New Insights into the Pros and Cons of the Clinical Use of Vitamin K Antagonists (VKAs) Versus Direct Oral Anticoagulants (DOACs)



Abstract
:1. Arterial and Venous Thrombosis

2. The Discovery of Oral Anticoagulant Drugs
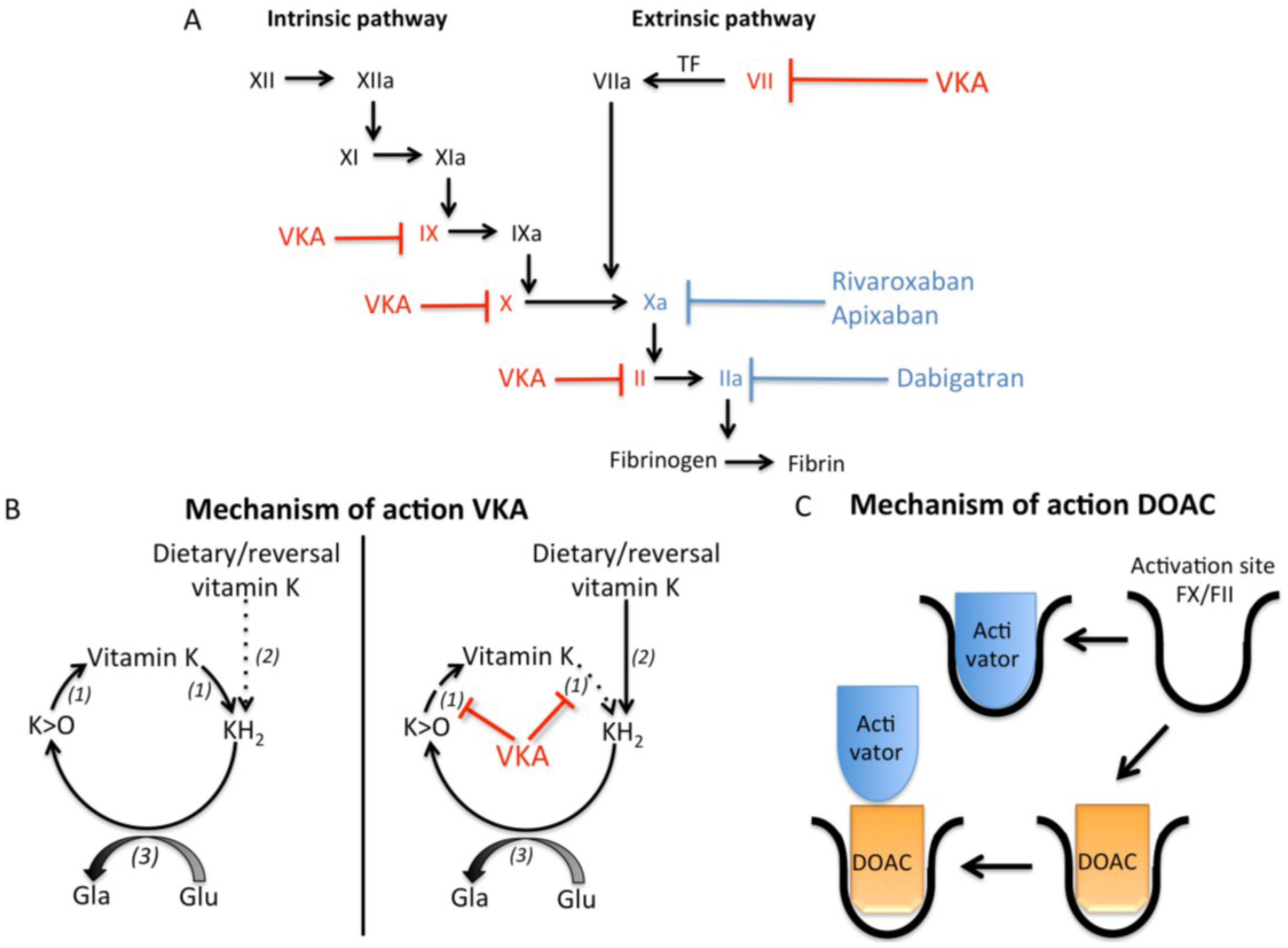
2.1. Vitamin K and Vitamin K Antagonists

{kind=link}
{kind=link}
{kind=link}
| Drug | Characterization | Dietary Sources | |
|---|---|---|---|
| Phylloquinone (vitamin K1) | Phytyl side chain | Leafy green vegetables | |
| Menaquinones (vitamin K2) | Isoprenoid side chain | MK-4 | Meat, eggs |
| MK-7 | Natto, Cheese | ||
| MK-9 | Cheese, curd, sauerkraut | ||
| Menadione (vitamin K3) | 2-methyl-1,4- naphthoquinone | Non-dietary metabolite. Precursor of MK-4 | |
2.2. Direct Thrombin Inhibitors
| VKA [10] | Dabigatran Etexilate [24,27,28] | Rivaroxaban [30,31] | Apixaban [32] | |||
|---|---|---|---|---|---|---|
| Warfarin | Acenocoumarol | Phenprocoumon | ||||
| Target | Vitamin K epoxide reductase | Vitamin K epoxide reductase | Vitamin K epoxide reductase | Thrombin | Factor Xa | Factor Xa |
| Pro-drug | No | No | No | Yes, active metabolite is dabigatran | No | No |
| Half-life (hours) | 20–60 | 8–11 | 120–144 | 12–17 | 5–9 | 9–14 |
| Onset time peak effect (hours) | 72–96 | 36–48 | 48–72 | 2 | 2–3 | 3 |
| Duration of action | 2–5 days | <48 h | 7–14 days | 24–36 h | 24 h | 24 h |
| Metabolism | Via cytochrome P 450 | Via cytochrome P 450 | Via cytochrome P 450 | Via P-Glucoprotein transporter | Via cytochrome P450 (30%), and P-Glucoprotein transporter | Via cytochrome P450 (15%), and P-Glucoprotein transporter |
| Elimination | Hepatical metabolized | 60% Renal | 63% Renal | 85% Renal | 66% Renal | 25% Renal |
| 29% Fecal | 33% Fecal | 6% Fecal | 28% Fecal | |||
| Bioavailability | 79%–100% | 60% | >99% | 6.5% | 80% | 66% |
2.3. Factor Xa Inhibitors
3. Clinical Trials with Oral Anticoagulation Drugs
3.1. Vitamin K Antagonists (VKA)
3.2. DOACs
3.2.1. Dabigatran Etexilate
3.2.2. Rivaroxaban
3.2.3. Apixaban
4. Advantages and Disadvantages of VKA and DOACs
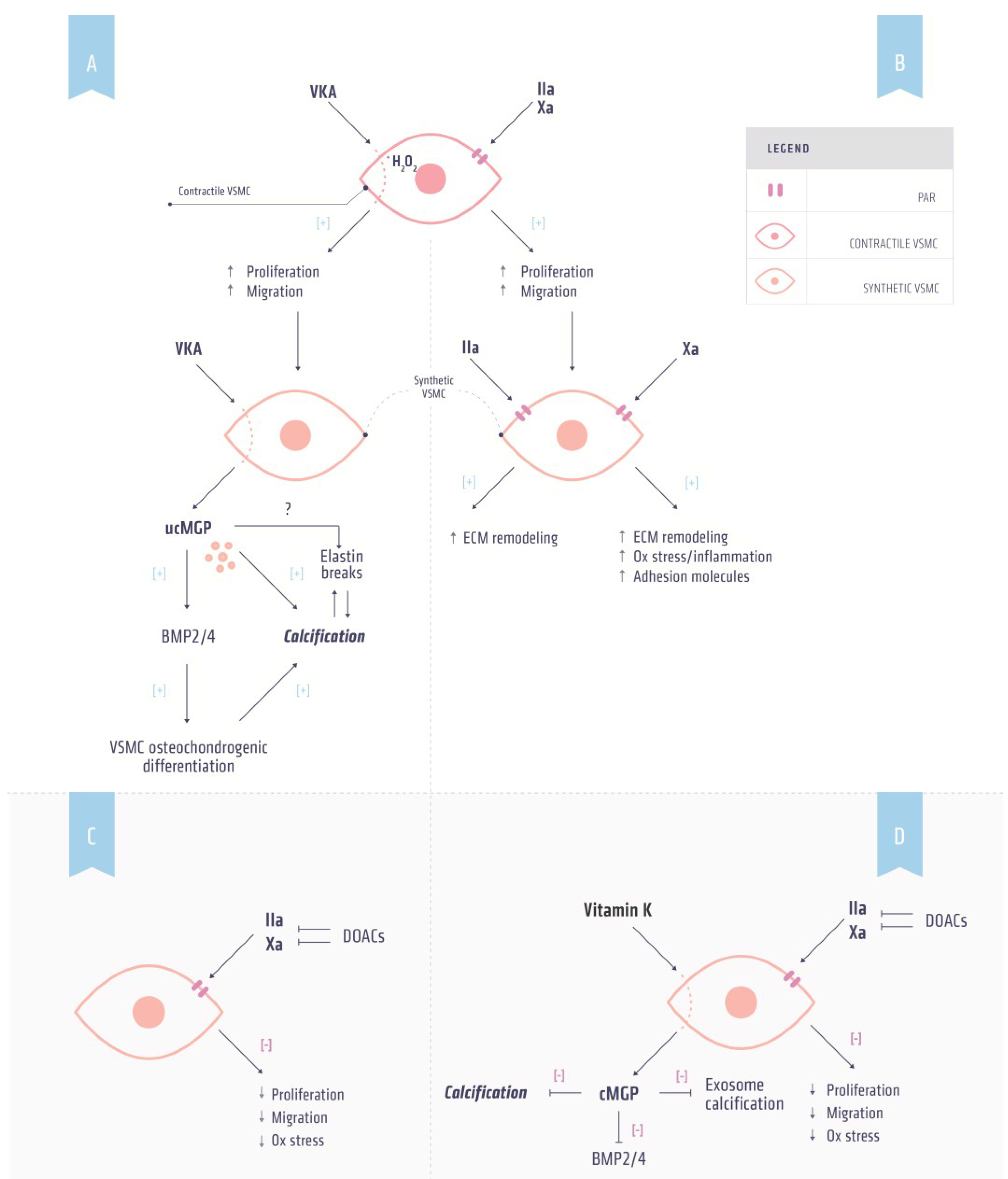
5. Vitamin K Dependent Proteins and Atherosclerosis
5.1. Coagulation and Atherosclerosis
5.2. Thrombin and Atherosclerosis

5.3. Factor Xa and Atherosclerosis
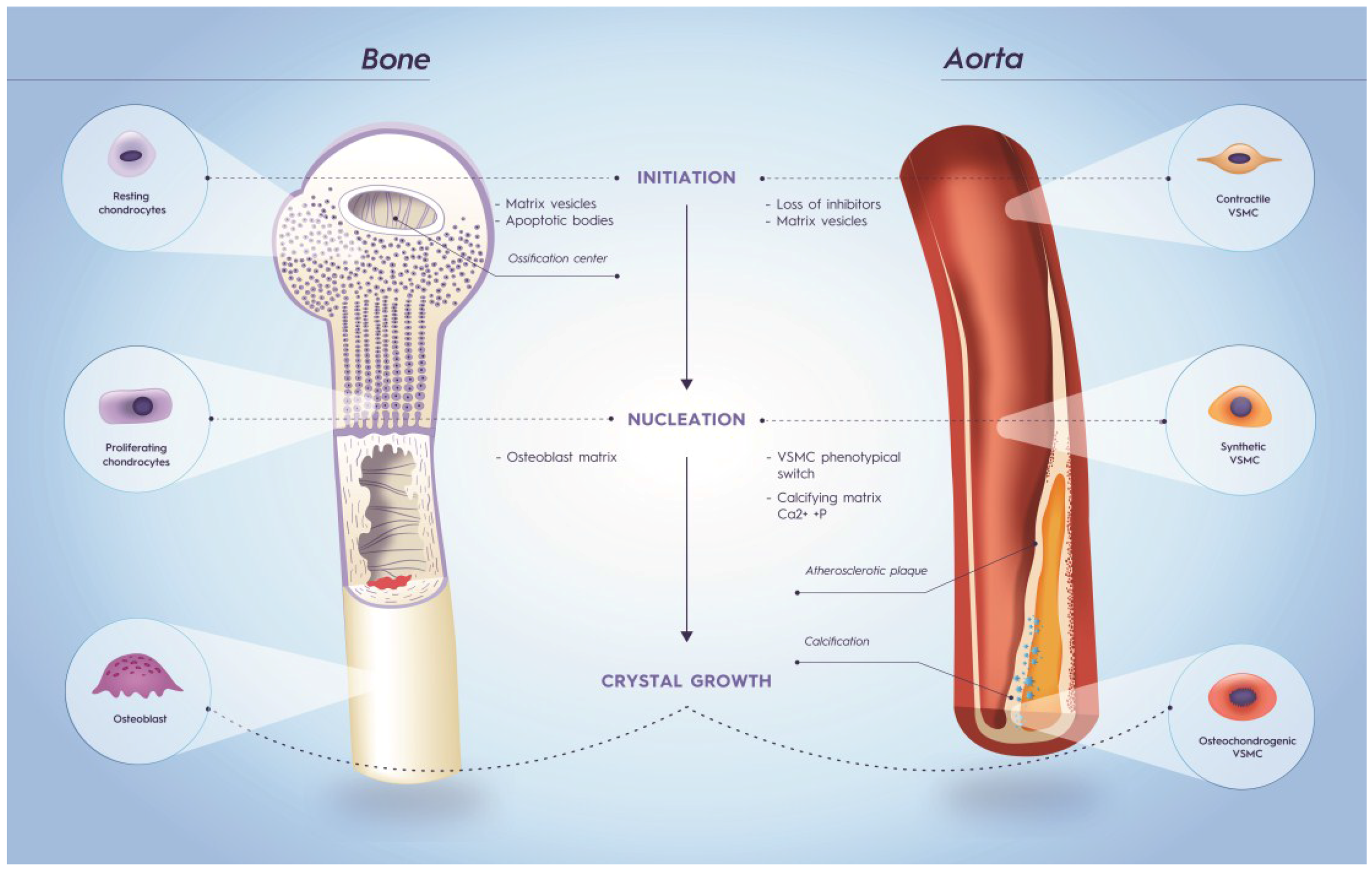
6. Vitamin K Dependent Proteins and Calcification
6.1. Osteocalcin

6.2. Matrix Gla Protein
6.3. Gla Rich Protein
7. Role for Vitamin K in Cardiovascular Disease
8. Vitamin K and Direct oral Anticoagulation
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mann, K.G. Biochemistry and physiology of blood coagulation. Thromb. Haemost. 1999, 82, 165–174. [Google Scholar] [PubMed]
- Macfarlane, R.G. An enzyme cascade in the blood clotting mechanism, and its function as a biochemical amplifier. Nature 1964, 202, 498–499. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.W. Are hemostasis and thrombosis two sides of the same coin? J. Exp. Med. 2006, 203, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L. Atherosclerosis and thrombosis: Lessons from animal models. Thromb. Haemost. 2001, 86, 356–365. [Google Scholar] [PubMed]
- Fuster, V.; Stein, B.; Ambrose, J.A.; Badimon, L.; Badimon, J.J.; Chesebro, J.H. Atherosclerotic plaque rupture and thrombosis. Evolving concepts. Circulation 1990, 82, II47–II59. [Google Scholar] [PubMed]
- Rosendaal, F.R. Venous thrombosis: A multicausal disease. Lancet 1999, 353, 1167–1173. [Google Scholar] [CrossRef]
- Schofield, F.W. A brief account of a disease in cattle simulating hemorrhagic septicaemia due to feeding sweet clover. Can. Vet. J. Rev. Vétérinaire Can. 1984, 25, 453–455. [Google Scholar]
- Link, K. The discovery of dicumarol and its sequels. Circulation 1959, 19, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, S.; Ciferri, F.E. Intramuscular administration of the anticoagulant warfarin (coumadin) sodium. J. Am. Med. Assoc. 1957, 165, 1377–1380. [Google Scholar] [CrossRef] [PubMed]
- Beinema, M.; Brouwers, J.R.B.J.; Schalekamp, T.; Wilffert, B. Pharmacogenetic differences between warfarin, acenocoumarol and phenprocoumon. Thromb. Haemost. 2008, 100, 1052–1057. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Booth, S.L.; van den Heuvel, E.G.H.M.; Stoecklin, E.; Baka, A.; Vermeer, C. The role of menaquinones (vitamin K2) in human health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Vermeer, C. Determination of phylloquinone and menaquinones in food. Eff. Food Matrix Circ. Vitam. K Conc. 2000, 30, 298–307. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Vermeer, C. Differential lipoprotein transport pathways of K-vitamins in healthy subjects. Biochim. Biophys. Acta 2002, 1570, 27–32. [Google Scholar] [CrossRef]
- Schurgers, L.J.; Teunissen, K.J.F.; Hamulyak, K.; Knapen, M.H.J.; Vik, H.; Vermeer, C. Vitamin K-containing dietary supplements: Comparison of synthetic vitamin K1 and natto-derived menaquinone-7. Blood 2007, 109, 3279–3283. [Google Scholar] [CrossRef] [PubMed]
- Thijssen, H.H.W.; Vervoort, L.M.T.; Schurgers, L.J.; Shearer, M.J. Menadione is a metabolite of oral vitamin K. Br. J. Nutr. 2006, 95, 260–266. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.; Sadowski, J.; Suttie, J. A new carboxylation reaction. The vitamin K-dependent incorporation of H-14-CO3- into prothrombin. J. Biol. Chem. 1975, 250, 4744–4748. [Google Scholar] [PubMed]
- Willems, B.A.G.; Vermeer, C.; Reutelingsperger, C.P.M.; Schurgers, L.J. The realm of vitamin K dependent proteins: Shifting from coagulation toward calcification. Mol. Nutr. Food Res. 2014, 58, 1620–1635. [Google Scholar] [CrossRef] [PubMed]
- Van Horn, W.D. Structural and functional insights into human vitamin K epoxide reductase and vitamin K epoxide reductase-like1. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Stafford, D. The vitamin K cycle. J. Thromb. Haemost. 2005, 3, 1873–1878. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I.; Hirsh, J. New antithrombotic agents. Chest 1998, 114, 715S–727S. [Google Scholar] [CrossRef] [PubMed]
- Van Aken, H.; Bode, C.; Darius, H.; Diehm, C.; Encke, A.; Gulba, D.C.; Haas, S.; Hacke, W.; Puhl, W.; Quante, M.; et al. State-of-the-art review: Anticoagulation: The present and future. Clin. Appl. Thromb. Hemost. 2001, 7, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Greinacher, A.; Warkentin, T.E. The direct thrombin inhibitor hirudin. Thromb. Haemost. 2008, 99, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.I.; Wille-Jørgensen, P.; Kälebo, P.; Mouret, P.; Rosencher, N.; Bösch, P.; Baur, M.; Ekman, S.; Bach, D.; Lindbratt, S.; et al. A comparison of recombinant hirudin with a low-molecular-weight heparin to prevent thromboembolic complications after total hip replacement. N. Engl. J. Med. 1997, 337, 1329–1335. [Google Scholar] [CrossRef] [PubMed]
- Investigators, G.L. Randomized trial of intravenous heparin versus recombinant hirudin for acute coronary syndromes. The Global Use of Strategies to Open Occluded Coronary Arteries (GUSTO) IIa Investigators. Circulation 1994, 90, 1631–1637. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Kearon, C.; Kakkar, A.K.; Mismetti, P.; Schellong, S.; Eriksson, H.; Baanstra, D.; Schnee, J.; Goldhaber, S.Z. RE-COVER Study Group Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N. Engl. J. Med. 2009, 361, 2342–2352. [Google Scholar] [CrossRef] [PubMed]
- Schulman, S.; Wåhlander, K.; Lundström, T.; Clason, S.B.; Eriksson, H. THRIVE III Investigators Secondary prevention of venous thromboembolism with the oral direct thrombin inhibitor ximelagatran. N. Engl. J. Med. 2003, 349, 1713–1721. [Google Scholar] [CrossRef] [PubMed]
- Laizure, S.C.; Parker, R.B.; Herring, V.L.; Hu, Z.-Y. Identification of carboxylesterase-dependent dabigatran etexilate hydrolysis. Drug Metab. Dispos. Biol. Fate Chem. 2014, 42, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Akwaa, F.; Spyropoulos, A.C. The potential of target-specific oral anticoagulants for the acute and long-term treatment of venous thromboembolism. Curr. Med. Res. Opin. 2014, 30, 2179–2190. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Hogg, K.; Weitz, J.I. Overview of the New Oral Anticoagulants: Opportunities and Challenges. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Becka, M.; Wensing, G.; Voith, B.; Zuehlsdorf, M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59–7939—An oral, direct Factor Xa inhibitor—After multiple dosing in healthy male subjects. Eur. J. Clin. Pharmacol. 2005, 61, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Haas, S. Novel factor Xa inhibitors for prevention and treatment of thromboembolic diseases. Expert Opin. Investig. Drugs 2006, 15, 843–855. [Google Scholar] [CrossRef] [PubMed]
- Weitz, J.I. Emerging anticoagulants for the treatment of venous thromboembolism. Thromb. Haemost. 2006, 96, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Nutt, E.M.; Jain, D.; Lenny, A.B.; Schaffer, L.; Siegl, P.K.; Dunwiddie, C.T. Purification and characterization of recombinant antistasin: A leech-derived inhibitor of coagulation factor Xa. Arch. Biochem. Biophys. 1991, 285, 37–44. [Google Scholar] [CrossRef]
- Yeh, C.H.; Fredenburgh, J.C.; Weitz, J.I. Oral direct factor Xa inhibitors. Circ. Res. 2012, 111, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Perzborn, E.; Strassburger, J.; Wilmen, A.; Pohlmann, J.; Roehrig, S.; Schlemmer, K.-H.; Straub, A. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—An oral, direct Factor Xa inhibitor. J. Thromb. Haemost. 2005, 3, 514–521. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Spronk, H.M.H. Differential cellular effects of old and new oral anticoagulants: Consequences to the genesis and progression of atherosclerosis. Thromb. Haemost. 2014, 112, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, R. A clinical and pharmacologic assessment of once-daily versus twice-daily dosing for rivaroxaban. J. Thromb. Thrombolysis 2014, 38, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Barritt, D.W.; Jordan, S.C. Anticoagulant drugs in the treatment of pulmonary embolism. A controlled trial. Lancet 1960, 1, 1309–1312. [Google Scholar] [CrossRef]
- Duxbury, B.M.; Poller, L. The oral anticoagulant saga: Past, present, and future. Clin. Appl. Thromb. Hemost. 2001, 7, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.; Hirsh, J.; Jay, R.; Carter, C.; England, C.; Gent, M.; Turpie, A.G.; McLoughlin, D.; Dodd, P.; Thomas, M.; et al. Different intensities of oral anticoagulant therapy in the treatment of proximal-vein thrombosis. N. Engl. J. Med. 1982, 307, 1676–1681. [Google Scholar] [CrossRef] [PubMed]
- Guyatt, G.H.; Akl, E.A.; Crowther, M.; Gutterman, D.D.; Schuünemann, H.J. Executive summary: Antithrombotic therapy and prevention of thrombosis, 9th ed.: American college of chest physicians evidence-based clinical practice guidelines. Chest 2012, 141, 7S–47S. [Google Scholar] [CrossRef] [PubMed]
- Connolly, S.J.; Ezekowitz, M.D.; Yusuf, S.; Eikelboom, J.; Oldgren, J.; Parekh, A.; Pogue, J.; Reilly, P.A.; Themeles, E.; Varrone, J.; et al. RE-LY Steering Committee and Investigators Dabigatran versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2009, 361, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.I.; Dahl, O.E.; Rosencher, N.; Kurth, A.A.; van Dijk, C.N.; Frostick, S.P.; Kälebo, P.; Christiansen, A.V.; Hantel, S.; Hettiarachchi, R.; et al. RE-MODEL Study Group Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: The RE-MODEL randomized trial. J. Thromb. Haemost. 2007, 5, 2178–2185. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Mahaffey, K.W.; Garg, J.; Pan, G.; Singer, D.E.; Hacke, W.; Breithardt, G.; Halperin, J.L.; Hankey, G.J.; Piccini, J.P.; et al. ROCKET AF Investigators Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N. Engl. J. Med. 2011, 365, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.I.; Borris, L.C.; Friedman, R.J.; Haas, S.; Huisman, M.V.; Kakkar, A.K.; Bandel, T.J.; Beckmann, H.; Muehlhofer, E.; Misselwitz, F.; et al. RECORD1 Study Group Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N. Engl. J. Med. 2008, 358, 2765–2775. [Google Scholar] [CrossRef] [PubMed]
- EINSTEIN Investigators; Bauersachs, R.; Berkowitz, S.D.; Brenner, B.; Büller, H.R.; Decousus, H.; Gallus, A.S.; Lensing, A.W.; Misselwitz, F.; Prins, M.H.; et al. Oral rivaroxaban for symptomatic venous thromboembolism. N. Engl. J. Med. 2010, 363, 2499–2510. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, B.I.; Kakkar, A.K.; Turpie, A.G.G.; Gent, M.; Bandel, T.-J.; Homering, M.; Misselwitz, F.; Lassen, M.R. Oral rivaroxaban for the prevention of symptomatic venous thromboembolism after elective hip and knee replacement. J. Bone Joint Surg. Br. 2009, 91, 636–644. [Google Scholar] [CrossRef] [PubMed]
- Granger, C.B.; Alexander, J.H.; McMurray, J.J.V.; Lopes, R.D.; Hylek, E.M.; Hanna, M.; Al-Khalidi, H.R.; Ansell, J.; Atar, D.; Avezum, A.; et al. ARISTOTLE Committees and Investigators Apixaban versus warfarin in patients with atrial fibrillation. N. Engl. J. Med. 2011, 365, 981–992. [Google Scholar] [CrossRef] [PubMed]
- Lassen, M.R.; Gallus, A.; Raskob, G.E.; Pineo, G.; Chen, D.; Ramirez, L.M. ADVANCE-3 Investigators Apixaban versus enoxaparin for thromboprophylaxis after hip replacement. N. Engl. J. Med. 2010, 363, 2487–2498. [Google Scholar] [CrossRef] [PubMed]
- Van Es, N.; Coppens, M.; Schulman, S.; Middeldorp, S.; Büller, H.R. Direct oral anticoagulants compared with vitamin K antagonists for acute venous thromboembolism: Evidence from phase 3 trials. Blood 2014, 124, 1968–1975. [Google Scholar] [CrossRef] [PubMed]
- Haas, S. Medical indications and considerations for future clinical decision making. Thromb. Res. 2003, 109 (Suppl. 1), S31–S37. [Google Scholar] [CrossRef]
- McCabe, K.M.; Booth, S.L.; Fu, X.; Shobeiri, N.; Pang, J.J.; Adams, M.A.; Holden, R.M. Dietary vitamin K and therapeutic warfarin alter the susceptibility to vascular calcification in experimental chronic kidney disease. Kidney Int. 2013, 83, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Chatrou, M.L.L.; Winckers, K.; Hackeng, T.M.; Reutelingsperger, C.P.; Schurgers, L.J. Vascular calcification: The price to pay for anticoagulation therapy with vitamin K-antagonists. Blood Rev. 2012, 26, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Rosenhek, R.; Binder, T.; Porenta, G.; Lang, I.; Christ, G.; Schemper, M.; Maurer, G.; Baumgartner, H. Predictors of outcome in severe, asymptomatic aortic stenosis. N. Engl. J. Med. 2000, 343, 611–617. [Google Scholar] [CrossRef] [PubMed]
- Rennenberg, R.J.M.W.; Kessels, A.G.H.; Schurgers, L.J.; van Engelshoven, J.M.A.; de Leeuw, P.W.; Kroon, A.A. Vascular calcifications as a marker of increased cardiovascular risk: A meta-analysis. Vasc. Health Risk Manag. 2009, 5, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Spronk, H.M.; Soute, B.A.; Schiffers, P.; DeMey, J.G.R.; Vermeer, C. Regression of warfarin-induced medial elastocalcinosis by high intake of vitamin K in rats. Blood 2007, 109, 2823–2831. [Google Scholar] [CrossRef] [PubMed]
- Mac-Way, F.; Poulin, A.; Utescu, M.S.; De Serres, S.A.; Marquis, K.; Douville, P.; Desmeules, S.; Larivière, R.; Lebel, M.; Agharazii, M. The impact of warfarin on the rate of progression of aortic stiffness in hemodialysis patients: A longitudinal study. Nephrol. Dial. Transplant. 2014, 29, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Cohen, D. Dabigatran: How the drug company withheld important analyses. BMJ (Clin. Res. ed.) 2014, 349. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.J.; Cohen, M.R.; Mattison, D.R. Dabigatran, bleeding, and the regulators. BMJ (Clin. Res. ed.) 2014, 349. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, E.J.; Lippi, G. Laboratory testing in the era of direct or non-vitamin K antagonist oral anticoagulants: A practical guide to measuring their activity and avoiding diagnostic errors. Semin. Thromb. Hemost. 2015, 41, 208–227. [Google Scholar] [PubMed]
- Lippi, G.; Favaloro, E.J.; Mattiuzzi, C. Combined administration of antibiotics and direct oral anticoagulants: A renewed indication for laboratory monitoring? Semin. Thromb. Hemost. 2014, 40, 756–765. [Google Scholar] [PubMed]
- Bauer, K.A. Targeted Anti-Anticoagulants. N. Engl. J. Med. 2015, 373, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J.; Newman, P. Metabolism and cell biology of vitamin K. Thromb. Haemost. 2008, 100, 530–547. [Google Scholar] [CrossRef] [PubMed]
- Crowther, M.; Crowther, M.A. Antidotes for Novel Oral Anticoagulants: Current Status and Future Potential. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Pollack, C.V.; Reilly, P.A.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Huisman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for Dabigatran Reversal. N. Engl. J. Med. 2015, 373, 511–520. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.; Stouffer, G.A.; Madamanchi, N.; Runge, M.S. New tricks for old dogs: Nonthrombotic effects of thrombin in vessel wall biology. Circ. Res. 2001, 88, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Borissoff, J.I.; Spronk, H.M.H.; Ten Cate, H. The hemostatic system as a modulator of atherosclerosis. N. Engl. J. Med. 2011, 364, 1746–1760. [Google Scholar] [PubMed]
- Ma, L.; Dorling, A. The roles of thrombin and protease-activated receptors in inflammation. Semin. Immunopathol. 2012, 34, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Kalz, J.; Ten Cate, H.; Spronk, H.M.H. Thrombin generation and atherosclerosis. J. Thromb. Thrombolysis 2014, 37, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Alberelli, M.A.; De Candia, E. Functional role of protease activated receptors in vascular biology. Vasc. Pharmacol. 2014, 62, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Martorell, L.; Martínez-González, J.; Rodríguez, C.; Gentile, M.; Calvayrac, O.; Badimon, L. Thrombin and protease-activated receptors (PARs) in atherothrombosis. Thromb. Haemost. 2008, 99, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Cheung, W.M.; D'Andrea, M.R.; Andrade-Gordon, P.; Damiano, B.P. Altered vascular injury responses in mice deficient in protease-activated receptor-1. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 3014–3024. [Google Scholar] [CrossRef] [PubMed]
- Dabbagh, K.; Laurent, G.J.; McAnulty, R.J.; Chambers, R.C. Thrombin stimulates smooth muscle cell procollagen synthesis and mRNA levels via a PAR-1 mediated mechanism. Thromb. Haemost. 1998, 79, 405–409. [Google Scholar] [PubMed]
- Flynn, P.; Byrne, C.; Baglin, T.; Weissberg, P.; Bennett, M. Thrombin generation by apoptotic vascular smooth muscle cells. Blood 1997, 89, 4378–4384. [Google Scholar] [PubMed]
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Spronk, H.M.; ten Cate, H.; Hofstra, L. Accelerated In Vivo Thrombin Formation Independently Predicts the Presence and Severity of CT Angiographic Coronary Atherosclerosis. JACC Cardiovasc. Imaging 2012, 5, 1201–1210. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Soto, A.G.; Coronel, L.J.; Goss, A.; van Ryn, J.; Trejo, J. Characterization of Thrombin-Bound Dabigatran Effects on Protease-Activated Receptor-1 Expression and Signaling In Vitro. Mol. Pharmacol. 2015, 88, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Pingel, S.; Tiyerili, V.; Mueller, J.; Werner, N.; Nickenig, G.; Mueller, C. Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice. Arch. Med. Sci. 2014, 10, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Kadoglou, N.P.E.; Moustardas, P.; Katsimpoulas, M.; Kapelouzou, A.; Kostomitsopoulos, N.; Schafer, K.; Kostakis, A.; Liapis, C.D. The beneficial effects of a direct thrombin inhibitor, dabigatran etexilate, on the development and stability of atherosclerotic lesions in apolipoprotein E-deficient mice: Dabigatran etexilate and atherosclerosis. Cardiovasc. Drugs Ther. Spons. Int. Soc. Cardiovasc. Pharmacother. 2012, 26, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Borensztajn, K.; Peppelenbosch, M.P.; Spek, C.A. Factor Xa: At the crossroads between coagulation and signaling in physiology and disease. Trends Mol. Med. 2008, 14, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Hamilton, J.R. Physiology, pharmacology, and therapeutic potential of protease-activated receptors in vascular disease. Pharmacol. Ther. 2012, 134, 246–259. [Google Scholar] [CrossRef] [PubMed]
- Esmon, C.T. Targeting factor Xa and thrombin: Impact on coagulation and beyond. Thromb. Haemost. 2014, 111, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Böhm, A.; Flößer, A.; Ermler, S.; Fender, A.C.; Lüth, A.; Kleuser, B.; Schrör, K.; Rauch, B.H. Factor-Xa-induced mitogenesis and migration require sphingosine kinase activity and S1P formation in human vascular smooth muscle cells. Cardiovasc. Res. 2013, 99, 505–513. [Google Scholar] [CrossRef] [PubMed]
- Borensztajn, K.; Spek, C.A. Blood coagulation factor Xa as an emerging drug target. Expert Opin. Ther. Targets 2011, 15, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Ferrell, W.R.; Lockhart, J.C.; Kelso, E.B.; Dunning, L.; Plevin, R.; Meek, S.E.; Smith, A.J.H.; Hunter, G.D.; McLean, J.S.; McGarry, F.; et al. Essential role for proteinase-activated receptor-2 in arthritis. J. Clin. Investig. 2003, 111, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.; Hirano, M.; Kanaide, H.; Hirano, K. Upregulation of proteinase-activated receptor-2 and increased response to trypsin in endothelial cells after exposure to oxidative stress in rat aortas. J. Vasc. Res. 2010, 47, 494–506. [Google Scholar] [CrossRef] [PubMed]
- Cicala, C.; Pinto, A.; Bucci, M.; Sorrentino, R.; Walker, B.; Harriot, P.; Cruchley, A.; Kapas, S.; Howells, G.L.; Cirino, G. Protease-activated receptor-2 involvement in hypotension in normal and endotoxemic rats in vivo. Circulation 1999, 99, 2590–2597. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Bea, F.; Preusch, M.; Wang, H.; Isermann, B.; Shahzad, K.; Katus, H.A.; Blessing, E. Evaluation of plaque stability of advanced atherosclerotic lesions in apo E-deficient mice after treatment with the oral factor Xa inhibitor rivaroxaban. Mediat. Inflamm. 2011, 2011, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Roijers, R.B.; Debernardi, N.; Cleutjens, J.P.M.; Schurgers, L.J.; Mutsaers, P.H.A.; van der Vusse, G.J. Microcalcifications in early intimal lesions of atherosclerotic human coronary arteries. Am. J. Pathol. 2011, 178, 2879–2887. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Teunissen, K.J.F.; Knapen, M.H.J.; Kwaijtaal, M.; van Diest, R.; Appels, A.; Reutelingsperger, C.P.; Cleutjens, J.P.M.; Vermeer, C. Novel conformation-specific antibodies against matrix γ-carboxyglutamic acid (Gla) protein: Undercarboxylated matrix Gla protein as marker for vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.S.B.; Cavaco, S.; Williamson, M.K.; Price, P.A.; Cancela, M.L.; Simes, D.C. Gla-rich protein is a novel vitamin K-dependent protein present in serum that accumulates at sites of pathological calcifications. Am. J. Pathol. 2009, 175, 2288–2298. [Google Scholar] [CrossRef] [PubMed]
- Joshi, N.V.; Vesey, A.T.; Williams, M.C.; Shah, A.S.V.; Calvert, P.A.; Craighead, F.H.M.; Yeoh, S.E.; Wallace, W.; Salter, D.; Fletcher, A.M.; et al. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: A prospective clinical trial. Lancet 2014, 383, 705–713. [Google Scholar] [CrossRef]
- Ehara, S.; Kobayashi, Y.; Yoshiyama, M.; Shimada, K.; Shimada, Y.; Fukuda, D.; Nakamura, Y.; Yamashita, H.; Yamagishi, H.; Takeuchi, K.; et al. Spotty calcification typifies the culprit plaque in patients with acute myocardial infarction: An intravascular ultrasound study. Circulation 2004, 110, 3424–3429. [Google Scholar] [CrossRef] [PubMed]
- Bluestein, D.; Alemu, Y.; Avrahami, I.; Gharib, M.; Dumont, K.; Ricotta, J.J.; Einav, S. Influence of microcalcifications on vulnerable plaque mechanics using FSI modeling. J. Biomech. 2008, 41, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Vengrenyuk, Y.; Carlier, S.; Xanthos, S.; Cardoso, L.; Ganatos, P.; Virmani, R.; Einav, S.; Gilchrist, L.; Weinbaum, S. A hypothesis for vulnerable plaque rupture due to stress-induced debonding around cellular microcalcifications in thin fibrous caps. Proc. Natl. Acad. Sci. USA 2006, 103, 14678–14683. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, G.; Perego, S.; Luzi, L.; Banfi, G. A four-season molecule: Osteocalcin. Updates in its physiological roles. Endocrine 2014, 48, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Schlieper, G.; Schurgers, L.; Brandenburg, V.; Reutlingsperger, C.; Floege, J. Vascular calcification in chronic kidney disease: An update. Nephrol. Dial. Transplant. 2015. [Google Scholar] [CrossRef] [PubMed]
- Yamada, S.; Taniguchi, M.; Tokumoto, M.; Toyonaga, J.; Fujisaki, K.; Suehiro, T.; Noguchi, H.; Iida, M.; Tsuruya, K.; Kitazono, T. The antioxidant tempol ameliorates arterial medial calcification in uremic rats: Important role of oxidative stress in the pathogenesis of vascular calcification in chronic kidney disease. J. Bone Miner. Res. 2012, 27, 474–485. [Google Scholar] [CrossRef] [PubMed]
- Ndip, A.; Wilkinson, F.L.; Jude, E.B.; Boulton, A.J.; Alexander, M.Y. RANKL-OPG and RAGE modulation in vascular calcification and diabetes: Novel targets for therapy. Diabetologia 2014, 57, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Lei, Y.; Sinha, A.; Nosoudi, N.; Grover, A.; Vyavahare, N. Hydroxyapatite and calcified elastin induce osteoblast-like differentiation in rat aortic smooth muscle cells. Exp. Cell Res. 2014, 323, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Hunter, G.K.; Hauschka, P.V.; Poole, A.R.; Rosenberg, L.C.; Goldberg, H.A. Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem. J. 1996, 317, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Desbois, C.; Boyce, B.; Pinero, G.; Story, B.; Dunstan, C.; Smith, E.; Bonadio, J.; Goldstein, S.; Gundberg, C.; et al. Increased bone formation in osteocalcin-deficient mice. Nature 1996, 382, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Kavukcuoglu, N.B.; Patterson-Buckendahl, P.; Mann, A.B. Effect of osteocalcin deficiency on the nanomechanics and chemistry of mouse bones. J. Mech. Behav. Biomed. Mater. 2009, 2, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.E.; Nishimoto, S.K.; Rho, J.Y.; Bhattacharya, S.K.; Lin, J.S.; Pharr, G.M. Correlations between osteocalcin content, degree of mineralization, and mechanical properties of C. carpio rib bone. J. Biomed. Mater. Res. 2001, 54, 547–553. [Google Scholar] [CrossRef]
- Evrard, S.; Delanaye, P.; Kamel, S.; Cristol, J.-P.; Cavalier, E. SFBC/SN joined working group on vascular calcifications vascular calcification: From pathophysiology to biomarkers. Clin. Chim. Acta 2015, 438, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Otsuka, A.A.; Poser, J.W.; Kristaponis, J.; Raman, N. Characterization of a γ-carboxyglutamic acid-containing protein from bone. Proc. Natl. Acad. Sci. USA 1976, 73, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Van de Loo, P.G.F.; Soute, B.; van Haarlem, L.; Vermeer, C. The effect of Gla-containing proteins on the precipitation of insoluble salts. Biochem. Biophys. Res. Commun. 1987, 142, 113–119. [Google Scholar] [CrossRef]
- Kapustin, A.N.; Shanahan, C.M. Osteocalcin: A novel vascular metabolic and osteoinductive factor? Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2169–2171. [Google Scholar] [CrossRef] [PubMed]
- Aikawa, E.; Nahrendorf, M.; Sosnovik, D.; Lok, V.M.; Jaffer, F.A.; Aikawa, M.; Weissleder, R. Multimodality molecular imaging identifies proteolytic and osteogenic activities in early aortic valve disease. Circulation 2007, 115, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Okura, T.; Kurata, M.; Enomoto, D.; Jotoku, M.; Nagao, T.; Desilva, V.R.; Higaki, J. Undercarboxylated osteocalcin is a biomarker of carotid calcification in patients with essential hypertension. Kidney Blood Press. Res. 2010, 33, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Uitto, J.; Reutelingsperger, C.P. Vitamin K-dependent carboxylation of matrix Gla-protein: A crucial switch to control ectopic mineralization. Trends Mol. Med. 2013, 19, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Wajih, N.; Borras, T.; Xue, W.; Hutson, S.M.; Wallin, R. Processing and transport of matrix γ-carboxyglutamic acid protein and bone morphogenetic protein-2 in cultured human vascular smooth muscle cells: Evidence for an uptake mechanism for serum fetuin. J. Biochem. 2004, 279, 43052–43060. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Ducy, P.; McKee, M.; Pinero, G.; Loyer, E.; Behringer, R.; Karsenty, G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 385, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Murshed, M.; Schinke, T.; McKee, M.D.; Karsenty, G. Extracellular matrix mineralization is regulated locally; different roles of two gla-containing proteins. J. Cell Biol. 2004, 165, 625–630. [Google Scholar] [CrossRef] [PubMed]
- Cranenburg, E.C.M.; van Spaendonck-Zwarts, K.Y.; Bonafe, L.; Mittaz Crettol, L.; Rödiger, L.A.; Dikkers, F.G.; van Essen, A.J.; Superti-Furga, A.; Alexandrakis, E.; Vermeer, C.; et al. Circulating matrix γ-carboxyglutamate protein (MGP) species are refractory to vitamin K treatment in a new case of Keutel syndrome. J. Thromb. Haemost. 2011, 9, 1225–1235. [Google Scholar] [CrossRef] [PubMed]
- Price, P.; Faus, S.; Williamson, M. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler. Thromb. Vasc. Biol. 1998, 18, 1400–1407. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.L.; Joannides, A.J.; Skepper, J.N.; McNair, R.; Schurgers, L.J.; Proudfoot, D.; Jahnen-Dechent, W.; Weissberg, P.L.; Shanahan, C.M. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: A potential mechanism for accelerated vascular calcification in ESRD. J. Am. Soc. Nephrol. 2004, 15, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Steitz, S.A.; Speer, M.Y.; Curinga, G.; Yang, H.Y.; Haynes, P.; Aebersold, R.; Schinke, T.; Karsenty, G.; Giachelli, C.M. Smooth muscle cell phenotypic transition associated with calcification: Upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ. Res. 2001, 89, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Bennett, B.J.; Wang, X.; Rosenfeld, M.E.; Giachelli, C.; Lusis, A.J.; Boström, K.I. Inhibition of bone morphogenetic proteins protects against atherosclerosis and vascular calcification. Circ. Res. 2010, 107, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Boström, K.I.; Rajamannan, N.M.; Towler, D.A. The regulation of valvular and vascular sclerosis by osteogenic morphogens. Circ. Res. 2011, 109, 564–577. [Google Scholar] [CrossRef] [PubMed]
- O’Young, J.; Liao, Y.; Xiao, Y.; Jalkanen, J.; Lajoie, G.; Karttunen, M.; Goldberg, H.A.; Hunter, G.K. Matrix Gla protein inhibits ectopic calcification by a direct interaction with hydroxyapatite crystals. J. Am. Chem. Soc. 2011, 133, 18406–18412. [Google Scholar] [CrossRef] [PubMed]
- Schurgers, L.J.; Spronk, H.M.H.; Skepper, J.N.; Hackeng, T.M.; Shanahan, C.M.; Vermeer, C.; Weissberg, P.L.; Proudfoot, D. Post-translational modifications regulate matrix Gla protein function: Importance for inhibition of vascular smooth muscle cell calcification. J. Thromb. Haemost. 2007, 5, 2503–2511. [Google Scholar] [CrossRef] [PubMed]
- Khavandgar, Z.; Roman, H.; Li, J.; Lee, S.; Vali, H.; Brinckmann, J.; Davis, E.C.; Murshed, M. Elastin haploinsufficiency impedes the progression of arterial calcification in MGP-deficient mice. J. Bone Miner. Res. 2014, 29, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Price, P.A.; Toroian, D.; Lim, J.E. Mineralization by inhibitor exclusion: The calcification of collagen with fetuin. J. Biol. Chem. 2009, 284, 17092–17101. [Google Scholar] [CrossRef] [PubMed]
- Cancela, M.L.; Conceição, N.; Laizé, V. Gla-rich protein, a new player in tissue calcification? Adv. Nutr. 2012, 3, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Viegas, C.S.B.; Rafael, M.S.; Enriquez, J.L.; Teixeira, A.; Vitorino, R.; Luís, I.M.; Costa, R.M.; Santos, S.; Cavaco, S.; Neves, J.; et al. Gla-rich protein acts as a calcification inhibitor in the human cardiovascular system. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Eitzinger, N.; Surmann-Schmitt, C.; Bösl, M.; Schett, G.; Engelke, K.; Hess, A.; von der Mark, K.; Stock, M. Ucma is not necessary for normal development of the mouse skeleton. Bone 2012, 50, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Geleijnse, J.M.; Vermeer, C.; Grobbee, D.E.; Schurgers, L.J.; Knapen, M.H.J.; van der Meer, I.M.; Hofman, A.; Witteman, J.C.M. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: The Rotterdam Study. J. Nutr. 2004, 134, 3100–3105. [Google Scholar] [PubMed]
- Juanola-Falgarona, M.; Salas-Salvadó, J.; Martínez-González, M.Á.; Corella, D.; Estruch, R.; Ros, E.; Fitó, M.; Arós, F.; Gómez-Gracia, E.; Fiol, M.; et al. Dietary intake of vitamin k is inversely associated with mortality risk. J. Nutr. 2014, 144, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.; Bots, M.L.; Atsma, F.; Bartelink, M.L.E.; Prokop, M.; Geleijnse, J.M.; Van Der Schouw, Y.T. High dietary menaquinone intake is associated with reduced coronary calcification. Atherosclerosis 2009, 203, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Westenfeld, R.; Krueger, T.; Schlieper, G.; Cranenburg, E.C.M.; Magdeleyns, E.J.; Heidenreich, S.; Holzmann, S.; Vermeer, C.; Jahnen-Dechent, W.; Ketteler, M.; et al. Effect of vitamin K2 supplementation on functional vitamin K deficiency in hemodialysis patients: A randomized trial. Am. J. Kidney Dis. 2012, 59, 186–195. [Google Scholar] [CrossRef] [PubMed]
- Theuwissen, E.; Teunissen, K.J.; Spronk, H.M.H.; Hamulyak, K.; ten Cate, H.; Shearer, M.J.; Vermeer, C.; Schurgers, L.J. Effect of low-dose supplements of menaquinone-7 (vitamin K2) on the stability of oral anticoagulant treatment: Dose-response relationship in healthy volunteers. J. Thromb. Haemost. 2013, 11, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Buitenhuis, H.C.; Soute, B.A.; Vermeer, C. Comparison of the vitamins K1, K2 and K3 as cofactors for the hepatic vitamin K-dependent carboxylase. Biochim. Biophys. Acta 1990, 1034, 170–175. [Google Scholar] [CrossRef]
- Schlieper, G.; Westenfeld, R.; Krüger, T.; Cranenburg, E.C.; Magdeleyns, E.J.; Brandenburg, V.M.; Djuric, Z.; Damjanovic, T.; Ketteler, M.; Vermeer, C.; et al. Circulating nonphosphorylated carboxylated matrix Gla protein predicts survival in ESRD. J. Am. Soc. Nephrol. 2011, 22, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Dahl, C.P.; Gullestad, L.; Aakhus, S.; Broch, K.; Skårdal, R.; Vermeer, C.; Aukrust, P.; Schurgers, L.J. Circulating levels of non-phosphorylated undercarboxylated matrix Gla protein are associated with disease severity in patients with chronic heart failure. Clin. Sci. 2011, 121, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Cranenburg, E.C.M.; Vermeer, C.; Koos, R.; Boumans, M.-L.; Hackeng, T.M.; Bouwman, F.G.; Kwaijtaal, M.; Brandenburg, V.M.; Ketteler, M.; Schurgers, L.J. The circulating inactive form of matrix Gla Protein (ucMGP) as a biomarker for cardiovascular calcification. J. Vasc. Res. 2008, 45, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, V.M.; Schurgers, L.J.; Kaesler, N.; Püsche, K.; van Gorp, R.H.; Leftheriotis, G.; Reinartz, S.; Koos, R.; Krüger, T. Prevention of vasculopathy by vitamin K supplementation: Can we turn fiction into fact? Atherosclerosis 2015, 240, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Morishima, Y.; Kamisato, C.; Honda, Y.; Furugohri, T.; Shibano, T. The effects of warfarin and edoxaban, an oral direct factor Xa inhibitor, on γcarboxylated (Gla-osteocalcin) and undercarboxylated osteocalcin (uc-osteocalcin) in rats. Thromb. Res. 2013, 131, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Krüger, T.; Oelenberg, S.; Kaesler, N.; Schurgers, L.J.; van de Sandt, A.M.; Boor, P.; Schlieper, G.; Brandenburg, V.M.; Fekete, B.C.; Veulemans, V.; et al. Warfarin induces cardiovascular damage in mice. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 2618–2624. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Gorp, R.H.; Schurgers, L.J. New Insights into the Pros and Cons of the Clinical Use of Vitamin K Antagonists (VKAs) Versus Direct Oral Anticoagulants (DOACs). Nutrients 2015, 7, 9538-9557. https://doi.org/10.3390/nu7115479
Van Gorp RH, Schurgers LJ. New Insights into the Pros and Cons of the Clinical Use of Vitamin K Antagonists (VKAs) Versus Direct Oral Anticoagulants (DOACs). Nutrients. 2015; 7(11):9538-9557. https://doi.org/10.3390/nu7115479
Chicago/Turabian StyleVan Gorp, Rick H., and Leon J. Schurgers. 2015. "New Insights into the Pros and Cons of the Clinical Use of Vitamin K Antagonists (VKAs) Versus Direct Oral Anticoagulants (DOACs)" Nutrients 7, no. 11: 9538-9557. https://doi.org/10.3390/nu7115479