Oncogenic Signaling Induced by HCV Infection
by
 , ,
, ,
Alessia Virzì
1,2 ,
,
Armando Andres Roca Suarez
1,2,
Thomas F. Baumert
1,2,3 and
Joachim Lupberger
1,2,* 1
Inserm, U1110, Institut de Recherche sur les Maladies Virales et Hépatiques, 67000 Strasbourg, France
2
Université de Strasbourg, 67000 Strasbourg, France
3
Pôle Hépato-digestif, Institut Hospitalo-universitaire, Hôpitaux Universitaires de Strasbourg, 67000 Strasbourg, France
*
Author to whom correspondence should be addressed.
Viruses 2018, 10(10), 538; https://doi.org/10.3390/v10100538
Submission received: 1 September 2018
/
Revised: 29 September 2018
/
Accepted: 30 September 2018
/
Published: 2 October 2018
(This article belongs to the Special Issue Cure of Hepatitis C Virus Infection and Hepatocellular Carcinoma)
{kind=link}
Abstract
:The liver is frequently exposed to toxins, metabolites, and oxidative stress, which can challenge organ function and genomic stability. Liver regeneration is therefore a highly regulated process involving several sequential signaling events. It is thus not surprising that individual oncogenic mutations in hepatocytes do not necessarily lead to cancer and that the genetic profiles of hepatocellular carcinomas (HCCs) are highly heterogeneous. Long-term infection with hepatitis C virus (HCV) creates an oncogenic environment by a combination of viral protein expression, persistent liver inflammation, oxidative stress, and chronically deregulated signaling events that cumulate as a tipping point for genetic stability. Although novel direct-acting antivirals (DAA)-based treatments efficiently eradicate HCV, the associated HCC risk cannot be fully eliminated by viral cure in patients with advanced liver disease. This suggests that HCV may persistently deregulate signaling pathways beyond viral cure and thereby continue to perturb cancer-relevant gene function. In this review, we summarize the current knowledge about oncogenic signaling pathways derailed by chronic HCV infection. This will not only help to understand the mechanisms of hepatocarcinogenesis but will also highlight potential chemopreventive strategies to help patients with a high-risk profile of developing HCC.
1. Introduction
Tumor-inducing viruses represent a considerable field of study for the comprehension of molecular carcinogenesis. Several oncogenes were first discovered in association with retroviruses and then associated with most forms of cancer [1,2]. The study of virus-coded oncogenes also led to the discovery of canonical signaling pathways and the understanding of elementary cellular processes. Several viruses are considered as oncogenic viruses as they are associated with human cancer, e.g., human papilloma virus (HPV), Epstein–Barr virus (EBV), human herpes virus 8 (HHV8), Merkel cell polyomavirus (MCPyV), human T-lymphotropic virus (HTLV-1), hepatitis B virus (HBV), and hepatitis C virus (HCV) [3].
Infection with oncogenic viruses generally leads to the disruption of genetic and epigenetic homeostasis and DNA repair mechanisms. In addition, some viruses stimulate the proliferation of cancer stem cells (CSCs), which are involved in cancer initiation, progression, and chemotherapy resistance [3]. Oncogenic viruses have a direct and indirect impact on carcinogenesis [4]. At least four HCV proteins (core, NS3, NS5A, and NS5B) seem to deregulate potentially oncogenic signaling pathways [5]. At the same time, it is beyond question that HCV creates a procarcinogenic environment in the liver by inducing a chronic inflammatory state [6]. In addition, liver disease progression can be favored by several cofactors, including alcohol consumption and coinfection with other viruses such as HBV and human immunodeficiency virus (HIV) [7]. Moreover, HCV infection is implicated in extrahepatic cancers, including B-cell non-Hodgkin lymphomas (NHL) [8] and cancers of the oral cavity, oropharynx, intrahepatic bile duct, pancreas, and kidney [9,10,11,12,13,14,15]. Although the molecular links between HCV and extrahepatic cancers are not well understood, it has been suggested that some of the possible mechanisms behind this association could be related to a chronic immune stimulation in the presence of HCV or to the infection of extrahepatic cell types [16].
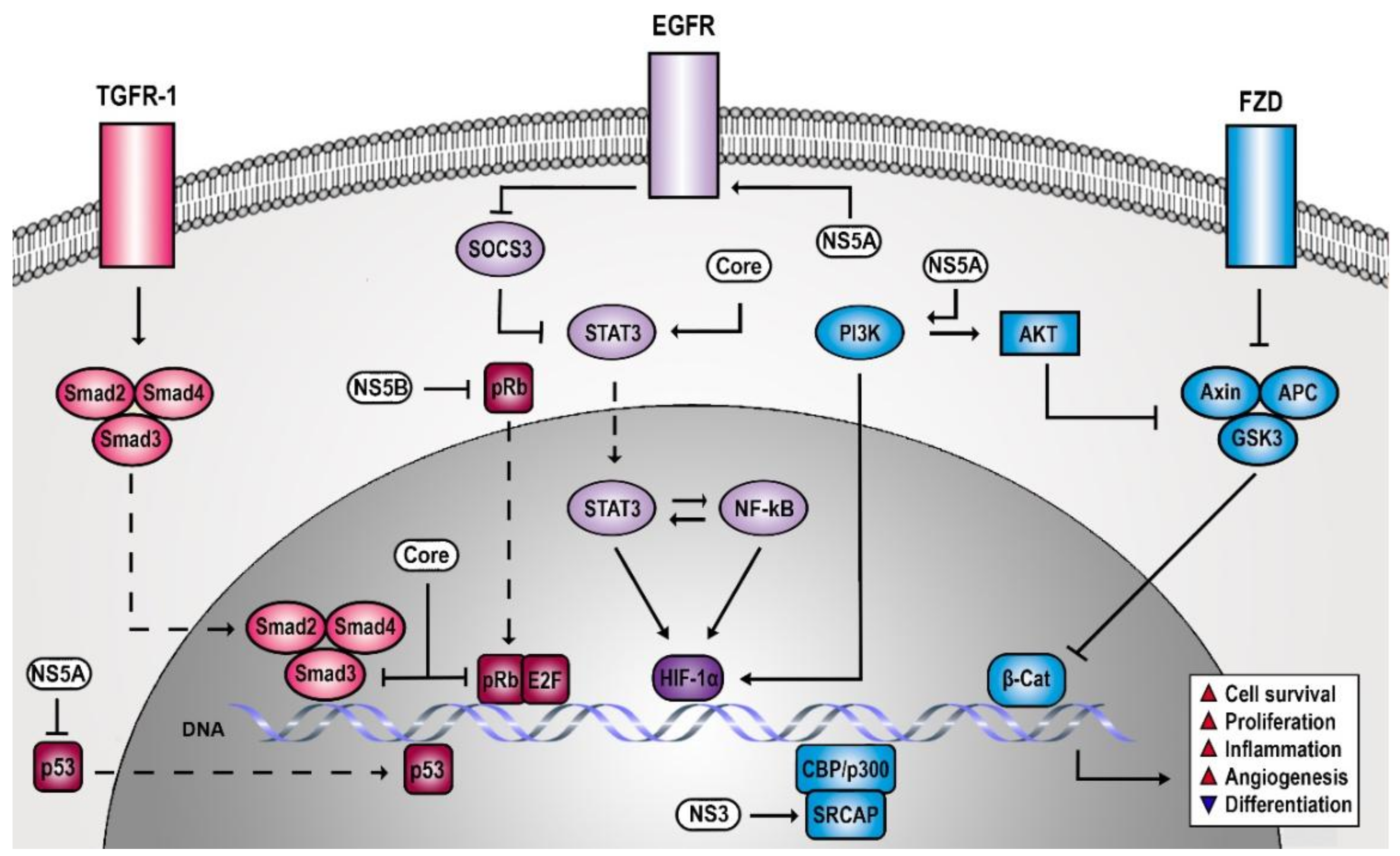
The study of the HCV life cycle revealed several host dependencies of the virus that involve signaling molecules [17,18,19,20,21]. However, it soon became evident that HCV not only requires signaling processes but also actively manipulates host signal transduction with considerable impact on liver pathogenesis. Numerous studies have described signaling cascades that are altered by chronic HCV infection and are potentially involved in carcinogenesis (Figure 1). In the present review, we classify these pathways in three cancer-relevant categories according to their role in cell proliferation/survival, differentiation/adhesion/angiogenesis, inflammatory response, and dissect potential clinical strategies for hepatocellular carcinomas (HCC) chemoprevention and therapy.
2. HCV Creates a Persistent Proliferative and Anti-Apoptotic Signaling Environment
Proliferative signaling pathways of mammalian cells are modulated by extracellular factors that engage precise programs of gene transcription and protein regulation [22,23]. Contact inhibition, controlled availability of growth factors, and other physiological feedback systems ensure a tight regulation of the proliferative signaling pathways. Excessive cell proliferation is the key feature of most types of cancers [24]. In general, growth factor and cytokine signaling pathways essentially induce all the primary steps of tumor progression, which include clonal expansion, invasion, angiogenesis, and metastatic formation [25]. Tumor suppressors, such as the cellular tumor antigen p53 and the retinoblastoma-associated protein (pRb), regulate cell proliferation, and their perturbation promotes a persistent activation of the cell cycle machinery [24]. Although HCC proliferative index is generally low, which is one of the reasons why most cytostatics are considered inefficient, there is a clear correlation of HCC risk and proliferative signals in a pretumor state [26].
2.1. HCV-Induced Receptor Tyrosine Kinase Signaling Contributes to Liver Cancer Risk
Growth factors like epidermal growth factor (EGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), and insulin growth factor (IGF) trigger downstream signal transduction by binding to their specific receptor tyrosine kinase receptors [27]. The cascade of events that follow epidermal growth factor receptor (EGFR) is one of the most widely studied signal transduction pathways [28,29,30]. ErbB-1 and three additional homologous members of the EGFR family (ErbB-2, ErbB-3, ErbB-4), regulate cell proliferation, differentiation, and migration under normal physiological conditions [29]. EGFR itself is critical in epithelial development, and other members of the family have a crucial role in cardiac, mammary glands, and nervous system development and disorders [28,31,32,33]. The EGFR signaling pathway plays a central role also in embryonic development and in the regeneration of stem cells in skin, liver, and gut [34,35]. Moreover, the EGFR signaling pathway is in the spotlight as a driver of cancer risk and progression [26,36,37].
Viruses have developed sophisticated strategies to manipulate EGFR functions (i.e., perturbing EGFR expression, activity, or recycling) [38]. EGFR is a host factor for HCV entry into hepatocytes by regulating the assembly of the coreceptor complex [17,21], viral internalization [39], and membrane fusion [17]. Furthermore, EGFR signaling pathway tempers the antiviral activity of interferon-alpha (IFN-α) by maintaining phosphorylation of signal transducer and activator of transcription 3 (STAT3) through the suppression of a negative feedback regulator (i.e., suppressor of cytokine signaling 3, SOCS3) [40]. It is evident that HCV has a vital interest in maintaining EGFR signaling. Indeed, HCV not only requires EGFR signaling but also actively induces the activation of this pathway during HCV binding and infection [41,42] and prolongs EGFR signaling by perturbing EGFR degradation via NS5A, as reported upon its ectopic expression [43]. This leads to an increased HCC risk in infected patients as persistent EGF signaling is a key driver of liver disease [26].
2.2. HCV Increases Cell Survival by Cytoplasmic Retention of p53 and pRb
Proliferative signals seem beneficial for HCV to avoid stress-induced growth arrest and apoptosis, both of which would oppose viral replication and survival [44,45]. The tumor suppressors pRb and p53 regulate cell growth control via their action on cell cycle checkpoints and apoptosis programs [22]. Therefore, pRb, p107, and p130 proteins cooperate with various proteins, including transcription factors of the E2F family required for cellular DNA replication [46,47,48]. The downstream interaction between pRb and E2F causes the inhibition of gene expression by the recruitment of histone deacetylases (HDACs) [49] and other chromatin remodeling factors [50,51,52]. pRb constitutively inhibits the transcriptional activity of E2Fs, whereas it is deactivated after phosphorylation by cyclin-dependent kinases (CDKs). G1 phase CDKs phosphorylate pRb family proteins, which leads to the activation of genes required for S phase entry (i.e., cyclin E) [22,24]. In contrast, p53 maintains genetic integrity of cells by blocking cell proliferation in response to stress and DNA damage by activating cyclin-dependent kinase inhibitors (CKIs) [24]. Therefore, p53 accumulates in the nucleus, where it acts as a transcription factor for cyclin-dependent kinase inhibitor 1 (CDKN1A) that codes for p21 [53,54]. Thus, it is not surprising that deregulation of p53 function or signaling is associated with many cancers [24]. For example, pRb is a target of viral oncoproteins encoded by adenovirus [55] and HPV [56]. In addition, HCV has developed strategies to suppress pRb [57,58,59]. During HCV infection, NS5B protein retains pRb in the cytoplasm of the hepatocyte, leading to its proteasomal degradation via E6-associated protein (E6AP) recruitment and polyubiquitination [57,59]. The isolated expression of HCV core protein impairs pRb expression in immortalized rat embryo fibroblasts and thereby promotes a E2F-1 activity with impact on cell proliferation and apoptosis [60]. The frequency and the geographic distribution of TP53 (p53) mutations presumably depend on the variability of aetiological and host susceptibility factors [61,62]. HCV and other viruses have sophisticated strategies to modulate or inhibit p53 signaling [63]. HCV core proteins, NS5A, and NS3 associate with p53 and repress its function without initiating its degradation. HCV core protein, however, seems to act as both activator and a repressor of p53 pathway [64,65,66]. This dual role of core protein may reflect a dose-dependent impact on p53 signaling, depending on the infection model used [67]. In vitro data suggest that the effect of NS3 protein on p53 depends on the HCV genotype [68,69]. Like pRb, virus-induced perturbation of p53 function involves a forced retention in the cytoplasm, which prevents DNA binding of p53. HCV NS5A colocalizes with p53 in the cytoplasmic perinuclear region and sufficiently reduces nuclear p53 concentration to suppress apoptosis. In addition, NS5A expression enforces p53 inhibition via binding to hTAFII32, which is an essential p53 coactivator [70]. In a more indirect manner, HCV proteins perturb the function of essential cofactors of p53 transcriptional activity. Core interacts with DEAD-Box Helicase 3 X-Linked (DDX3X), as observed in an isolated core-expression context [71,72,73]. DDX3X is a target of p53 [74] and modulates CDKN1A promoter activity. Furthermore, NS5B binds and relocalizes p53 coactivator DEAD-Box Helicase 5 (DDX5) to the cytoplasm [75,76,77]. However, the findings on p53 signaling during HCV infection have to be interpreted with caution as many of the immortalized cell lines used to study HCV present defects in p53 signaling [6]. For example, Huh7-derived cell lines, which are commonly used due to their high permissiveness towards HCV, accumulate a functionally damaged p53 mutant in the nucleus [78].
2.3. HCV Impairs TGF-β Signaling Promoting Epithelial Mesenchymal Transition (EMT)
Cytokines of the transforming growth factor β (TGF-β) superfamily are dimers with conserved structures and exert pleiotropic effects [79]. In physiological conditions, TGF-β acts as a potent growth inhibitor for several types of cells [80,81,82,83,84] and promotes apoptosis in epithelial cells [85]. Consequently, impaired TGF-β may result in cellular hyperproliferation and cancer [86]. In addition, these cytokines stimulate the expression of extracellular matrix components, which promote in vivo fibrosis in different tissues [85,87]. In the liver, TGF-β seems to contribute to all stages of disease development, from early injury through inflammation, fibrosis towards cirrhosis and HCC [88,89]. TGF-β presumably acts as tumor suppressor during the early stage of cancer development but promotes tumor progression, migration, and invasion in advanced HCCs once the tumor cells have acquired resistance to its suppressive proprieties [89,90,91]. Members of the TGF-β superfamily interact with two different receptor types, called type I and type II receptors, which are both required for cellular signaling [85,92,93]. TGF-β binds directly to receptor II, which is constitutively active. This event induces the recruitment of receptor I into the complex that subsequently becomes phosphorylated by receptor II and activate downstream signals [92], which includes SMAD proteins [94,95]. Particularly, the activated type I receptor phosphorylates the intracellular substrate R-SMAD (Smad 2/3 or Smad 1/5/8) that crosses the nuclear membrane after binding co-SMAD (Smad4) [85,89]. Smad4 is a critical effector of intracellular signaling and, like TGF-β, has a dual role as tumor suppressor and promoter of HCC [96]. Once in the nucleus, the SMAD complex regulates the transcription of TGF-β-induced target genes together with essential transcriptional cofactors. The SMAD complex induces a specific gene signature by the canonical TGF-β signaling pathway [97], which provokes growth arrest and proapoptotic signals in an early stage. Later, proliferative and antiapoptotic responses gain the upper hand by crosstalk with growth signaling. This noncanonical TGF-β pathway includes modulation of EGFR, mitogen-activated protein kinase (MAPK), phosphoinositide 3 kinase (PI3K)/Akt, Ras, and Rho-like small GTPases signaling pathways [98,99]. TGF-β can induce epithelial to mesenchymal transition (EMT) in human primary hepatocytes, a program that promotes cell invasion and metastasis [100]. During EMT, the epithelial cells lose their phenotypic features and gain invasive properties to become mesenchymal cells. Physiologically, EMT is indispensable in the context of embryonic development. However, there is increasing evidence that it also plays a role in pathological conditions, probably contributing to metastatic carcinoma development as well [101].
HCV has developed strategies targeting TGF-β signaling, presumably to maintain a proliferative antiapoptotic signaling environment that stimulate the HCV life cycle and prevent stress-induced cell death. HCV infection induces unfolded protein response (UPR), which upregulates TGF-β expression via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) [102,103]. Mainly, the HCV core protein seems to modulate TGF-β signaling (i.e., via an interaction with Smad3) [104,105]. However, HCV does not affect the nuclear translocation of the Smad3/4 complex, suggesting a transient nuclear localization of HCV core protein [105]. An interesting hypothesis suggests that chronic infection provokes the selection of protumorigenic HCV variants in the liver, which strongly interfere with TGF-β signaling. This is supported by the isolation of HCV core variants from HCCs that better resist TGF-β-mediated antiproliferative effects and more intensely promote cell transformation compared to HCV core variants isolated from tissue adjacent to the tumor [105]. Beside its association with SMAD, HCV core expression induces endoglin (CD105) expression on the surface of infected hepatocytes. As a component of the TGF-β receptor complex, endoglin abundance stimulates fibrogenesis and promotes tumor growth and metastasis [106]. Endoglin induces inhibitor of DNA binding 1 (ID1) function via stimulation of ALK-1/SMAD1/5 signaling, which acts as proliferative and antiapoptotic and is a central regulator of CSC development [107]. HCV infection or ectopic expression of viral core enhances the expression of ID1-related markers for survival, proliferation, and CSCs (i.e., BCL2, CyclinD1, HES1, NOTCH1, NANOG, and SOX2 proteins) [106]. Furthermore, endoglin is an angiogenesis marker in patients with HCC [108,109].
3. HCV Manipulates Signaling Circuits of Differentiation, Adhesion, and Angiogenesis
A hallmark of HCC development is the dedifferentiation of hepatocytes, which is accompanied by important changes in intracellular communication and nutrients supply. The identification and understanding of stem cell-like cells in cancers has significantly contributed to the current understanding of tumor formation [110]. Even though CSCs share a few key features of normal tissue stem cells (e.g., unlimited proliferative and differentiation ability), they are potentially able to reproduce many of the elements related to cancer initiation, metastasis, and recurrence after therapy [111,112,113]. For HCC, a rare population of CSCs, called liver cancer stem cells (LCSCs), is abundant in tumor tissues and support self-renewal malignant transformation and resistance to chemotherapy [114]. Several LCSCs markers have been identified that have impact on the signaling circuitry, and some of them have been proposed as therapeutic targets for liver cancer treatment [115].
3.1. HCV Infection of Hepatocytes Provokes Stem Cell-Like Characteristics
During HCV infection, the virus predisposes cells towards the acquisition of CSC characteristics by the dysregulation of several signaling pathways [116,117]. Many of the characteristic CSC markers (i.e., CD133, CD90, CD44, and EpCAM) are also modulators of signaling pathways, including MAPK pathway, TGF-β mediated EMT, Wnt signaling, which are required to maintain CSC properties [115,118,119,120,121,122,123,124,125,126,127]. Other CSCs markers, such as doublecortin-like kinase 1 (DCLK1), impact microtubule filaments, polarized polymers of α and β tubulin heterodimers that are essential for cellular transport, cell division, and differentiation. DCLK1 is overexpressed in the liver of patients with HCV-associated HCC, while its level is very low or absent in normal hepatocytes. Interestingly, HCV replication, inflammation, and cirrhosis contribute to DCLK1 accumulation in the perinuclear region of the hepatocytes, where it colocalizes with NS5A and microtubule filaments [117]. This suggests that HCV-induced DCLK1 activity promotes microtubule filament polymerization and stabilization [117,128]. The maintenance of the CSC state is principally driven by reactivation of embryonic differentiation programs. Of these, especially Wnt, Notch, and Hedgehog signaling pathways potentially play a role in HCV-induced carcinogenesis [129,130,131,132].
3.2. HCV Causes Wnt Upregulation and β-Catenin Accumulation
Wnt pathway is a crucial component for embryonic development and tissue homeostasis [133]. Activation of the pathway starts when Wnt ligands bind to Frizzled (FZD) receptor, a seven transmembrane protein containing an extracellular cysteine-rich ligand-binding domain. When FZD receptor is activated, it inhibits the degradation of β-catenin. This leads to β-catenin accumulation and translocation to the nucleus, where it activates regulators of cell proliferation [134], such as WISP-1, c-MYC, and CCND1 [135,136,137]. Absence of FZD stimulation causes degradation of cytosolic β-catenin by a complex that consists of Axin, adenomatous polyposis coli protein (APC), and two serine/threonine kinases (GSK3β and CK1). Moreover, β-catenin potentiates the expression of ΔN-p73, a repressor of p53 and Tap73 proteins, conferring antiapoptotic and chemoresistance proprieties to HCC cells [134,138,139,140]. Components of the Wnt signaling are frequently mutated in liver cancer [141], which mostly result in β-catenin stabilization [142]. HCV infection manipulates Wnt signaling in multiple ways via its structural and nonstructural viral proteins. Isolated expression of NS5A has been reported to directly promote Wnt signaling by its interaction with PI3K and subsequent activation of Akt. This induces the phosphorylation and inhibition of glycogen synthase kinase 3β (GSK3β), a key component of β-catenin degradation complex [143]. Furthermore, ectopic expression of HCV core protein induces cell proliferation by forcing the expression of Wnt-1 and its downstream target gene WISP2, which induce Wnt signaling [144].
3.3. HCV Enhances Notch Signaling by Coactivating Hes-1 Promoter
Notch signaling suppresses cell differentiation, and it is involved in the maintenance of CSCs [145,146]. Notch ligands and receptors are both EGF-homologous transmembrane proteins mediating intercellular communication, cell proliferation, differentiation, and apoptosis [147]. Its impact on the cell is defined by the cellular microenvironment and its crosstalk with different signaling pathways [148]. To be activated, Notch receptors undergo a sequence of proteolytical cleavage upon interaction to a cell-bound ligand exposed on the surface of neighboring cells. Subsequently, this leads to the release of the Notch intracellular domain (NICD) and its translocation into the nucleus. Nuclear NICD associates with numerous cofactors and repressors, fine-tuning its transcriptional activity [147]. The complex orchestrates transcription of Notch target genes that regulate cell differentiation, such as hairy enhancer of split (HES1) [149], HES-related proteins (HEY), Notch-regulated ankyrin repeat protein (NRARP) [150], cyclin D1 (CCND1) [151], c-MYC [152,153,154,155], and receptor tyrosine-protein kinase erbB-2 (ERBB-2) [156]. In addition, Notch influences inflammation and metabolism by contributing to the activation of NF-κB [157] and peroxisome proliferator-activated receptor (PPAR) [148].
HCV infection interferes with Notch signaling and thereby contributes to hepatocarcinogenesis. Under isolated expression condition, NS3 protein binds to Snf2-related CBP activator protein (SRCAP) and cooperatively enhances Hes-1 promoter activity [158]. This leads to increased Notch-induced HES1 expression [159], a transcriptional repressor of cell differentiation [160], suggesting that HCV promotes a dedifferentiated CSC-like state of infected hepatocytes.
3.4. HCV-Induced Liver Damage Promotes Hedgehog Signaling
The Hedgehog pathway (Hh) is involved in the regulation of several morphogenic key functions, such as proliferation, survival, migration, and differentiation [161]. The Hedgehog ligands are essential during morphogenesis and embryogenesis processes as well as for the maintenance of stem cell homeostasis during adulthood [162]. Importantly, Hh pathway plays an essential role in adult liver repair and regeneration [163] and is implicated in several types of liver cancer, such as gallbladder cancer [164], cholangiocarcinoma [165,166,167], hepatoblastoma [168], and HCC [169]. Probably, the production of Hh ligands is favored by the accumulation of liver damage markers (i.e., platelet-derived growth factor (PDGF), TGF-β, and EGF) [170,171,172].
In patients with viral hepatitis, the Hh pathway is found to be induced [173], which presumably reflects tissue damage and liver regeneration during chronic infection. Interestingly, the permissiveness of cells to HCV replication seems to positively correlate with Hh pathway activity [174], suggesting that liver regeneration and a profibrotic environment may promote HCV infection. This is supported by the identification of additional key regulators of liver regeneration that are activated by HCV infection, including EGFR [17,41,42] and IL-6/STAT3 [175] signaling. Moreover, the presence of Hh activity promotes EMT in crosstalk with TGF-β and Wnt signaling [176], which once more highlights the relevance of EMT induction for HCV and its consequences for HCV-associated liver pathogenesis and HCC development.
3.5. HCV Promotes Angiogenesis via VEGF and HIF-1α Stabilization
Angiogenesis is a complex growth factor-dependent process responsible for the formation of new vessels from existing vascular trees [177,178]. Physiological angiogenesis is maintained by the balance between proangiogenic and antiangiogenic factors [179]. In pathological conditions, new growth in the vascular web is relevant as the proliferation of cancer cells and metastasis depend on a satisfactory source of oxygen and nutrients as well as waste removal from organs and tissues [180]. Several angiogenic growth factors are elevated in HCC patients, i.e., vascular endothelial growth factor A (VEGF-A), angiopoietin-2 and PDGF [181,182]. HCV infection leads to the development of hepatic angiogenesis, which significantly contributes to HCC progression and invasion [183]. This proangiogenic state is reversed in the livers of patients after viral clearance [184]. Vascular endothelial growth factor (VEGF) is a key regulator of angiogenesis in both normal and neoplastic tissues. Its expression and function are modulated by cytokines and other factors, such as the hypoxia-inducible factor 1α (HIF-1α) [182,185,186]. HCV infection leads to the stabilization of HIF-1α, mediated via oxidative stress and the induction of hypoxia [187]. In addition, the activation of PI3-K/Akt, Erk1/2, NF-κB, and STAT3 is necessary for hypoxia-inducible factor 1-alpha (HIF-1α) stabilization, which leads to the stimulation of VEGF [187]. HCV core protein triggers hepatic angiogenesis by a mechanism that involves crosstalk of multiple pathways, which is reflected by altered marker expression for hepatic angiogenesis, including TGF-β2, VEGF, and CD34 expression [185].
4. HCV Tweaks Signaling of the Inflammatory Response
Inflammation is an essential physiological response to several distressing stimuli, including infection. Inflammation is also tightly linked to the mechanisms of tissue regeneration and cancer. During chronic inflammation, NF-κB and STAT3 are central regulators of liver inflammation and are frequently associated with increased risk of cancer [188,189]. As part of the immune system, NF-κB contributes to the elimination of transformed cells. In support of this, NF-κB activation during the acute inflammatory response is highly associated with cytotoxic immune cell response [190]. The activation of NF-κB is induced by the IκB kinase (IKK) complex, which mediates phosphorylation and proteasomal degradation of IκB. This allows NF-κB dimers to translocate into the nucleus, where they induce an inflammatory and antiapoptotic response [191]. NF-κB is constitutively active in many types of cancer, promoting tumorigenic processes [192,193,194]. This suggests a dual role of NF-κB as a tumor suppressor and a tumor promoter, depending on the duration and intensity of tissue inflammation. NF-κB is a transcription factor and a central regulator of inflammation and cell survival. In quiescent cells, NF-κB is inactive, blocked by a tight association with inhibitor of NF-κB (IκB). NF-κB is further regulated by post-translational modifications (e.g., phosphorylation), which are important for its activation and crosstalk with other signaling pathways [195]. Moreover, NF-κB activity is influenced by dynamic protein–protein interactions, forming a tight network of feedback loops and interconnections [196]. In addition, STAT3 possesses a dual role as tumor suppressor and oncogene. It is not only a pivotal transcription factor in acute inflammation, but it is also a key element of liver regeneration [197] by regulating cell proliferation, survival, angiogenesis, and chemotaxis [198,199]. STAT3 is induced by a variety of different ligands, including interleukin 6 (IL-6), cardiotrophin-1 (CT-1), leukemia inhibitory factor (LIF), EGF, oncostatin M (OSM), IFN-α, and IFN-β [200]. Engagement of these ligands to their receptors leads to a subsequent recruitment of Janus kinases (JAK1, 2 and 3) and tyrosine kinase 2 (TYK2) that phosphorylate STAT3 [92,201,202,203]. Once phosphorylated, STAT3 forms homo- or heterodimers with STAT1 or STAT5 that translocate to the nucleus and bind specific DNA sequences. Without a doubt, STAT3 phosphorylation is necessary for its transcriptional activity. However, unphosphorylated STAT3 also presents biological functions, such as the expression of cell cycle progression genes [204,205]. NF-κB and STAT3 signaling are closely linked. NF-κB-mediated inflammation induces hepatic IL-6 production and STAT3 signaling [206]. Activated STAT3 in cancer cells binds to the NF-κB complex proteins RelA/p65 and the histone acetyltransferase p300 in the nucleus. As a consequence, p300 reversibly acetylates RelA/p65 dimers [207], which cause its nuclear retention [208]. At the same time, NF-κB can also impair oxidative stress, which is an activator of STAT3 [209]. In most HCC tumors, however, STAT3 activity does not coincide with NF-κB activation [210].
HCV Affects the STAT3/NF-κB Circuitry to Maintain a Pro-Inflammatory State
One of the most important examples of inflammation-associated cancers is HCC succeeding chronic HCV infection [211]. Compared to HBV infection, where viral genome integration accounts for the majority of HCCs, HCV-induced HCC is linked to liver disease progression from nonalcoholic fatty liver disease (NAFLD), chronic inflammation, fibrosis, and cirrhosis. This therefore suggests that HCV-induced signals promote liver fibrosis and disease progression following a similar disease pattern observed for other aetiologies. Indeed, HCV causes hepatic inflammation and induces complex alterations in host signal transduction [212]. These include deregulation of cytokine, metabolic, and oxidative stress pathways [213]. HCV-encoded proteins also cover an important role in initiating and maintaining this chronic inflammatory state. For instance, NS5A upregulates the expression of cyclooxygenase-2 (COX-2) [213], which promotes chronic inflammation by the synthesis of prostaglandins. It is therefore not surprising that HCV manipulates regulatory signaling of the inflammatory response, including NF-κB [189] and STAT3 [214], and thereby increases the risk of HCC development. HCV induces chronic hepatic inflammation that is mediated by elevated NF-κB activity. However, the question is whether this is simply a consequence of the cellular defense against infection by HCV or whether the virus has an interest in maintaining an inflammatory state for its own benefit. Several lines of evidence suggest that HCV indeed gains from tweaking the outcome of the inflammatory response. For example, HCV infection enhances tumor necrosis factor alpha (TNF-α)-induced cell death by suppression of NF-κB activation involving a mechanism dependent on core, NS4B, and NS5B [215]. At the same time, HCV makes use of parts of the NF-κB signaling by activating IKKα which, independent of NF-κB, induces the expression of lipogenic genes that contribute to core-associated lipid droplet formation [20]. The same is true for STAT3, which is a mediator of inflammation and part of the interferon response against viral infection. STAT3 transcriptional activity is elevated upon HCV infection in livers of patients and in cell culture [175] and is associated with poor prognosis in HCCs [189]. STAT3 is activated by HCV-induced oxidative stress via core, NS2, and NS3 proteins [216] and by the innate antiviral immune response in hepatocytes [40]. Additionally, the presence of HCV not only affects the infected hepatocytes but equally affects the liver microenvironment. Exosomes secreted from HCV-infected cells carrying miR-19a induce STAT3 activation in hepatic stellate cells and favor fibrotic gene expression [217]. STAT3 activation in the context of HCV infection has also been linked to the presence of myeloid-derived suppressor cells (MDSCs), a cell type that favors the expansion of Treg lymphocytes and has been associated with an increased tumor burden in HCC patients [218]. The question then arises as to whether the elevated STAT3 signaling is simply a consequence of infection or whether it is beneficial to the virus. Interestingly, HCV core protein also directly associates and activates STAT3 function, which promotes cell transformation [219], suggesting an important role of STAT3 for HCV. Indeed, HCV has a vital interest in maintaining a persistent STAT3 signaling as STAT3 is a cofactor for HCV infection and tempers the antiviral impact of the interferon response [40].
5. Clinical Relevance and Perspectives
Chronic HCV infection is a major cause of HCC, the second most deadly cancer worldwide with only very limited treatment options. HCV-related HCC will remain a major health problem for the next decades, despite the recent development of direct-acting antivirals (DAAs) and their deployment in therapy [220]. Especially in patients with advanced liver disease, the HCC risk cannot be fully reversed after viral cure [221]. This is similar to alcohol-induced liver disease, where the HCC risk during abstinence persists for several years [222]. Although the oncogenic mechanism of alcohol and its carcinogenic metabolite acetaldehyde differ from that of viral hepatitis, it has been suggested that, similar to alcohol [223,224], HCV infection may leave an epigenetic footprint in the host genome. An interesting question is whether this also creates persistent alterations in the host signaling network that maintain an oncogenic pressure to the hepatocyte, like an echo from the chronic infection.
Another point worth mentioning is a suggested increase in tumor recurrence rates in HCC patients after DAA-induced sustained virological response and tumor resection [225,226]. However, these results remain controversial as other groups could not confirm this observation [227,228]. Therefore, whether antiviral treatment in HCC patients leads to a long-term survival benefit is currently unknown, and current guidelines suggest a close surveillance and imaging in these patients [229]. The treatment of HCC is particularly challenging for patient cohorts with moderate and severe liver dysfunction (Child–Pugh Class B or C) in term of toxicity and efficacy as the use of sorafenib for the treatment of Child–Pugh B patients has been questioned [230]. Moreover, the HCC proliferative index is low, which is one of the reasons most cytostatics and small molecules are considered inefficient.
By hijacking the host signaling network, HCV generates a proliferative and antiapoptotic environment, which promotes hepatocyte dedifferentiation and EMT. This forms an optimal environment for the virus to persist but with serious consequences to the host. The signaling pathways deregulated by chronic HCV infection resemble the hallmarks of cancer [231,232], suggesting that HCV-induced oncogenic signaling likely contributes to liver disease progression and hepatocarcinogenesis. Targeting signaling components with therapeutic antibodies or clinical kinase inhibitors in cancer therapy is widely established. The current pharmacological therapy for HCC is essentially based on the multikinase inhibitor sorafenib [233], which is able to increase survival rates of selected HCC patients. Other kinase inhibitors clinically tested include linifanib (VEGFR and PDGFR inhibitor) [234] and erlotinib (EGFR inhibitor) [235]; the latter failed in phase 3 of its clinical trial [236]. The identification of therapeutic targets in established HCCs is difficult because genetic alterations in tumors are highly heterogeneous [237]. Nevertheless, such approach holds promise in the framework of a personalized treatment, and targeting derailed signaling pathways in patients at risk of developing HCCs can be part of novel chemopreventive strategies. In support of this, an important proof-of-concept was demonstrated in 2014 by Bryan Fuchs and colleagues as erlotinib-attenuated fibrogenesis and HCC development in a rat model [26]. Other HCV-modulated signaling pathways (i.e., NF-κB and STAT3) offer interesting opportunities to therapeutic intervention, as well as prevention, especially in the pathological context of HCC [189].
However, this requires new and well-tolerated compounds that allow a long-term administration of kinase inhibitors to patients with advanced liver disease. A deeper understanding of the signaling network of HCV infection will also contribute to a better understanding of general signaling events involved in liver disease progression, given the gene expression profiles in patients at risk of HCC seem to be independent of the underlying aetiology [238]. In future, well-established HCV infection models will be instrumental in highlighting additional deregulated and druggable signaling pathways that are associated with HCC risk. This will help to overcome the lack of appropriate study models of HCC development and contribute to the discovery of novel drivers and drug targets of liver disease and HCC development.
Funding
This research was funded by the European Union (ERC-AdG-2014 HEPCIR to T.F.B., EU H2020 HEPCAR 667273 to T.F.B. and J.L.), the French Cancer Agency (ARC IHU201301187 to T.F.B.), the US Department of Defense (W81XWH-16-1-0363) to T.F.B., the National Institutes (NCI 1R21CA209940-01A1, NIAID R03AI131066, NIAID 5U19AI123862-02 to T.F.B.), and the French Agence Nationale de Recherche sur le Sida et les Hépatites Virales (ANRS) (ECTZ4236; ECTZ4446 to J.L. and A.A.R.S). This work has benefitted from the support of the Initiative of excellence IDEX-Unistra (ANR-10-IDEX-0002-02 to J.L. and A.V.) and has been published under the framework of the LABEX ANR-10-LAB-28. IDEX and LABEX are initiatives from the French program “Investments for the future”.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Coffin, J.M.; Varmus, H.E.; Bishop, J.M.; Essex, M.; Hardy, W.D., Jr.; Martin, G.S.; Rosenberg, N.E.; Scolnick, E.M.; Weinberg, R.A.; Vogt, P.K. Proposal for naming host cell-derived inserts in retrovirus genomes. J. Virol. 1981, 40, 953–957. [Google Scholar] [PubMed]
- Vogt, P.K. Retroviral oncogenes: A historical primer. Nat. Rev. Cancer 2012, 12, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.G.; Ou, J.H. OncogeniC Viruses and cancer. Virol. Sin. 2015, 30, 83–84. [Google Scholar] [CrossRef] [PubMed]
- Lemon, S.M.; McGivern, D.R. Is hepatitis C Virus carcinogenic? Gastroenterology 2012, 142, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Ray, R.B.; Ray, R. Oncogenic potential of hepatitis C Virus proteins. Viruses 2010, 2, 2108–2133. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Lemon, S.M. Virus-specific mechanisms of carcinogenesis in hepatitis C Virus associated liver cancer. Oncogene 2011, 30, 1969–1983. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C Virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Gisbert, J.P.; Garcia-Buey, L.; Pajares, J.M.; Moreno-Otero, R. Prevalence of hepatitis C Virus infection in b-cell non-hodgkin’s lymphoma: Systematic review and meta-analysis. Gastroenterology 2003, 125, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Su, F.H.; Chang, S.N.; Chen, P.C.; Sung, F.C.; Huang, S.F.; Chiou, H.Y.; Su, C.T.; Lin, C.C.; Yeh, C.C. Positive association between hepatitis c infection and oral cavity cancer: A nationwide population-based cohort study in taiwan. PLoS ONE 2012, 7, e48109. [Google Scholar] [CrossRef] [PubMed]
- Mahale, P.; Sturgis, E.M.; Tweardy, D.J.; Ariza-Heredia, E.J.; Torres, H.A. Association between hepatitis C Virus and head and neck cancers. J. Natl. Cancer Inst. 2016, 108, djw035. [Google Scholar] [CrossRef] [PubMed]
- El-Serag, H.B.; Engels, E.A.; Landgren, O.; Chiao, E.; Henderson, L.; Amaratunge, H.C.; Giordano, T.P. Risk of hepatobiliary and pancreatic cancers after hepatitis C Virus infection: A population-based study of U.S. Veterans. Hepatology 2009, 49, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Magnusson, M.; Torner, A.; Ye, W.; Duberg, A.S. Risk of pancreatic cancer among individuals with hepatitis c or hepatitis b virus infection: A nationwide study in sweden. Br. J. Cancer 2013, 109, 2917–2923. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.C.; Moonka, D.; Brown, K.A.; Rogers, C.; Huang, M.A.; Bhatt, N.; Lamerato, L. Risk for renal cell carcinoma in chronic hepatitis c infection. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Mahale, P.; Torres, H.A.; Kramer, J.R.; Hwang, L.Y.; Li, R.; Brown, E.L.; Engels, E.A. Hepatitis C Virus infection and the risk of cancer among elderly us adults: A registry-based case-control study. Cancer 2017, 123, 1202–1211. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, A.H. The association of extrahepatic cancers with chronic hepatitis C Virus infection. Gastroenterol. Hepatol. (N. Y.) 2016, 12, 185–187. [Google Scholar]
- Pol, S.; Vallet-Pichard, A.; Hermine, O. Extrahepatic cancers and chronic HCV infection. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Zeisel, M.B.; Xiao, F.; Thumann, C.; Fofana, I.; Zona, L.; Davis, C.; Mee, C.J.; Turek, M.; Gorke, S.; et al. Egfr and EPHA2 are host factors for hepatitis C Virus entry and possible targets for antiviral therapy. Nat. Med. 2011, 17, 589–595. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Casanova, C.; Fischer, B.; Weiss, A.; Fofana, I.; Fontaine, N.; Fujiwara, T.; Renaud, M.; Kopp, A.; Schuster, C.; et al. PI4K-β and MKNK1 are regulators of hepatitis C Virus IRES-dependent translation. Sci. Rep. 2015, 5, 13344. [Google Scholar] [CrossRef] [PubMed]
- Paul, D.; Madan, V.; Bartenschlager, R. Hepatitis C Virus RNA replication and assembly: Living on the fat of the land. Cell Host Microbe 2014, 16, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhang, Y.Y.; Chiu, S.; Hu, Z.; Lan, K.H.; Cha, H.; Sodroski, C.; Zhang, F.; Hsu, C.S.; Thomas, E.; et al. Integrative functional genomics of hepatitis C Virus infection identifies host dependencies in complete viral replication cycle. PLoS Pathog. 2014, 10, e1004163. [Google Scholar] [CrossRef] [PubMed]
- Zona, L.; Lupberger, J.; Sidahmed-Adrar, N.; Thumann, C.; Harris, H.J.; Barnes, A.; Florentin, J.; Tawar, R.G.; Xiao, F.; Turek, M.; et al. Hras signal transduction promotes hepatitis C Virus cell entry by triggering assembly of the host tetraspanin receptor complex. Cell Host Microbe 2013, 13, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2002, 2, 103–112. [Google Scholar] [CrossRef] [Green Version]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef] [PubMed]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiology (Bethesda) 2010, 25, 85–101. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, B.C.; Hoshida, Y.; Fujii, T.; Wei, L.; Yamada, S.; Lauwers, G.Y.; McGinn, C.M.; DePeralta, D.K.; Chen, X.; Kuroda, T.; et al. Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcinoma. Hepatology 2014, 59, 1577–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pennycuick, A.; Villanueva, A. Signaling in Hepatocellular Carcinoma, 3rd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2015. [Google Scholar]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: Towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ERBB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Bublil, E.M.; Yarden, Y. The EGF receptor family: Spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 2007, 19, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, J.; Baselga, J. Epidermal growth factor receptor targeting in cancer. Semin. Oncol. 2006, 33, 369–385. [Google Scholar] [CrossRef] [PubMed]
- Salomon, D.S.; Kim, N.; Saeki, T.; Ciardiello, F. Transforming growth factor-alpha: An oncodevelopmental growth factor. Cancer Cells 1990, 2, 389–397. [Google Scholar] [PubMed]
- Sunnerhagen, M.S.; Persson, E.; Dahlqvist, I.; Drakenberg, T.; Stenflo, J.; Mayhew, M.; Robin, M.; Handford, P.; Tilley, J.W.; Campbell, I.D.; et al. The effect of aspartate hydroxylation on calcium binding to epidermal growth factor-like modules in coagulation factors IX and X. J. Biol. Chem. 1993, 268, 23339–23344. [Google Scholar] [PubMed]
- Huang, W.C.; Chan, S.H.; Jang, T.H.; Chang, J.W.; Ko, Y.C.; Yen, T.C.; Chiang, S.L.; Chiang, W.F.; Shieh, T.Y.; Liao, C.T.; et al. MiRNA-491-5p and GIT1 serve as modulators and biomarkers for oral squamous cell carcinoma invasion and metastasis. Cancer Res. 2014, 74, 751–764. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Takeda, T.; Sasaki, Y.; Sakon, M.; Yamada, T.; Ishiguro, S.; Imaoka, S.; Tsujimoto, M.; Higashiyama, S.; Monden, M.; et al. Expression and clinical significance of the ERBB family in intrahepatic cholangiocellular carcinoma. Pathol. Res. Pract. 2001, 197, 95–100. [Google Scholar] [CrossRef] [PubMed]
- Zheng, K.; Kitazato, K.; Wang, Y. Viruses exploit the function of epidermal growth factor receptor. Rev. Med. Virol. 2014, 24, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Baktash, Y.; Madhav, A.; Coller, K.E.; Randall, G. Single particle imaging of polarized hepatoma organoids upon hepatitis C Virus infection reveals an ordered and sequential entry process. Cell Host Microbe 2018, 23, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Lupberger, J.; Duong, F.H.; Fofana, I.; Zona, L.; Xiao, F.; Thumann, C.; Durand, S.C.; Pessaux, P.; Zeisel, M.B.; Heim, M.H.; et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-α. Hepatology 2013, 58, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Diao, J.; Pantua, H.; Ngu, H.; Komuves, L.; Diehl, L.; Schaefer, G.; Kapadia, S.B. Hepatitis C Virus induces epidermal growth factor receptor activation via CD81 binding for viral inteRNAlization and entry. J. Virol. 2012, 86, 10935–10949. [Google Scholar] [CrossRef] [PubMed]
- Mailly, L.; Xiao, F.; Lupberger, J.; Wilson, G.K.; Aubert, P.; Duong, F.H.T.; Calabrese, D.; Leboeuf, C.; Fofana, I.; Thumann, C.; et al. Clearance of persistent hepatitis C Virus infection in humanized mice using a claudin-1-targeting monoclonal antibody. Nat. Biotechnol. 2015, 33, 549–554. [Google Scholar] [CrossRef] [PubMed]
- Igloi, Z.; Kazlauskas, A.; Saksela, K.; Macdonald, A.; Mankouri, J.; Harris, M. Hepatitis C Virus NS5A protein blocks epidermal growth factor receptor degradation via a proline motif- dependent interaction. J. Gen. Virol. 2015, 96, 2133–2144. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.T.D.; Tran, G.V.Q.; Shin, D.J.; Lim, Y.S.; Hwang, S.B. Hepatitis C Virus exploits death receptor 6-mediated signaling pathway to facilitate viral propagation. Sci. Rep. 2017, 7, 6445. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C Virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998, 12, 2245–2262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nevins, J.R. The RB/E2F pathway and cancer. Hum. Mol. Genet. 2001, 10, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Trimarchi, J.M.; Lees, J.A. Sibling rivalry in the E2F family. Nat. Rev. Mol. Cell Biol. 2002, 3, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Ren, B.; Cam, H.; Takahashi, Y.; Volkert, T.; Terragni, J.; Young, R.A.; Dynlacht, B.D. E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev. 2002, 16, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Harbour, J.W.; Dean, D.C. The RB/E2F pathway: Expanding roles and emerging paradigms. Genes Dev. 2000, 14, 2393–2409. [Google Scholar] [CrossRef] [PubMed]
- Rayman, J.B.; Takahashi, Y.; Indjeian, V.B.; Dannenberg, J.H.; Catchpole, S.; Watson, R.J.; Te Riele, H.; Dynlacht, B.D. E2F mediates cell cycle-dependent transcriptional repression in vivo by recruitment of an HDAC1/MSIN3B corepressor complex. Genes Dev. 2002, 16, 933–947. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, H.; Ishiguro, K.; Gaubatz, S.; Livingston, D.M.; Nakatani, Y. A complex with chromatin modifiers that occupies E2F- and MYC-responsive genes in g0 cells. Science 2002, 296, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. P53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. Waf1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Whyte, P.; Buchkovich, K.J.; Horowitz, J.M.; Friend, S.H.; Raybuck, M.; Weinberg, R.A.; Harlow, E. Association between an oncogene and an anti-oncogene: The adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 1988, 334, 124–129. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Howley, P.M.; Munger, K.; Harlow, E. The human papilloma virus-16 E7 oncoprotein is able to bind to the retinoblastoma gene product. Science 1989, 243, 934–937. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Liang, Y.; Kim, S.; McGivern, D.R.; Huibregtse, J.; Nomoto, A.; Lemon, S.M. Hepatitis C Virus induces E6AP-dependent degradation of the retinoblastoma protein. PLoS Pathog. 2007, 3, 1335–1347. [Google Scholar] [CrossRef] [PubMed]
- McGivern, D.R.; Villanueva, R.A.; Chinnaswamy, S.; Kao, C.C.; Lemon, S.M. Impaired replication of hepatitis C Virus containing mutations in a conserved NS5B retinoblastoma protein-binding motif. J. Virol. 2009, 83, 7422–7433. [Google Scholar] [CrossRef] [PubMed]
- Munakata, T.; Nakamura, M.; Liang, Y.; Li, K.; Lemon, S.M. Down-regulation of the retinoblastoma tumor suppressor by the hepatitis C Virus NS5B RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 2005, 102, 18159–18164. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.; Baek, W.; Yang, S.; Chang, J.; Sung, Y.C.; Suh, M. HCV core protein modulates RB pathway through pRB down-regulation and E2F-1 up-regulation. Biochim. Biophys. Acta 2001, 1538, 59–66. [Google Scholar] [CrossRef]
- Hussain, S.P.; Schwank, J.; Staib, F.; Wang, X.W.; Harris, C.C. Tp53 mutations and hepatocellular carcinoma: Insights into the etiology and pathogenesis of liver cancer. Oncogene 2007, 26, 2166. [Google Scholar] [CrossRef] [PubMed]
- Olivier, M.; Hussain, S.P.; Caron de Fromentel, C.; Hainaut, P.; Harris, C.C. Tp53 mutation spectra and load: A tool for generating hypotheses on the etiology of cancer. IARC Sci. Publ. 2004, 247–270. [Google Scholar]
- Sato, Y.; Tsurumi, T. Genome guardian p53 and viral infections. Rev. Med. Virol. 2013, 23, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Lo, S.Y.; Chen, M.; Wu, K.; Fung, Y.K.; Ou, J.H. Activation of p53 tumor suppressor by hepatitis C Virus core protein. Virology 1999, 264, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Otsuka, M.; Kato, N.; Lan, K.; Yoshida, H.; Kato, J.; Goto, T.; Shiratori, Y.; Omata, M. Hepatitis C Virus core protein enhances p53 function through augmentation of DNA binding affinity and transcriptional ability. J. Biol. Chem. 2000, 275, 34122–34130. [Google Scholar] [CrossRef] [PubMed]
- Ray, R.B.; Steele, R.; Meyer, K.; Ray, R. Hepatitis C Virus core protein represses p21WAF1/CIP1/SID1 promoter activity. Gene 1998, 208, 331–336. [Google Scholar] [CrossRef]
- Kao, C.F.; Chen, S.Y.; Chen, J.Y.; Wu Lee, Y.H. Modulation of p53 transcription regulatory activity and post-translational modification by hepatitis C Virus core protein. Oncogene 2004, 23, 2472–2483. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Nagano-Fujii, M.; Tanaka, M.; Nomura-Takigawa, Y.; Ikeda, M.; Kato, N.; Sada, K.; Hotta, H. NS3 protein of hepatitis C Virus associates with the tumour suppressor p53 and inhibits its function in an NS3 sequence-dependent manner. J. Gen. Virol. 2006, 87, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Nagano-Fujii, M.; Deng, L.; Ishido, S.; Sada, K.; Hotta, H. Single-point mutations of hepatitis C Virus NS3 that impair p53 interaction and anti-apoptotic activity of NS3. Biochem. Biophys. Res. Commun. 2006, 340, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Lan, K.H.; Sheu, M.L.; Hwang, S.J.; Yen, S.H.; Chen, S.Y.; Wu, J.C.; Wang, Y.J.; Kato, N.; Omata, M.; Chang, F.Y.; et al. Hcv NS5A interacts with p53 and inhibits p53-mediated apoptosis. Oncogene 2002, 21, 4801–4811. [Google Scholar] [CrossRef] [PubMed]
- You, L.R.; Chen, C.M.; Yeh, T.S.; Tsai, T.Y.; Mai, R.T.; Lin, C.H.; Lee, Y.H. Hepatitis C Virus core protein interacts with cellular putative RNA helicase. J. Virol. 1999, 73, 2841–2853. [Google Scholar] [PubMed]
- Mamiya, N.; Worman, H.J. Hepatitis C Virus core protein binds to a dead box RNA helicase. J. Biol. Chem. 1999, 274, 15751–15756. [Google Scholar] [CrossRef] [PubMed]
- Owsianka, A.M.; Patel, A.H. Hepatitis C Virus core protein interacts with a human dead box protein DDX3. Virology 1999, 257, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Chao, C.H.; Chen, C.M.; Cheng, P.L.; Shih, J.W.; Tsou, A.P.; Lee, Y.H. DDX3, a dead box RNA helicase with tumor growth-suppressive property and transcriptional regulation activity of the p21WAF1/CIP1 promoter, is a candidate tumor suppressor. Cancer Res. 2006, 66, 6579–6588. [Google Scholar] [CrossRef] [PubMed]
- Goh, P.Y.; Tan, Y.J.; Lim, S.P.; Tan, Y.H.; Lim, S.G.; Fuller-Pace, F.; Hong, W. Cellular RNA helicase p68 relocalization and interaction with the hepatitis C Virus (HCV) NS5B protein and the potential role of p68 in hcv RNA replication. J. Virol. 2004, 78, 5288–5298. [Google Scholar] [CrossRef] [PubMed]
- Nicol, S.M.; Bray, S.E.; Black, H.D.; Lorimore, S.A.; Wright, E.G.; Lane, D.P.; Meek, D.W.; Coates, P.J.; Fuller-Pace, F.V. The RNA helicase p68 (DDX5) is selectively required for the induction of p53-dependent p21 expression and cell-cycle arrest after DNA damage. Oncogene 2013, 32, 3461–3469. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.J.; Nicol, S.M.; Wilson, B.J.; Jacobs, A.M.; Bourdon, J.C.; Wardrop, J.; Gregory, D.J.; Lane, D.P.; Perkins, N.D.; Fuller-Pace, F.V. The dead box protein p68: A novel transcriptional coactivator of the p53 tumour suppressor. EMBO J. 2005, 24, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Bressac, B.; Galvin, K.M.; Liang, T.J.; Isselbacher, K.J.; Wands, J.R.; Ozturk, M. Abnormal structure and expression of p53 gene in human hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 1990, 87, 1973–1977. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, M.; Imamura, T.; Miyazono, K. Signal transduction by bone morphogenetic proteins. Cytokine Growth Factor Rev. 1998, 9, 49–61. [Google Scholar] [CrossRef]
- Chaouchi, N.; Arvanitakis, L.; Auffredou, M.T.; Blanchard, D.A.; Vazquez, A.; Sharma, S. Characterization of transforming growth factor-β 1 induced apoptosis in normal human b cells and lymphoma b cell lines. Oncogene 1995, 11, 1615–1622. [Google Scholar] [PubMed]
- Lomo, J.; Blomhoff, H.K.; Beiske, K.; Stokke, T.; Smeland, E.B. TGF-β 1 and cyclic amp promote apoptosis in resting human b lymphocytes. J. Immunol. 1995, 154, 1634–1643. [Google Scholar] [PubMed]
- Wahl, S.M.; Hunt, D.A.; Wong, H.L.; Dougherty, S.; McCartney-Francis, N.; Wahl, L.M.; Ellingsworth, L.; Schmidt, J.A.; Hall, G.; Roberts, A.B.; et al. Transforming growth factor-β is a potent immunosuppressive agent that inhibits Il-1-dependent lymphocyte proliferation. J. Immunol. 1988, 140, 3026–3032. [Google Scholar] [PubMed]
- Roberts, A.B.; Sporn, M.B. Physiological actions and clinical applications of transforming growth factor-β (TGF-β). Growth Factors 1993, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Roberts, A.B. Molecular and cell biology of TGF-β. Miner. Electrolyte Metab. 1998, 24, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Shinozaki, M.; Hara, T.; Furuya, T.; Miyazono, K. Two major SMAD pathways in TGF-β superfamily signalling. Genes Cells 2002, 7, 1191–1204. [Google Scholar] [CrossRef] [PubMed]
- Meindl-Beinker, N.M.; Matsuzaki, K.; Dooley, S. TGF-β signaling in onset and progression of hepatocellular carcinoma. Dig. Dis. 2012, 30, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Branton, M.H.; Kopp, J.B. TGF-β and fibrosis. Microbes Infect. 1999, 1, 1349–1365. [Google Scholar] [CrossRef]
- Dooley, S.; ten Dijke, P. TGF-β in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, I.; Moreno-Caceres, J.; Sanchez, A.; Dooley, S.; Dewidar, B.; Giannelli, G.; Ten Dijke, P.; Consortium, I.-L. TGF-β signalling and liver disease. FEBS J. 2016, 283, 2219–2232. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Giannelli, G.; Rani, B.; Dituri, F.; Cao, Y.; Palasciano, G. Moving towards personalised therapy in patients with hepatocellular carcinoma: The role of the microenvironment. Gut 2014, 63, 1668–1676. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L.; Attisano, L.; Wieser, R.; Ventura, F.; Massague, J. Mechanism of activation of the TGF-β receptor. Nature 1994, 370, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-β signalling from cell membrane to nucleus through smad proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Massaous, J.; Hata, A. TGF-β signalling through the SMAD pathway. Trends Cell Biol. 1997, 7, 187–192. [Google Scholar] [CrossRef]
- Schuster, N.; Krieglstein, K. Mechanisms of TGF-β-mediated apoptosis. Cell Tissue Res. 2002, 307, 1–14. [Google Scholar] [CrossRef] [PubMed]
- HeRNAnda, P.Y.; Chen, K.; Das, A.M.; Sideras, K.; Wang, W.; Li, J.; Cao, W.; Bots, S.J.; Kodach, L.L.; de Man, R.A.; et al. Smad4 exerts a tumor-promoting role in hepatocellular carcinoma. Oncogene 2015, 34, 5055–5068. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Heldin, C.H. Non-smad TGF-β signals. J. Cell Sci. 2005, 118, 3573–3584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.E. Non-smad pathways in TGF-β signaling. Cell Res. 2009, 19, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, S.; Benzoubir, N.; Nobilet, S.; Charneau, P.; Samuel, D.; Zignego, A.L.; Atfi, A.; Brechot, C.; Bourgeade, M.F. Liver cancer-derived hepatitis C Virus core proteins shift TGF-β responses from tumor suppression to epithelial-mesenchymal transition. PLoS ONE 2009, 4, e4355. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Chusri, P.; Kumthip, K.; Hong, J.; Zhu, C.; Duan, X.; Jilg, N.; Fusco, D.N.; Brisac, C.; Schaefer, E.A.; Cai, D.; et al. Hcv induces transforming growth factor β1 through activation of endoplasmic reticulum stress and the unfolded protein response. Sci. Rep. 2016, 6, 22487. [Google Scholar] [CrossRef] [PubMed]
- Asselah, T.; Bieche, I.; Mansouri, A.; Laurendeau, I.; Cazals-Hatem, D.; Feldmann, G.; Bedossa, P.; Paradis, V.; Martinot-Peignoux, M.; Lebrec, D.; et al. In vivo hepatic endoplasmic reticulum stress in patients with chronic hepatitis C. J. Pathol. 2010, 221, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, P.L.; Chang, M.H.; Chao, C.H.; Lee, Y.H. Hepatitis c viral proteins interact with SMAD3 and differentially regulate TGF-β/SMAD3-mediated transcriptional activation. Oncogene 2004, 23, 7821–7838. [Google Scholar] [CrossRef] [PubMed]
- Pavio, N.; Battaglia, S.; Boucreux, D.; Arnulf, B.; Sobesky, R.; Hermine, O.; Brechot, C. Hepatitis C Virus core variants isolated from liver tumor but not from adjacent non-tumor tissue interact with SMAD3 and inhibit the TGF-β pathway. Oncogene 2005, 24, 6119–6132. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.C.; Sasaki, R.; Meyer, K.; Ray, R. Hepatitis C Virus core protein modulates endoglin (CD105) signaling pathway for liver pathogenesis. J. Virol. 2017, 91, e01235-17. [Google Scholar] [CrossRef] [PubMed]
- Lasorella, A.; Benezra, R.; Iavarone, A. The id proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Pan, Y.; Chen, J.; Sun, X.; Qiu, Y.; Ding, Y. Endoglin (CD105) expression in angiogenesis of primary hepatocellular carcinomas: Analysis using tissue microarrays and comparisons with CD34 and VEGF. Ann. Clin. Lab. Sci. 2007, 37, 39–48. [Google Scholar] [PubMed]
- Ho, J.W.; Poon, R.T.; Sun, C.K.; Xue, W.C.; Fan, S.T. Clinicopathological and prognostic implications of endoglin (CD105) expression in hepatocellular carcinoma and its adjacent non-tumorous liver. World J. Gastroenterol. 2005, 11, 176–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bomken, S.; Fiser, K.; Heidenreich, O.; Vormoor, J. Understanding the cancer stem cell. Br. J. Cancer 2010, 103, 439–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marquardt, J.U.; Factor, V.M.; Thorgeirsson, S.S. Epigenetic regulation of cancer stem cells in liver cancer: Current concepts and clinical implications. J. Hepatol. 2010, 53, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.M.; Xu, Y.; Fan, J.; Zhou, J.; Yang, X.R.; Qiu, S.J.; Liao, Y.; Wu, W.Z.; Ji, Y.; Ke, A.W.; et al. Identification of side population cells in human hepatocellular carcinoma cell lines with stepwise metastatic potentials. J. Cancer Res. Clin. Oncol. 2008, 134, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Alison, M.R. Liver stem cells: Implications for hepatocarcinogenesis. Stem Cell Rev. 2005, 1, 253–260. [Google Scholar] [CrossRef]
- Song, K.; Wu, J.; Jiang, C. Dysregulation of signaling pathways and putative biomarkers in liver cancer stem cells (review). Oncol. Rep. 2013, 29, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nio, K.; Yamashita, T.; Kaneko, S. The evolving concept of liver cancer stem cells. Mol. Cancer 2017, 16, 4. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.C.; Bose, S.K.; Steele, R.; Meyer, K.; Di Bisceglie, A.M.; Ray, R.B.; Ray, R. Promotion of cancer stem-like cell properties in hepatitis C Virus-infected hepatocytes. J. Virol. 2015, 89, 11549–11556. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.; Allam, H.; May, R.; Sureban, S.M.; Bronze, M.S.; Bader, T.; Umar, S.; Anant, S.; Houchen, C.W. Hepatitis C Virus-induced cancer stem cell-like signatures in cell culture and murine tumor xenografts. J. Virol. 2011, 85, 12292–12303. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Chan, K.W.; Hu, L.; Lee, T.K.; Wo, J.Y.; Ng, I.O.; Zheng, B.J.; Guan, X.Y. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology 2007, 132, 2542–2556. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Li, J.; Hu, C.; Chen, X.; Yao, M.; Yan, M.; Jiang, G.; Ge, C.; Xie, H.; Wan, D.; et al. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int. J. Cancer 2007, 120, 1444–1450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, K.H.; Ma, S.; Lee, T.K.; Chan, Y.P.; Kwan, P.S.; Tong, C.M.; Ng, I.O.; Man, K.; To, K.F.; Lai, P.B.; et al. CD133(+) liver tumor-initiating cells promote tumor angiogenesis, growth, and self-renewal through neurotensin/interleukin-8/CXCl1 signaling. Hepatology 2012, 55, 807–820. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ho, D.W.; Ng, M.N.; Lau, C.K.; Yu, W.C.; Ngai, P.; Chu, P.W.; Lam, C.T.; Poon, R.T.; Fan, S.T. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell 2008, 13, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.F.; Ngai, P.; Ho, D.W.; Yu, W.C.; Ng, M.N.; Lau, C.K.; Li, M.L.; Tam, K.H.; Lam, C.T.; Poon, R.T.; et al. Identification of local and circulating cancer stem cells in human liver cancer. Hepatology 2008, 47, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Hao, X.; Yan, M.; Yao, M.; Ge, C.; Gu, J.; Li, J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int. J. Cancer 2010, 126, 2067–2078. [Google Scholar] [CrossRef] [PubMed]
- Mima, K.; Okabe, H.; Ishimoto, T.; Hayashi, H.; Nakagawa, S.; Kuroki, H.; Watanabe, M.; Beppu, T.; Tamada, M.; Nagano, O.; et al. CD44s regulates the TGF-β-mediated mesenchymal phenotype and is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. 2012, 72, 3414–3423. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Honda, M.; Nakamoto, Y.; Baba, M.; Nio, K.; Hara, Y.; Zeng, S.S.; Hayashi, T.; Kondo, M.; Takatori, H.; et al. Discrete nature of epcam+ and CD90+ cancer stem cells in human hepatocellular carcinoma. Hepatology 2013, 57, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Budhu, A.; Forgues, M.; Wang, X.W. Activation of hepatic stem cell marker epcam by wnt-β-catenin signaling in hepatocellular carcinoma. Cancer Res. 2007, 67, 10831–10839. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T.; Ji, J.; Budhu, A.; Forgues, M.; Yang, W.; Wang, H.Y.; Jia, H.; Ye, Q.; Qin, L.X.; Wauthier, E.; et al. Epcam-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology 2009, 136, 1012–1024. [Google Scholar] [CrossRef] [PubMed]
- Roohvand, F.; Maillard, P.; Lavergne, J.P.; Boulant, S.; Walic, M.; Andreo, U.; Goueslain, L.; Helle, F.; Mallet, A.; McLauchlan, J.; et al. Initiation of hepatitis C Virus infection requires the dynamic microtubule network: Role of the viral nucleocapsid protein. J. Biol. Chem. 2009, 284, 13778–13791. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, Q.; Fuhler, G.M.; Smits, R.; Peppelenbosch, M.P. Action and function of WNT/β-catenin signaling in the progression from chronic hepatitis C to hepatocellular carcinoma. J. Gastroenterol. 2017, 52, 419–431. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Xia, Y.; Chen, K.; Zheng, Y.; Wang, J.; Lu, W.; Yin, Q.; Wang, F.; Zhou, Y.; Guo, C. Oncogenic role of the notch pathway in primary liver cancer. Oncol. Lett. 2016, 12, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; He, J.; Zhang, X.; Bian, Y.; Yang, L.; Xie, G.; Zhang, K.; Tang, W.; Stelter, A.A.; Wang, Q.; et al. Activation of the hedgehog pathway in human hepatocellular carcinomas. Carcinogenesis 2006, 27, 1334–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pez, F.; Lopez, A.; Kim, M.; Wands, J.R.; Caron de Fromentel, C.; Merle, P. WNT signaling and hepatocarcinogenesis: Molecular targets for the development of innovative anticancer drugs. J. Hepatol. 2013, 59, 1107–1117. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The WNT signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Levrero, M. Viral hepatitis and liver cancer: The case of hepatitis C. Oncogene 2006, 25, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Corcoran, R.B.; Welsh, J.W.; Pennica, D.; Levine, A.J. WISP-1 is a WNT-1- and β-catenin-responsive oncogene. Genes Dev. 2000, 14, 585–595. [Google Scholar] [PubMed]
- He, T.C.; Sparks, A.B.; Rago, C.; Hermeking, H.; Zawel, L.; da Costa, L.T.; Morin, P.J.; Vogelstein, B.; Kinzler, K.W. Identification of C-MYC as a target of the APC pathway. Science 1998, 281, 1509–1512. [Google Scholar] [CrossRef] [PubMed]
- Shtutman, M.; Zhurinsky, J.; Simcha, I.; Albanese, C.; D’Amico, M.; Pestell, R.; Ben-Ze’ev, A. The cyclin D1 gene is a target of the β-catenin/LEF-1 pathway. Proc. Natl. Acad. Sci. USA 1999, 96, 5522–5527. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Schilling, T.; Sayan, A.E.; Kairat, A.; Lorenz, K.; Schulze-Bergkamen, H.; Oren, M.; Koch, A.; Tannapfel, A.; Stremmel, W.; et al. TAP73/delta NP73 influences apoptotic response, chemosensitivity and prognosis in hepatocellular carcinoma. Cell Death Differ. 2005, 12, 1564–1577. [Google Scholar] [CrossRef] [PubMed]
- Zaika, A.I.; Slade, N.; Erster, S.H.; Sansome, C.; Joseph, T.W.; Pearl, M.; Chalas, E.; Moll, U.M. Deltanp73, a dominant-negative inhibitor of wild-type p53 and tap73, is up-regulated in human tumors. J. Exp. Med. 2002, 196, 765–780. [Google Scholar] [CrossRef] [PubMed]
- Moll, U.M.; Slade, N. P63 and p73: Roles in development and tumor formation. Mol. Cancer Res. 2004, 2, 371–386. [Google Scholar] [PubMed]
- Polakis, P. WNT signaling in cancer. Cold Spring Harb. Perspect. Biol. 2012, 4, a008052. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Fujii, H.; Sankila, A.; Mahler-Araujo, B.M.; Matsuda, M.; Cathomas, G.; Ohgaki, H. β-catenin mutations are frequent in human hepatocellular carcinomas associated with hepatitis C Virus infection. Am. J. Pathol. 1999, 155, 1795–1801. [Google Scholar] [CrossRef]
- Street, A.; Macdonald, A.; Crowder, K.; Harris, M. The hepatitis C Virus NS5A protein activates a phosphoinositide 3-kinase-dependent survival signaling cascade. J. Biol. Chem. 2004, 279, 12232–12241. [Google Scholar] [CrossRef] [PubMed]
- Fukutomi, T.; Zhou, Y.; Kawai, S.; Eguchi, H.; Wands, J.R.; Li, J. Hepatitis C Virus core protein stimulates hepatocyte growth: Correlation with upregulation of WNT-1 expression. Hepatology 2005, 41, 1096–1105. [Google Scholar] [CrossRef] [PubMed]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Harris, P.J.; Warren, R.Q.; Ivy, S.P. Targeting cancer stem cells by inhibiting WNT, notch, and hedgehog pathways. Nat. Rev. Clin. Oncol. 2011, 8, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Kopan, R.; Ilagan, M.X. The canonical notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 2014, 141, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Kao, H.Y.; Ordentlich, P.; Koyano-Nakagawa, N.; Tang, Z.; Downes, M.; Kintner, C.R.; Evans, R.M.; Kadesch, T. A histone deacetylase corepressor complex regulates the notch signal transduction pathway. Genes Dev. 1998, 12, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Lamar, E.; Deblandre, G.; Wettstein, D.; Gawantka, V.; Pollet, N.; Niehrs, C.; Kintner, C. Nrarp is a novel intracellular component of the notch signaling pathway. Genes Dev. 2001, 15, 1885–1899. [Google Scholar] [CrossRef] [PubMed]
- Ronchini, C.; Capobianco, A.J. Induction of cyclin d1 transcription and cdk2 activity by notch(IC): Implication for cell cycle disruption in transformation by notch(IC). Mol. Cell Biol. 2001, 21, 5925–5934. [Google Scholar] [CrossRef] [PubMed]
- Weng, A.P.; Millholland, J.M.; Yashiro-Ohtani, Y.; Arcangeli, M.L.; Lau, A.; Wai, C.; Del Bianco, C.; Rodriguez, C.G.; Sai, H.; Tobias, J.; et al. C-MYC is an important direct target of notch1 in t-cell acute lymphoblastic leukemia/lymphoma. Genes Dev. 2006, 20, 2096–2109. [Google Scholar] [CrossRef] [PubMed]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. Notch1 directly regulates c-myc and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.M.; Calvo, J.A.; Draheim, K.M.; Cunningham, L.A.; Hermance, N.; Beverly, L.; Krishnamoorthy, V.; Bhasin, M.; Capobianco, A.J.; Kelliher, M.A. Notch1 contributes to mouse t-cell leukemia by directly inducing the expression of C-MYC. Mol. Cell Biol. 2006, 26, 8022–8031. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martin, M.; Ferrando, A. The notch1-MYC highway toward t-cell acute lymphoblastic leukemia. Blood 2017, 129, 1124–1133. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fischer, W.H.; Gill, G.N. Regulation of the ERBB-2 promoter by RBPJκ and notch. J. Biol. Chem. 1997, 272, 14110–14114. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Zlobin, A.; Volgina, V.; Gottipati, S.; Osborne, B.; Simel, E.J.; Miele, L.; Gabrilovich, D.I. Notch-1 regulates NF-κB activity in hemopoietic progenitor cells. J. Immunol. 2001, 167, 4458–4467. [Google Scholar] [CrossRef] [PubMed]
- Iwai, A.; Takegami, T.; Shiozaki, T.; Miyazaki, T. Hepatitis C Virus NS3 protein can activate the notch-signaling pathway through binding to a transcription factor, Srcap. PLoS ONE 2011, 6, e20718. [Google Scholar] [CrossRef] [PubMed]
- Eissenberg, J.C.; Wong, M.; Chrivia, J.C. Human srcap and drosophila melanogaster dom are homologs that function in the notch signaling pathway. Mol. Cell Biol. 2005, 25, 6559–6569. [Google Scholar] [CrossRef] [PubMed]
- Kageyama, R.; Ohtsuka, T.; Kobayashi, T. The hes gene family: Repressors and oscillators that orchestrate embryogenesis. Development 2007, 134, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Choi, S.; Michelotti, G.; Diehl, A.M. Hedgehog signaling in the liver. J. Hepatol. 2011, 54, 366–373. [Google Scholar] [CrossRef] [PubMed]
- Briscoe, J.; Therond, P.P. The mechanisms of hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.V.; Diehl, A.M. Hedgehog signalling in liver pathophysiology. J. Hepatol. 2018, 68, 550–562. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, T.; Lu, J.; Cao, Y.; Song, N.; Yang, T.; Dong, R.; Yang, Y.; Zang, L.; Du, X.; et al. Immunohistochemical evidence of the prognostic value of hedgehog pathway components in primary gallbladder carcinoma. Surg. Today 2012, 42, 770–775. [Google Scholar] [CrossRef] [PubMed]
- Razumilava, N.; Gradilone, S.A.; Smoot, R.L.; Mertens, J.C.; Bronk, S.F.; Sirica, A.E.; Gores, G.J. Non-canonical hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol. 2014, 60, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Jinawath, A.; Akiyama, Y.; Sripa, B.; Yuasa, Y. Dual blockade of the hedgehog and ERK1/2 pathways coordinately decreases proliferation and survival of cholangiocarcinoma cells. J. Cancer Res. Clin Oncol. 2007, 133, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Guicciardi, M.E.; Cazanave, S.C.; Mertens, J.C.; Sirica, A.E.; Gores, G.J. Myofibroblast-derived PDGF-BB promotes hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 2011, 54, 2076–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichenmuller, M.; Gruner, I.; Hagl, B.; Haberle, B.; Muller-Hocker, J.; von Schweinitz, D.; Kappler, R. Blocking the hedgehog pathway inhibits hepatoblastoma growth. Hepatology 2009, 49, 482–490. [Google Scholar] [CrossRef] [PubMed]
- Patil, M.A.; Zhang, J.; Ho, C.; Cheung, S.T.; Fan, S.T.; Chen, X. Hedgehog signaling in human hepatocellular carcinoma. Cancer Biol. Ther. 2006, 5, 111–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omenetti, A.; Yang, L.; Li, Y.X.; McCall, S.J.; Jung, Y.; Sicklick, J.K.; Huang, J.; Choi, S.; Suzuki, A.; Diehl, A.M. Hedgehog-mediated mesenchymal-epithelial interactions modulate hepatic response to bile duct ligation. Lab. Investig. 2007, 87, 499–514. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.; Brown, K.D.; Witek, R.P.; Omenetti, A.; Yang, L.; Vandongen, M.; Milton, R.J.; Hines, I.N.; Rippe, R.A.; Spahr, L.; et al. Accumulation of hedgehog-responsive progenitors parallels alcoholic liver disease severity in mice and humans. Gastroenterology 2008, 134, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, A.; Popov, Y.; Jung, Y.; Choi, S.S.; Witek, R.P.; Yang, L.; Brown, K.D.; Schuppan, D.; Diehl, A.M. The hedgehog pathway regulates remodelling responses to biliary obstruction in rats. Gut 2008, 57, 1275–1282. [Google Scholar] [CrossRef] [PubMed]
- Pereira, T.d.A.; Witek, R.P.; Syn, W.-K.; Choi, S.S.; Bradrick, S.; Karaca, G.F.; Agboola, K.M.; Jung, Y.; Omenetti, A.; Moylan, C.A.; et al. Viral factors induce hedgehog pathway activation in humans with viral hepatitis, cirrhosis, and hepatocellular carcinoma. Lab. Investig. 2010, 90, 1690. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.S.; Bradrick, S.; Qiang, G.; Mostafavi, A.; Chaturvedi, G.; Weinman, S.A.; Diehl, A.M.; Jhaveri, R. Up-regulation of hedgehog pathway is associated with cellular permissiveness for hepatitis C Virus replication. Hepatology 2011, 54, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Van Renne, N.; Roca Suarez, A.A.; Duong, F.H.T.; Gondeau, C.; Calabrese, D.; Fontaine, N.; Ababsa, A.; Bandiera, S.; Croonenborghs, T.; Pochet, N.; et al. Mir-135a-5p-mediated downregulation of protein tyrosine phosphatase receptor delta is a candidate driver of hcv-associated hepatocarcinogenesis. Gut 2018, 67, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tian, X.J.; Xing, J. Signal transduction pathways of EMT induced by TGF-β, SHH, and WNT and their crosstalks. J. Clin Med. 2016, 5, 41. [Google Scholar] [CrossRef] [PubMed]
- Paternostro, C.; David, E.; Novo, E.; Parola, M. Hypoxia, angiogenesis and liver fibrogenesis in the progression of chronic liver diseases. World J. Gastroenterol. 2010, 16, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Valfre di Bonzo, L.; Novo, E.; Cannito, S.; Busletta, C.; Paternostro, C.; Povero, D.; Parola, M. Angiogenesis and liver fibrogenesis. Histol. Histopathol. 2009, 24, 1323–1341. [Google Scholar] [PubMed]
- Semela, D.; Dufour, J.F. Angiogenesis and hepatocellular carcinoma. J. Hepatol. 2004, 41, 864–880. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Yano, H.; Nishida, T.; Kamura, T.; Kojiro, M. Angiogenesis in cancer. Vasc. Health Risk Manag. 2006, 2, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, S.; Marais, R.; Zhu, A.X. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene 2010, 29, 4989–5005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mas, V.R.; Maluf, D.G.; Archer, K.J.; Yanek, K.C.; Fisher, R.A. Angiogenesis soluble factors as hepatocellular carcinoma noninvasive markers for monitoring hepatitis C Virus cirrhotic patients awaiting liver transplantation. Transplantation 2007, 84, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; El-Khattouti, A.; Soell, M.; Ghozlan, H.; Haikel, Y.; Abdelkader, O.; Megahed, M. Hepatitis C Virus-mediated angiogenesis: Molecular mechanisms and therapeutic strategies. World J. Gastroenterol. 2014, 20, 15467–15475. [Google Scholar] [CrossRef] [PubMed]
- Salcedo Mora, X.; Sanz-Cameno, P.; Medina, J.; Martin-Vilchez, S.; Garcia-Buey, L.; Borque, M.J.; Moreno-Otero, R. Association between angiogenesis soluble factors and disease progression markers in chronic hepatitis c patients. Rev. Esp. Enferm. Dig. 2005, 97, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.; Selimovic, D.; Ghozlan, H.; Abdel-kader, O. Hepatitis C Virus core protein triggers hepatic angiogenesis by a mechanism including multiple pathways. Hepatology 2009, 49, 1469–1482. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. What is the evidence that tumors are angiogenesis dependent? J. Natl. Cancer Inst. 1990, 82, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Nasimuzzaman, M.; Waris, G.; Mikolon, D.; Stupack, D.G.; Siddiqui, A. Hepatitis C Virus stabilizes hypoxia-inducible factor 1α and stimulates the synthesis of vascular endothelial growth factor. J. Virol. 2007, 81, 10249–10257. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Hofseth, L.J.; Harris, C.C. Radical causes of cancer. Nat. Rev. Cancer 2003, 3, 276–285. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Karin, M. NF-κB and STAT3-key players in liver inflammation and cancer. Cell Res. 2011, 21, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L. Immune regulation of cancer. J. Clin. Oncol. 2010, 28, 4531–4538. [Google Scholar] [CrossRef] [PubMed]
- Kondylis, V.; Kumari, S.; Vlantis, K.; Pasparakis, M. The interplay of IKK, NF-κB and RIPK1 signaling in the regulation of cell death, tissue homeostasis and inflammation. Immunol. Rev. 2017, 277, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Shen, S.; Verma, I.M. NF-κB, an active player in human cancers. Cancer Immunol. Res. 2014, 2, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. NF-κB as a critical link between inflammation and cancer. Cold Spring Harb. Perspect. Biol. 2009, 1, a000141. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Ghosh, S. The NF-κB family of transcription factors and its regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef] [PubMed]
- Tieri, P.; Termanini, A.; Bellavista, E.; Salvioli, S.; Capri, M.; Franceschi, C. Charting the NF-kappab pathway interactome map. PLoS ONE 2012, 7, e32678. [Google Scholar] [CrossRef] [PubMed]
- Bohm, F.; Kohler, U.A.; Speicher, T.; Werner, S. Regulation of liver regeneration by growth factors and cytokines. EMBO Mol. Med. 2010, 2, 294–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Nishio, Y.; Inoue, M.; Wang, X.J.; Wei, S.; Matsusaka, T.; Yoshida, K.; Sudo, T.; Naruto, M.; Kishimoto, T. Molecular cloning of aprf, a novel ifn-stimulated gene factor 3 p91-related transcription factor involved in the GP130-mediated signaling pathway. Cell 1994, 77, 63–71. [Google Scholar] [CrossRef]
- Mackey-Lawrence, N.M.; Petri, W.A., Jr. Leptin and mucosal immunity. Mucosal. Immunol. 2012, 5, 472–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCartney, E.M.; Helbig, K.J.; Narayana, S.K.; Eyre, N.S.; Aloia, A.L.; Beard, M.R. Signal transducer and activator of transcription 3 is a proviral host factor for hepatitis C Virus. Hepatology 2013, 58, 1558–1568. [Google Scholar] [CrossRef] [PubMed]
- Wilks, A.F.; Harpur, A.G.; Kurban, R.R.; Ralph, S.J.; Zurcher, G.; Ziemiecki, A. Two novel protein-tyrosine kinases, each with a second phosphotransferase-related catalytic domain, define a new class of protein kinase. Mol. Cell Biol. 1991, 11, 2057–2065. [Google Scholar] [CrossRef] [PubMed]
- Firmbach-Kraft, I.; Byers, M.; Shows, T.; Dalla-Favera, R.; Krolewski, J.J. Tyk2, prototype of a novel class of non-receptor tyrosine kinase genes. Oncogene 1990, 5, 1329–1336. [Google Scholar] [PubMed]
- Kaptein, A.; Paillard, V.; Saunders, M. Dominant negative STAT3 mutant inhibits interleukin-6-induced JAK-STAT signal transduction. J. Biol. Chem. 1996, 271, 5961–5964. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chatterjee-Kishore, M.; Staugaitis, S.M.; Nguyen, H.; Schlessinger, K.; Levy, D.E.; Stark, G.R. Novel roles of unphosphorylated STAT3 in oncogenesis and transcriptional regulation. Cancer Res. 2005, 65, 939–947. [Google Scholar] [PubMed]
- Yang, J.; Liao, X.; Agarwal, M.K.; Barnes, L.; Auron, P.E.; Stark, G.R. Unphosphorylated STAT3 accumulates in response to Il-6 and activates transcription by binding to nfkappab. Genes Dev. 2007, 21, 1396–1408. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Kamata, H.; Luo, J.L.; Leffert, H.; Karin, M. Ikkβ couples hepatocyte death to cytokine-driven compensatory proliferation that promotes chemical hepatocarcinogenesis. Cell 2005, 121, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Herrmann, A.; Deng, J.H.; Kujawski, M.; Niu, G.; Li, Z.; Forman, S.; Jove, R.; Pardoll, D.M.; Yu, H. Persistently activated STAT3 maintains constitutive NF-kappab activity in tumors. Cancer Cell 2009, 15, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.G.; Bubici, C.; Zazzeroni, F.; Papa, S.; Jones, J.; Alvarez, K.; Jayawardena, S.; de Smaele, E.; Cong, R.; Beaumont, C.; et al. Ferritin heavy chain upregulation by NF-kappab inhibits TNFα-induced apoptosis by suppressing reactive oxygen species. Cell 2004, 119, 529–542. [Google Scholar] [CrossRef] [PubMed]
- He, G.; Yu, G.Y.; Temkin, V.; Ogata, H.; Kuntzen, C.; Sakurai, T.; Sieghart, W.; Peck-Radosavljevic, M.; Leffert, H.L.; Karin, M. Hepatocyte Ikkβ/NF-κB inhibits tumor promotion and progression by preventing oxidative stress-driven STAT3 activation. Cancer Cell 2010, 17, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Seeff, L.B. Introduction: The burden of hepatocellular carcinoma. Gastroenterology 2004, 127, S1–S4. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C Virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Marrone, A.; Restivo, L.; Guerrera, B.; Sellitto, A.; Rinaldi, L.; Romano, C.; Adinolfi, L.E. Chronic hcv infection and inflammation: Clinical impact on hepatic and extra-hepatic manifestations. World J. Hepatol. 2013, 5, 528–540. [Google Scholar] [CrossRef] [PubMed]
- Roca Suarez, A.A.; van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kang, W.; Ryu, S.W.; Kim, W.I.; Chang, D.Y.; Lee, D.H.; Park, D.Y.; Choi, Y.H.; Choi, K.; Shin, E.C.; et al. Hepatitis C Virus infection enhances TNFα-induced cell death via suppression of NF-κB. Hepatology 2012, 56, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Machida, K.; Cheng, K.T.; Lai, C.K.; Jeng, K.S.; Sung, V.M.; Lai, M.M. Hepatitis C Virus triggers mitochondrial permeability transition with production of reactive oxygen species, leading to DNA damage and stat3 activation. J. Virol. 2006, 80, 7199–7207. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.B.; Sasaki, R.; Shrivastava, S.; Di Bisceglie, A.M.; Ray, R.; Ray, R.B. Exosome-mediated intercellular communication between hepatitis C Virus-infected hepatocytes and hepatic stellate cells. J. Virol. 2017, 91, JVI–02225. [Google Scholar] [CrossRef] [PubMed]
- Wan, S.; Kuo, N.; Kryczek, I.; Zou, W.; Welling, T.H. Myeloid cells in hepatocellular carcinoma. Hepatology 2015, 62, 1304–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Hanada, T.; Tokuhisa, T.; Kosai, K.; Sata, M.; Kohara, M.; Yoshimura, A. Activation of STAT3 by the hepatitis C Virus core protein leads to cellular transformation. J. Exp. Med. 2002, 196, 641–653. [Google Scholar] [CrossRef] [PubMed]
- Baumert, T.F.; Juhling, F.; Ono, A.; Hoshida, Y. Hepatitis C-related hepatocellular carcinoma in the era of new generation antivirals. BMC Med. 2017, 15, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Meer, A.J.; Feld, J.J.; Hofer, H.; Almasio, P.L.; Calvaruso, V.; FeRNAndez-Rodriguez, C.M.; Aleman, S.; Ganne-Carrie, N.; D’Ambrosio, R.; Pol, S.; et al. Risk of cirrhosis-related complications in patients with advanced fibrosis following hepatitis C Virus eradication. J. Hepatol. 2017, 66, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Heckley, G.A.; Jarl, J.; Asamoah, B.O.; U, G.G. How the risk of liver cancer changes after alcohol cessation: A review and meta-analysis of the current literature. BMC Cancer 2011, 11, 446. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Seitz, H.K.; Bataller, R.; Cortez-Pinto, H.; Gao, B.; Gual, A.; Lackner, C.; Mathurin, P.; Mueller, S.; Szabo, G.; Tsukamoto, H. Publisher correction: Alcoholic liver disease. Nat. Rev. Dis. Primers 2018, 4, 18. [Google Scholar] [CrossRef] [PubMed]
- Conti, F.; Buonfiglioli, F.; Scuteri, A.; Crespi, C.; Bolondi, L.; Caraceni, P.; Foschi, F.G.; Lenzi, M.; Mazzella, G.; Verucchi, G.; et al. Early occurrence and recurrence of hepatocellular carcinoma in hcv-related cirrhosis treated with direct-acting antivirals. J. Hepatol. 2016, 65, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Reig, M.; Marino, Z.; Perello, C.; Inarrairaegui, M.; Ribeiro, A.; Lens, S.; Diaz, A.; Vilana, R.; Darnell, A.; Varela, M.; et al. Unexpected high rate of early tumor recurrence in patients with hcv-related hcc undergoing interferon-free therapy. J. Hepatol. 2016, 65, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Cheung, M.C.M.; Walker, A.J.; Hudson, B.E.; Verma, S.; McLauchlan, J.; Mutimer, D.J.; Brown, A.; Gelson, W.T.H.; MacDonald, D.C.; Agarwal, K.; et al. Outcomes after successful direct-acting antiviral therapy for patients with chronic hepatitis c and decompensated cirrhosis. J. Hepatol. 2016, 65, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Mettke, F.; Schlevogt, B.; Deterding, K.; Wranke, A.; Smith, A.; Port, K.; Manns, M.P.; Vogel, A.; Cornberg, M.; Wedemeyer, H. Interferon-free therapy of chronic hepatitis C with direct-acting antivirals does not change the short-term risk for de novo hepatocellular carcinoma in patients with liver cirrhosis. Aliment. Pharmacol. Ther. 2018, 47, 516–525. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. European Association for the Study of the, L. Easl recommendations on treatment of hepatitis c 2018. J. Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [PubMed]
- Pinter, M.; Trauner, M.; Peck-Radosavljevic, M.; Sieghart, W. Cancer and liver cirrhosis: Implications on prognosis and management. ESMO Open 2016, 1, e000042. [Google Scholar] [CrossRef] [PubMed]
- Bandiera, S.; Billie Bian, C.; Hoshida, Y.; Baumert, T.F.; Zeisel, M.B. Chronic hepatitis C Virus infection and pathogenesis of hepatocellular carcinoma. Curr. Opin. Virol. 2016, 20, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M. Sorafenib: A review in hepatocellular carcinoma. Target. Oncol. 2017, 12, 243–253. [Google Scholar] [CrossRef] [PubMed]
- Cainap, C.; Qin, S.; Huang, W.T.; Chung, I.J.; Pan, H.; Cheng, Y.; Kudo, M.; Kang, Y.K.; Chen, P.J.; Toh, H.C.; et al. Linifanib versus sorafenib in patients with advanced hepatocellular carcinoma: Results of a randomized phase iii trial. J. Clin. Oncol. 2015, 33, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Rosmorduc, O.; Evans, T.R.; Ross, P.J.; Santoro, A.; Carrilho, F.J.; Bruix, J.; Qin, S.; Thuluvath, P.J.; Llovet, J.M.; et al. Search: A phase III, randomized, double-blind, placebo-controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J. Clin. Oncol. 2015, 33, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; HeRNAndez-Gea, V. Hepatocellular carcinoma: Reasons for phase III failure and novel perspectives on trial design. Clin. Cancer Res. 2014, 20, 2072–2079. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, S.; Wei, L.; Song, W.M.; Higashi, T.; Ghoshal, S.; Kim, R.S.; Bian, C.B.; Yamada, S.; Sun, X.; Venkatesh, A.; et al. Molecular liver cancer prevention in cirrhosis by organ transcriptome analysis and lysophosphatidic acid pathway inhibition. Cancer Cell 2016, 30, 879–890. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hepatitis C Virus (HCV)-induced oncogenic signaling. HCV infection creates a procarcinogenic effect through the simultaneous dysregulation of cell survival, proliferation, inflammatory, angiogenic, and differentiation signaling pathways. The tight control of target genes involved in transcriptional regulation and cell cycle progression is altered by HCV via different strategies. Forcing p53 in the cytoplasm, NS5A prevents the gene expression of cyclin-dependent kinase inhibitor p21 (not shown). This cytoplasmic-retention strategy is also shared by NS5B, which traps pRb in the cytoplasm. Consequently, E2F is free to act as transcriptional activator for cell proliferation target genes. Core protein, which is preferentially localized in the cytoplasm, translocates to the nucleus, where it interferes with transforming growth factor-beta (TGF-β) signaling via Smad3 interaction. Epidermal growth factor receptor (EGFR) and mitogen-activated protein kinase (MAPK) signaling are not only required for HCV entry but also represent oncogenic targets for HCV-encoded proteins. Both NS5A and core protein induce the activation of signal transducer and activator of transcription 3 (STAT3) by indirect (inhibiting the suppressor of cytokine signaling 3, SOCS3) and direct mechanisms, respectively. Following its translocation to the nucleus, STAT3 strongly promotes a proinflammatory environment in cooperation with nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling. Furthermore, STAT3 and NF-κB, together with PI3K, induce hypoxia-inducible factor 1-alpha (HIF-1α) stabilization, which mediates the transcription of several proangiogenic factors (e.g., vascular endothelial growth factor, VEGF). HCV impairs cell differentiation programs by manipulating Wnt and Notch signaling pathways. NS5A induces a sustained Wnt signaling activation through the PI3K/Akt axis. This leads to the inactivation of a downstream degradation complex and the consequent accumulation of β-catenin in the nucleus, where it activates the expression of cell proliferation-related genes. NS3 stimulates downstream components of Notch pathway by the recruitment of CREB-binding protein (CBP)/p300 complex on Snf2-related CBP activator (SRCAP), repressing cell differentiation programs. TGFR-1: TGF-β receptor 1; FZD: Wnt receptor (Frizzled).
Figure 1.
Hepatitis C Virus (HCV)-induced oncogenic signaling. HCV infection creates a procarcinogenic effect through the simultaneous dysregulation of cell survival, proliferation, inflammatory, angiogenic, and differentiation signaling pathways. The tight control of target genes involved in transcriptional regulation and cell cycle progression is altered by HCV via different strategies. Forcing p53 in the cytoplasm, NS5A prevents the gene expression of cyclin-dependent kinase inhibitor p21 (not shown). This cytoplasmic-retention strategy is also shared by NS5B, which traps pRb in the cytoplasm. Consequently, E2F is free to act as transcriptional activator for cell proliferation target genes. Core protein, which is preferentially localized in the cytoplasm, translocates to the nucleus, where it interferes with transforming growth factor-beta (TGF-β) signaling via Smad3 interaction. Epidermal growth factor receptor (EGFR) and mitogen-activated protein kinase (MAPK) signaling are not only required for HCV entry but also represent oncogenic targets for HCV-encoded proteins. Both NS5A and core protein induce the activation of signal transducer and activator of transcription 3 (STAT3) by indirect (inhibiting the suppressor of cytokine signaling 3, SOCS3) and direct mechanisms, respectively. Following its translocation to the nucleus, STAT3 strongly promotes a proinflammatory environment in cooperation with nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling. Furthermore, STAT3 and NF-κB, together with PI3K, induce hypoxia-inducible factor 1-alpha (HIF-1α) stabilization, which mediates the transcription of several proangiogenic factors (e.g., vascular endothelial growth factor, VEGF). HCV impairs cell differentiation programs by manipulating Wnt and Notch signaling pathways. NS5A induces a sustained Wnt signaling activation through the PI3K/Akt axis. This leads to the inactivation of a downstream degradation complex and the consequent accumulation of β-catenin in the nucleus, where it activates the expression of cell proliferation-related genes. NS3 stimulates downstream components of Notch pathway by the recruitment of CREB-binding protein (CBP)/p300 complex on Snf2-related CBP activator (SRCAP), repressing cell differentiation programs. TGFR-1: TGF-β receptor 1; FZD: Wnt receptor (Frizzled).

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Virzì, A.; Roca Suarez, A.A.; Baumert, T.F.; Lupberger, J. Oncogenic Signaling Induced by HCV Infection. Viruses 2018, 10, 538. https://doi.org/10.3390/v10100538
AMA Style
Virzì A, Roca Suarez AA, Baumert TF, Lupberger J. Oncogenic Signaling Induced by HCV Infection. Viruses. 2018; 10(10):538. https://doi.org/10.3390/v10100538
Chicago/Turabian StyleVirzì, Alessia, Armando Andres Roca Suarez, Thomas F. Baumert, and Joachim Lupberger. 2018. "Oncogenic Signaling Induced by HCV Infection" Viruses 10, no. 10: 538. https://doi.org/10.3390/v10100538
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.