of cell types susceptible to infection. Such mechanisms are not unprecedented among viral or even bacterial pathogens. For example, polyoma virus middle T protein dysregulates Src-family kinases to facilitate transformation48. Likewise, enteropathogenic E. coli take advantage of redundancy in both tyrosine kinases and downstream adaptors to form actin-filled membranous pedestals26,49. Notably, enteropathogenic E. coli uses c-Abl, Arg and other tyrosine kinases to phosphorylate the virulence factor Tir, a molecule that has protein sequence homology with vaccinia virus strain A36R (ref. 20). Finally, retroviruses such as Rous sarcoma virus and Ableson leukemia virus have long been recognized to harbor oncogenes that activate mammalian signaling pathways regulating cell division, as a means of indirectly facilitating viral replication50. Given the widespread utilization of cellular signaling molecules by microbial pathogens, the possibility exists that inhibitors developed against host signaling molecules dysregulated in cancer may prove effective as antimicrobial agents against a wide variety of pathogens.
The identification of the Src- and Abl-family kinases as participants in dissemination of vaccinia virus has important implications for treating poxvirus infections. Variola infections or severe adverse reactions associated with vaccination against smallpox are primarily treated with vaccinia immune globulin and the nucleotide analog cidofovir7,51. Vaccinia immune globulin has only limited efficacy in controlling symptoms associated with severe adverse reactions5,7. Cidofovir is effective against poxviruses but is extremely toxic5,7. Moreover, protocols used in the 1970s to control smallpox outbreaks5,7 may not be valid today because vaccination may prove lethal in individuals with acquired or congenital immunocompromising conditions, and rapid identification of such individuals may be impractical6.
STI-571 is a well-established therapy for chronic myelogenous leukemia and stromal tumors, and has relatively few serious side effects29. In the event of a poxvirus outbreak, administration of STI-571 in either a therapeutic or prophylactic capacity may prove to be an effective alternative. The use of STI-571 to treat poxvirus infections still requires that the infection be cleared by an effective immune response, and more experimental work will be required to determine whether the drug can protect immunocompromised individuals from side effects associated with vaccination, and whether STI-571 affects immune responses required for clearance of poxviruses or for the acquisition of memory. In this regard, however, no serious immunosuppressive effects have been reported with long-term (months) treatment of individuals with CML with STI-571 (ref. 29), and our data show that lethally infected mice treated with STI-571 fully recover. Our data showing that STI-571 limits EEV production and reduces spread of vaccinia virus to adjacent organs (e.g., the ovaries) is in accordance with reports suggesting that EEV mediate virulence in vivo. Antibodies against EEV and CEV protect against virus challenge in vivo and in vitro44,45, and viral strains with deletions in A36R form fewer CEV and EEV and show attenuated virulence in mice17. Finally, EEV are less immunogenic than IMV43.
Smallpox infections commence with respiratory inoculation with small numbers of variola particles (∼10; ref. 4), which then disseminate in two stages of viremia. Data presented here suggest that STI-571 will probably prove effective both as a prophylactic and when administered before systemic dissemination. Consistent with this idea, STI-571 prevented dissemination to ovaries after intraperitoneal infection, and promoted survival in a lethal challenge. Drugs such as PD-166326 may prove useful once the virus has disseminated, a prospect we are currently testing. Finally, use of STI-571 in conjunction with cidofovir may permit use of the latter at less toxic doses.
The strategy of using STI-571 to treat a poxvirus infection suggests an important general solution to the capacity of infectious microbes to develop drug resistance. Resistance to drugs directed against microbial targets occurs by mutation of binding sites for the drug52, or by drug inactivation or removal53. But STI-571 disables host proteins essential for the virus life cycle. To develop resistance, a microbe would have to considerably alter its pathogenic strategy without strong growth selection, making the likelihood of engendering resistance much lower than with conventional antibiotics or antimicrobials54. That STI-571 inhibits release of EEV from vaccinia virus strain IHD-J suggests that mutations in viral proteins cannot easily overcome an inhibitor of EEV release directed at a host target.
In summary, we propose the use of STI-571, a drug developed to treat cancer without substantial side effects, as a therapeutic for infections caused by poxviruses. Our approach of interfering with host targets may have general utility in developing new drugs, or new uses for existing drugs, to combat a variety of microbial pathogens that acquire drug resistance.
Methods
Cell culture.
BSC-40 cells from ATCC, 3T3 cells and 3T3 cells derived from Abl1−/−Abl2−/− mice55, or 3T3 cells derived from Src−/−Fyn−/−Yes1−/− mice (ATCC CRL-2459) were maintained in DMEM supplemented with 10% FBS, penicillin and streptomycin as described26. For microscopy, we grew cells on glass coverslips in DMEM containing serum and incubated for 16 h at 37 °C with 104 PFU vaccinia virus strain WR or vaccinia virus GFP-B5R31. For some experiments, we transfected cells 1–2 d before infection with plasmid vectors using Fugene-6 (Roche). Abl-T315I and Arg-T314I were constructed using Quik-Change site-directed mutagenesis technology (Stratagene) as described26. PD compounds (e.g., PD-166326 and SKI-DV1-10; Supplementary Fig. 3 online) were synthesized as described33,56, and were indistinguishable in their effects in all assays. PD-166326 and the mesylate salt of STI-571 (Supplementary Fig. 4 online) was synthesized as described33,56. PD compounds and PP2 (Calbiochem) were dissolved in 100% DMSO. STI-571 was dissolved in water. For most experiments, PD-166326, PP2 or DMSO was added to cells immediately before infection. For post-infection treatment experiments, we added compound or DMSO to cells 8–14 h after addition of vaccinia virus, and we fixed the cells 10 min to 2 h subsequently. Although the Ki of PD-166326 for Abl is about 5–10 nM as measured by in vitro kinase assay, micromolar concentrations of PD-166326 are required to block actin tails or plaques in cells. We surmise that this discrepancy arose because the drug, when applied to the medium, may not achieve the same concentration in the cell. In this regard, we have now synthesized more soluble derivatives of PD-166326, which have the same Ki for wild-type Abl, but which block actin tails at slightly lower concentrations (i.e., 1 μM; data not shown). Additionally, the ATP concentration within the cell may increase the effective Kiin vivo. We carried out in vitro kinase assays at nanomolar to micromolar concentrations of ATP, whereas the free ATP concentration inside the cell has been estimated to be around 2–5 mM. Because ATP and PD-166326 compete for the same binding site, it is not at all unreasonable to expect that the effective Ki increases. In vitro kinase assays with high concentrations of ATP seem consistent with this prediction. Moreover, in systems where Src family kinases have been suggested to be involved, PP1 concentrations in the micromolar range are routinely used despite a measured Kiin vitro of 5–50 nM depending on the kinase.
Immunofluorescence staining.
For immunofluorescence analysis, we fixed cells in 2% formaldehyde and permeablized them in Triton-X-100 as described25,57. Vaccinia virus was recognized by staining with DAPI (1 μg/ml; Sigma), and actin by staining with 488-phalloidin, 566-phalloidin or 594-phalloidin (1 μg/ml; Molecular Probes). The primary antibodies and concentrations used in this study were as follows: α-Wiskott-Aldrich Syndrome protein (α-WASP) polyclonal antibody (affinity purified, 1:200 dilution), α-hemagglutinin (α-HA) monoclonal antibody (3F10; 0.01 μg/ml, Roche), α-Nck monoclonal antibody (1 μg/ml; Oncogene Research), α-Abl monoclonal antibody (AB3; 0.5 μg/ml for overexpressed Abl proteins; 50 μg/ml for endogenous Abl proteins; 8E9; 0.05 μg/ml; Pharmingen), α-Src polyclonal antibody (0.1 μg/ml; Santa Cruz), α-Fyn monoclonal antibody (0.1 μg/ml; ABCAM), α-Yes polyclonal antibody (0.1 μg/ml; Cell Signaling), α-Arg polyclonal antibody (1:200; UBI), α-pY412 (0.1 μg/ml; Cell Signaling) and α-TW2.3 monoclonal antibody (ascites, 1:2,000 for microscopy30). We determined the specificity of kinase-specific antibodies using the staining of cell lines lacking particular kinases26. Cells expressing exogenous wild-type c-Abl were distinguished by relatively high fluorescence intensity with lower α-Abl monoclonal antibody concentrations. Thus images were acquired with much shorter exposures than those used to detect endogenous c-Abl–like protein. We obtained secondary antibodies from Jackson Immunochemicals.
Microscopy.
Images were acquired with a scientific-grade cooled charge-coupled device (Cool-Snap HQ with ORCA-ER chip) on a multi-wavelength wide-field three-dimensional microscopy system (Intelligent Imaging Innovations) based on a Zeiss 200M inverted microscope using a 63× N.A.1.4 lens (Zeiss). We imaged immunofluorescent samples at room temperature (22–25 °C) using a standard Sedat filter set (Chroma) in successive 0.20 μm focal planes through the samples, and out-of-focus light was removed with a constrained iterative deconvolution algorithm58. Actin tails were recognized by intense phalloidin staining associated with DAPI or GFP fluorescence objects measuring ∼200 nm in diameter. Fluorescence at the end of actin tails which colocalized with DAPI staining or with GFP fluorescence in cells infected with GFP-labeled vaccinia virus was used to indicate localization of kinases or other cellular molecules to virions. Colocalization was assessed by coincidence of fluorescent staining of kinases in the Cy5 and Cy3 channels. We calibrated the microscope filters with multicolored fluorescent beads to insure coincidence of fluorescent signals in all channels to within a pixel (100 nm for the 63× N.A. 1.4 lens). For quantitative measurements of kinase localization, approximately 500 tails in at least seven cells were assessed for each kinase.
Plaque assays.
For plaque assays, we seeded cells in 6-well dishes, grew them to confluence, and incubated them with serial dilutions of vaccinia virus strain WR or IHD-J. After 1 h, we washed the cells to remove excess virus and replaced DMEM; the cells were incubated for an additional 3–4 d. We then fixed cells and stained them with 15% ethanol and 1% crystal violet to visualize plaques. For measurements of secreted EEV, cells were incubated in 2% FBS DMEM, media was removed 24 h after infection, centrifuged for 15 min at 400g, dilutions made and added to uninfected BSC-40 cell monolayers, and the number of plaques assessed 2–4 d subsequently. For some experiments, the supernatant collected from infected cells was incubated with α-IMV monoclonal antibody (2D5; at various dilutions of ascites ranging from 1:100 to 1:1,000; Supplementary Fig. 6 online)40, which has been shown to inactivate IMV but not EEV. Plaque assays were used to determine if different cell lines were infected with equal efficacy. We used single-step growth curves to determine yield of virus in different cell lines. To do this, cell monolayers were infected at an MOI of 0.1 or 5. We harvested monolayers 1, 2 or 3 d after infection, froze them and thawed them three times to release viral particles in the cell, and centrifuged them to remove cellular debris. We then serially diluted the supernatant and added it to monolayers of uninfected BSC-40 cells, and assessed the number of plaques after 2 d.
Mouse assays
For mouse experiments, we dissolved the mesylate salt of STI-571 in saline and loaded it into Alzet pumps (1007D or 2002) capable of dispensing a continuous flow of drug (100 mg/kg/d). We inserted the pumps subcutaneously into anesthetized 6-week-old female C57/Bl6 mice (Jackson Laboratories). We did not determine the level of STI-571 in the blood. At these drug concentrations, we observed no weight loss or other adverse effects in uninfected animals, and such concentrations have been used to treat cancer in mice over prolonged periods (up to 3 months)46. Thus the drug seemed nontoxic. We infected some of the mice by intraperitoneal inoculation with 104 PFU vaccinia virus 24 h after insertion of the pump59. After 4 d, the mice were killed and their ovaries extracted for analysis of viral genome copies by real-time PCR. For survival experiments, we inoculated mice intranasally with 2 × 104 PFU 1 d after implantation of pumps containing drug or carrier. Weight was monitored daily and mice were killed if their weight declined by more than 30%. All experiments with mice were carried out in accordance with Institutional Animal Care and Use Committee regulations (Emory University protocol number AD07-1156-03R04).
Viral copy number measurements.
The probe and primers for real-time PCR were designed by the use of Primer Express software (Applied Biosystems) within the conserved region of the vaccinia virus UDG gene. The TaqMan Probe 5′-CGAGACGAGACGTCGCCTATTCCTG-3′ (Applied Biosystems) was labeled at 5′ end with the reporter dye FAM (6-carboxyfluorescein) and at the 3′ end with the quencher dye TAMRA (6-carboxytetramethyl-rhodamine) with a melting temperature (Tm) of 68 °C. The primer sequences were as follows:
5′-GGTAGAGTTTTATAACGAAGTAGCCAGTT-3′ (sense; length, 29 bases, Tm = 58 °C) and 5′-CTCGTTTATTTCTAAGCGGTTGTTT-3′ (antisense; length, 25 bases, Tm = 58 °C). We performed real-time PCR using the ABI Prism 7900HT sequence detection system (Applied Biosystems) with TaqMan Gold kit under the following conditions: the 50 μl reactions contained 5 μl of 10× TaqMan buffer A; 4 mM MgCl2; 200 μM dNTPs; 200 nM of each primer, 125 nM fluorogenic probe, and 1.25 units of AmpliTaq Gold DNA polymerase. Universal thermal cycling conditions consisted of 10 min at 95 °C, followed by 45 cycles of 15 s at 95 °C and 1 min at 60 °C. We analyzed 250 ng DNA isolated from ovaries using DNAeasy Kit (Qiagen) as a template for amplification and expressed results as copies of vaccinia virus per 250 ng of DNA. We carried out each reaction in duplicate. For statistical analysis, mice with a viral copy number of greater than 104 were scored as clinically ill. By this criterion, all of the mice in the carrier group, and one of the twelve mice in the group treated with STI-571 were ranked clinically ill. This difference was judged statistically significant by a two-sided Fisher exact test (P < 10−6).
Note: Supplementary information is available on the Nature Medicine website.
References
Esposito, J. & Fenner, F. in Fields Virology 4th edn. Vol. 2 (eds. Knipe, D.M. & Howley, P.M.) Ch. 85, 2885–2921 (Lippincott, Williams and Wilkins, Philadelphia, 2001.)
Moss, B. in Fields Virology 4th edn. Vol. 2 (eds. Knipe, D.M. and Howley, P.M.) Ch. 84, 2849–2883 (Lippincott, Williams and Wilkins, Philadelphia, 2001).
Crotty, S. et al. Cutting edge: long-term B cell memory in humans after smallpox vaccination. J. Immunol. 171, 4969–4973 (2003).
National Research Council, Institute of Medicine. Assessment of Future Scientific Needs for Live Variola Virus 126 (National Academy Press, Washington, DC, 1999).
Harrison, S.C. et al. Discovery of antivirals against smallpox. Proc. Natl. Acad. Sci. USA 101, 11178–11192 (2004).
Amorosa, V.K. & Isaacs, S.N. Separate worlds set to collide: smallpox, vaccinia virus vaccination, and human immunodeficiency virus and acquired immunodeficiency syndrome. Clin. Infect. Dis. 37, 426–432 (2003).
Rotz, L.D., Dotson, D.A., Damon, I.K., Becher, J.A. Advisory Committee on Immunization Practices. Vaccinia (smallpox) vaccine recommendations of the Advisory Committee on Immunization Practices (ACIP), 2001. MMWR Recomm. Rep. 50, 1–25 (2001).
Cono, J., Casey, C.G., Bell, D.M. Centers for Disease Control and Prevention. Smallpox vaccination and adverse reactions guidance for clinicians. MMWR Recomm. Rep. 52, 1–28 (2003).
Smith, J.N. & Ahmer, B.M. Detection of other microbial species by Salmonella: expression of the SdiA regulon. J. Bacteriol. 185, 1357–1366 (2003).
Smith, G.L., Murphy, B.J. & Law, M. Vaccinia virus motility. Annu. Rev. Microbiol. 57, 323–342 (2003).
Carter, G.C. et al. Vaccinia virus cores are transported on microtubules. J. Gen. Virol. 84, 2443–2458 (2003).
Hollinshead, M. et al. Vaccinia virus utilizes microtubules for movement to the cell surface. J. Cell Biol. 154, 389–402 (2001).
Rietdorf, J. et al. Kinesin-dependent movement on microtubules precedes actin-based motility of vaccinia virus. Nat. Cell Biol. 3, 992–1000 (2001).
Ward, B.M. & Moss, B. Vaccinia virus intracellular movement is associated with microtubules and independent of actin tails. J. Virol. 75, 11651–11663 (2001).
Smith, G.L., Vanderplasschen, A. & Law, M. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83, 2915–2931 (2002).
Wolffe, E.J., Weisberg, A.S. & Moss, B. Role for the vaccinia virus A36R outer envelope protein in the formation of virus-tipped actin-containing microvilli and cell-to-cell virus spread. Virology 244, 20–26 (1998).
Parkinson, J.E. & Smith, G.L. Vaccinia virus gene A36R encodes a M(r) 43-50 K protein on the surface of extracellular enveloped virus. Virology 204, 376–390 (1994).
Frischknecht, F. et al. Actin-based motility of vaccinia virus mimics receptor tyrosine kinase signalling. Nature 401, 926–929 (1999).
Newsome, T.P., Scaplehorn, N. & Way, M. SRC mediates a switch from microtubule- to actin-based motility of vaccinia virus. Science 306, 124–129 (2004).
Frischknecht, F. & Way, M. Surfing pathogens and the lessons learned for actin polymerization. Trends Cell Biol. 11, 30–38 (2001).
Moreau, V. et al. A complex of N-WASP and WIP integrates signalling cascades that lead to actin polymerization. Nat. Cell Biol. 2, 441–448 (2000).
Scaplehorn, N. et al. Grb2 and Nck act cooperatively to promote actin-based motility of vaccinia virus. Curr. Biol. 12, 740–745 (2002).
Nataro, J.P. & Kaper, J.B. Diarrheagenic Escherichia coli . Clin. Microbiol. Rev. 11, 142–201 (1998).
Kenny, B. et al. Enteropathogenic E. coli (EPEC) transfers its receptor for intimate adherence into mammalian cells. Cell 91, 511–520 (1999).
Kalman, D. et al. Enteropathogenic E. coli acts through WASP and Arp2/3 complex to form actin pedestals. Nat. Cell Biol. 1, 389–391 (1999).
Swimm, A. et al. Enteropathogenic Escherichia coli use redundant tyrosine kinases to form actin pedestals. Mol. Biol. Cell 15, 3520–3529 (2004).
Schindler, T. et al. Structural mechanism for STI-571 inhibition of abelson tyrosine kinase. Science 289, 1938–1942 (2000).
Druker, B.J. et al. Chronic myelogenous leukemia. Hematology (Am Soc Hematol Educ Program) 87–112 (2001).
Goldman, J.M. & Druker, B.J. Chronic myeloid leukemia: current treatment options. Blood 98 2039–2042 (2001).
Yuwen, H. et al. Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology 195, 732–744 (1993).
Ward, B.M. & Moss, B. Visualization of intracellular movement of vaccinia virus virions containing a green fluorescent protein-B5R membrane protein chimera. J. Virol. 75, 4802–4813 (2001).
Pluk, H., Dorey, K. & Superti-Furga, G. Autoinhibition of c-Abl. Cell 108, 247–259 (2002).
Kraker, A.J. et al. Biochemical and cellular effects of c-Src kinase-selective pyrido[2, 3- d]pyrimidine tyrosine kinase inhibitors. Biochem. Pharmacol. 60, 885–898 (2000).
Dorsey, J.F. et al. The pyrido[2,3-d]pyrimidine derivative PD180970 inhibits p210Bcr-Abl tyrosine kinase and induces apoptosis of K562 leukemic cells. Cancer Res. 60, 3127–3131 (2000).
Wisniewski, D. et al. Characterization of potent inhibitors of the Bcr-Abl and the c-kit receptor tyrosine kinases. Cancer Res. 62, 4244–4255 (2002).
Liu, Y. et al. Structural basis for selective inhibition of Src family kinases by PP1. Chem. Biol. 6, 671–678 (1999).
Tatton, L. et al. The Src-selective kinase inhibitor PP1 also inhibits Kit and Bcr-Abl tyrosine kinases. J. Biol. Chem. 278, 4847–4853 (2003).
Ward, B.M., Weisberg, A.S. & Moss, B. Mapping and functional analysis of interaction sites within the cytoplasmic domains of the vaccinia virus A33R and A36R envelope proteins. J. Virol. 77, 4113–4126 (2003).
Law, M. & Smith, G.L. Antibody neutralization of the extracellular enveloped form of vaccinia virus. Virology 280, 132–142 (2001).
Law, M., Hollinshead, R. & Smith, G.L. Antibody-sensitive and antibody-resistant cell-to-cell spread by vaccinia virus: role of the A33R protein in antibody-resistant spread. J. Gen. Virol. 83, 209–222 (2002).
Blasco, R., Sisler, J.R. & Moss, B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J. Virol. 67, 3319–3325 (1993).
Ichihashi, Y. & Oie, M. Neutralizing epitope on penetration protein of vaccinia virus. Virology 220, 491–494 (1996).
Vanderplasschen, A. et al. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. USA 95, 7544–7549 (1998).
Boulter, E.A. & Appleyard, G. Differences between extracellular and intracellular forms of poxvirus and their implications. Prog. Med. Virol. 16, 86–108 (1973).
Appleyard, G., Hapel, A.J. & Boulter, E.A. An antigenic difference between intracellular and extracellular rabbitpox virus. J. Gen. Virol. 13, 9–17 (1971).
Wolff, N.C. & Ilaria, R.L. Jr. Establishment of a murine model for therapy-treated chronic myelogenous leukemia using the tyrosine kinase inhibitor STI571. Blood 98, 2808–2816 (2001).
Chahroudi, A., Chavan, R., Kozyr, N., Silvestri, G. & Feinberg, M.B. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J. Virol. in the press (2005).
Ichaso, N. & Dilworth, S.M. Cell transformation by the middle T-antigen of polyoma virus. Oncogene 20, 7908–7916 (2001).
Gruenheid, S. et al. Enteropathogenic E. coli Tir binds Nck to initiate actin pedestal formation in host cells. Nat. Cell Biol. 3, 856–859 (2001).
Bishop, J.M. Molecular themes in oncogenesis. Cell 64, 235–248 (1991).
Baker, R.O., Bray, M. & Huggins, J.W. Potential antiviral therapeutics for smallpox, monkeypox and other orthopoxvirus infections. Antiviral Res. 57, 13–23 (2003).
Idemyor, V. Bacterial resistance to antimicrobial agents--the time for concern. Ann. Pharmacother. 27, 1285 (1993).
Li, X.Z. & Nikaido, H. Efflux-mediated drug resistance in bacteria. Drugs 64, 159–204 (2004).
Gould, I.M. Antibiotic policies and control of resistance. Curr. Opin. Infect. Dis. 15, 395–400 (2002).
Koleske, A.J. et al. Essential roles for the Abl and Arg tyrosine kinases in neurulation. Neuron 21, 1259–1272 (1998).
Nagar, B. et al. Crystal structures of the kinase domain of c-Abl in complex with the small molecule inhibitors PD173955 and imatinib (STI-571). Cancer Res. 62, 4236–4243 (2002).
Kalman, D. et al. Ras family GTPases control growth of astrocyte processes. Mol. Biol. Cell 10, 1665–1683 (1999).
Swedlow, J.R., Sedat, J.W. & Agard, D.A. Deconvolution in Optical Microscopy, in Deconvolution of Images and Spectra (ed. Jansson, P.A.) 284–307 (Academic Press, San Diego, 1997).
Ramirez, J.C. et al. Tissue distribution of the Ankara strain of vaccinia virus (MVA) after mucosal or systemic administration. Arch. Virol. 148, 827–839 (2003).
Acknowledgements
The authors thank D. Kalman, D. Steinhauer, O. Weiner, J. Taunton and K. Saxe for discussions; A. Family, S. Staprans, N. Kozyr and R. Griffith for assistance and advice; B. Meyer and J. Wang for Abl cDNAs; T. Koleske for Abl1−/−Abl2−/− cells, Abl1 and Abl2 cells, and c-Arg-YFP cDNA; B. Moss and J. Yudell for α-TW2.3 mAb; R. Blasco for GFP-VV; C. Lowell for Src−/− cells, Src−/−Fyn−/− cells, and Src−/−Yes1−/− cells; G. Smith for α-IMV mAb; and L. Burleigh, D. Steinhauer, and C. Moran for commenting on the manuscript. The work was supported by US National Institutes of Health grant PO1 AI 46007 (to M.B.F.), and by grants from the University Research Council, Emory University, the Southeastern Regional Center for Excellence in Bioterrorism (SERCEB) Pilot Project Feasibility Award, grant R01-AI056067-01 from the N.I.A.I.D., and an award from the Emtech Biotechnology Foundation (all to D.K.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Fig. 1
Protein sequences surrounding phosphorylation sites in VV A36R and enteropathogenic E. coli Tir. (PDF 53 kb)
Supplementary Fig. 2
Identification of VV-infected cells. (PDF 352 kb)
Supplementary Fig. 3
Effects of PD-166326 and STI-571. (PDF 339 kb)
Supplementary Fig. 4
Characterization of kinases sufficient for VV actin motility. (PDF 166 kb)
Supplementary Fig. 5
Abl-family kinases do not affect viral entry or replication. (PDF 356 kb)
Supplementary Fig. 6
Distinguishing the effects of Abl and Arg on EEV and IMV. (PDF 62 kb)
Rights and permissions
About this article
Cite this article
Reeves, P., Bommarius, B., Lebeis, S. et al. Disabling poxvirus pathogenesis by inhibition of Abl-family tyrosine kinases. Nat Med 11, 731–739 (2005). https://doi.org/10.1038/nm1265
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/nm1265
This article is cited by
-
Mpox (formerly monkeypox): pathogenesis, prevention, and treatment
Signal Transduction and Targeted Therapy (2023)
-
Antivirals in medical biodefense
Virus Genes (2020)
-
Vaccinia virus-mediated cancer immunotherapy: cancer vaccines and oncolytics
Journal for ImmunoTherapy of Cancer (2019)
-
Preclinical pharmacokinetic evaluation to facilitate repurposing of tyrosine kinase inhibitors nilotinib and imatinib as antiviral agents
BMC Pharmacology and Toxicology (2018)
-
NPF motifs in the vaccinia virus protein A36 recruit intersectin-1 to promote Cdc42:N-WASP-mediated viral release from infected cells
Nature Microbiology (2016)
