Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections
by
 , , and
, , and
Sébastien Lhomme
1,2,3,* ,
,
Olivier Marion
2,3,4 ,
,
Florence Abravanel
1,2,3,
Jacques Izopet
1,2,3 and
Nassim Kamar
2,3,4,* 1
Virology Laboratory, National Reference Center for Hepatitis E Virus, Toulouse Purpan University Hospital, 31300 Toulouse, France
2
INSERM UMR1043, Center for Pathophysiology of Toulouse Purpan, 31300 Toulouse, France
3
Université Toulouse III Paul Sabatier, 31330 Toulouse, France
4
Department of Nephrology and Organs Transplantation, Toulouse Rangueil University Hospital, 31400 Toulouse, France
*
Authors to whom correspondence should be addressed.
J. Clin. Med. 2020, 9(2), 331; https://doi.org/10.3390/jcm9020331
Submission received: 18 December 2019
/
Revised: 14 January 2020
/
Accepted: 22 January 2020
/
Published: 24 January 2020
(This article belongs to the Special Issue Immunobiology Pathogenesis and Therapeutic Perspectives in Viral Hepatitis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Hepatitis E virus (HEV) is the most common cause of acute viral hepatitis throughout the world. Most infections are acute but they can become chronic in immunocompromised patients, such as solid organ transplant patients, patients with hematologic malignancy undergoing chemotherapy and those with a human immunodeficiency virus (HIV) infection. Extra-hepatic manifestations, especially neurological and renal diseases, have also been described. To date, four main genotypes of HEV (HEV1-4) were described. HEV1 and HEV2 only infect humans, while HEV3 and HEV4 can infect both humans and animals, like pigs, wild boar, deer and rabbits. The real epidemiology of HEV has been underestimated because most infections are asymptomatic. This review focuses on the recent advances in our understanding of the pathophysiology of acute HEV infections, including severe hepatitis in patients with pre-existing liver disease and pregnant women. It also examines the mechanisms leading to chronic infection in immunocompromised patients and extra-hepatic manifestations. Acute infections are usually self-limiting and do not require antiviral treatment. Conversely, a chronic HEV infection can be cleared by decreasing the dose of immunosuppressive drugs or by treating with ribavirin for 3 months. Nevertheless, new drugs are needed for those cases in which ribavirin treatment fails.
1. Introduction
The hepatitis E virus (HEV) is the leading cause of acute viral hepatitis worldwide. HEV, which is mainly transmitted enterically, is responsible for outbreaks in developing countries and zoonotic cases in both developing and developed countries [1]. HEV genotypes 1 (HEV1) and 2 (HEV2) are mainly found in developing countries and are restricted to humans. Other genotypes, including HEV3 and HEV4, have been detected in both humans and animals, with pigs being the main reservoir. While most infections are asymptomatic, they can cause acute hepatitis, including severe forms in patients with pre-existing liver disease and in pregnant women living in developing countries. An HEV infection may also trigger extra-hepatic manifestations and can lead to chronic infections in immunocompromised patients. Its pathogenesis is still unclear but our knowledge has greatly improved in the past few years.
2. HEV Genome and Classification
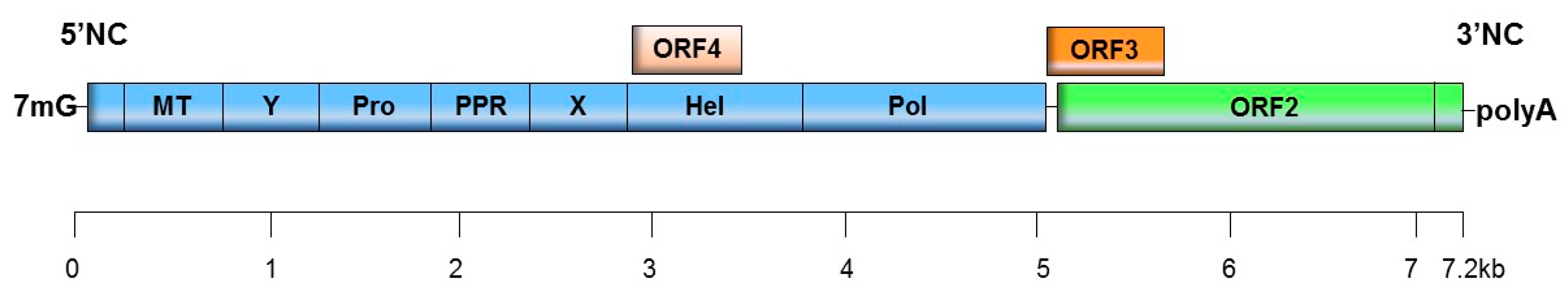
HEV is a small virus with the positive-sense, single-stranded ~7.2 kb RNA genome that contains three open reading frames (ORF), ORF1, ORF2 and ORF3 (Figure 1). ORF1 encodes a non-structural protein about 1693 amino acids (aa) long, with at least four putative functional domains: methyltransferase, cysteine protease, helicase and RNA-dependent RNA polymerase (RdRp). Other domains homologous with those of other plant and animal positive-stranded RNA viruses have been described: the Y domain, the polyproline region (PPR), also called the hypervariable region (HVR), and a macro domain, also called the X domain [2]. Sequence analyses suggest that the Y domain is an integral part of the methyltransferase [3]. It is still unclear whether the ORF1 polyprotein is processed into individual proteins [2]. HEV1 was recently shown to have an additional reading frame, ORF4, which overlaps with ORF1. Stress on the endoplasmic reticulum induces synthesis of the ORF4 protein, which is required for the proper functioning of HEV RNA polymerase [4]. ORF2 and ORF3 also overlap, and their corresponding proteins are translated from a bicistronic subgenomic RNA. ORF2 encodes the 660 aa capsid protein, which has been divided into three domains: S (shell), M (middle) and P (protruding). It was recently shown that ORF2 also encodes a secreted free form of the capsid protein (ORF2s) that differs from the actual capsid protein, ORF2i (for infectious). ORF2i translation is initiated at a previously unrecognized internal start codon [5]. ORF2s is secreted into the extracellular space as an O, N glycosylated and sialylated dimer. Lastly, ORF3 encodes a 113 or 114 aa phosphoprotein, depending on the genotype. This protein has ion channel activity and is involved in virus egress from infected cells [6]. The HEV circulating in the blood is quasi-enveloped, as it is cloaked in host cell membranes, but it is shed into the feces as un-enveloped virions because lipids have been removed by the action of bile salts [7,8].
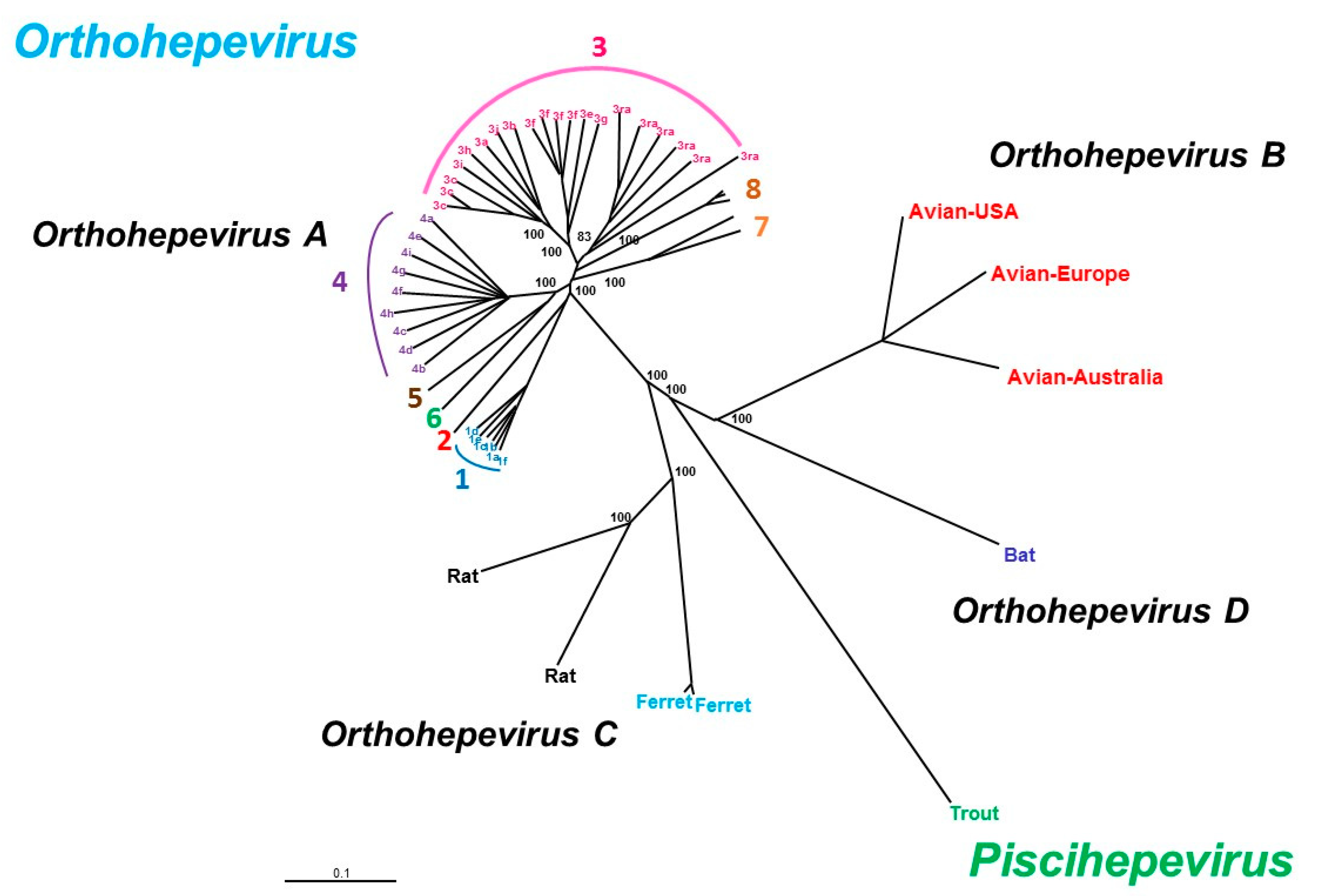
HEV belongs to the Hepeviridae family, which has two genera: Piscihepevirus (cutthroat trout virus) and Orthohepevirus (mammalian and avian strains) with four species (A–D) (Figure 2). The Orthohepevirus A HEV species infect humans and other mammals, Orthohepevirus B infects chickens, Orthohepevirus C infects rats and ferrets, Orthohepevirus D infects bats and Piscihepevirus A infects the cutthroat trout. The largest species, Orthohepevirus A, consists of at least eight distinct HEV genotypes that infect human (HEV1, 2, 3, 4 and 7), pigs (HEV3 and 4), wild boar (HEV3, 4, 5 and 6), rabbits (HEV3), mongooses (HEV3), deer (HEV3), yaks (HEV4) and camels (HEV7 and HEV8) [9]. Only one serotype has been described. While several subgenotypes are described, no consistent criteria have yet been defined to discriminate between virus subgenotypes. HEV3 variants are arranged in three major clades: HEV3abjkchi, HEV3efg, and HEV-3rabbit (ra) based on phylogenetic groupings [10,11]. Two of the four major genotypes, HEV1 and HEV2, only infect humans and are found in developing countries. HEV3 is widely distributed around the world and HEV4 is found mainly in Asia. The HEV3 and HEV4 genotypes are transmitted zoonotically from pigs, wild boar, deer and mongooses [12]. Rabbit strains that are close to HEV3 have been identified in humans [13,14,15]. Camel HEV7 has been described in a liver transplant recipient who had consumed camel meat and milk [16]. HEV5 and HEV6 have been described in wild boar in Japan but not yet in humans. However, Cynomolgus monkeys have been experimentally infected with an HEV5 strain [17]. Cynomolgus macaques are also susceptible to HEV8 [18].
3. Clinical Course of HEV Infection
While most HEV infections are asymptomatic, any illness that is caused is usually self-limiting and lasts just a few weeks in the majority of patients. Acute icteric hepatitis, the classic presentation of hepatitis E, occurs in 5%–30% of infected patients. The prodromal phase lasts up to one week and its non-specific symptoms include fever and nausea, vomiting, anorexia or malaise. Dark urine and jaundice mark the onset of the icteric phase. Symptoms usually resolve spontaneously after a few days to a week, but mortality rates can vary from 0.5% to 4.0% of infections during an outbreak [21].
HEV1 and HEV2 mainly infect young adult males (15–30 years) in developing countries and can be asymptomatic, cause mild systemic illness, or icteric acute hepatitis that can be fulminant or lead to acute liver failure. Pregnant women are particularly at risk and a large proportion of those in their second and third trimester of pregnancy can progress to acute liver failure. The mortality rate may reach 25% during the third trimester [22]. Pregnant women die of obstetric complications such as hemorrhage or eclampsia. Fulminant liver failure were also described. Stillbirths are common, as is vertical transmission to infants, which leads to increased neonatal morbidity and mortality [23]. One Indian study found that mortality rates of HEV-related and non-HEV-related acute liver failure in pregnant women were similar, although HEV-related acute liver failure was more common during pregnancy [24]. HEV1 infection during pregnancy is also associated with more frequent miscarriages, preterm deliveries, stillbirths and perinatal mortality.
In developed countries, patients infected with HEV are usually middle-aged or elderly men (>55 years). Severe HEV infections were not described in pregnant women. Patients with underlying liver disease have a poor prognosis in both developing and developed countries [25,26]. In Europe, 5%–33% of patients infected with HEV3 or HEV4 develop symptoms, including jaundice [27,28,29]. The occurrence of symptoms could be linked to the virus load, as recently suggested by a study showing that symptomatic patients had a higher HEV RNA concentration than did HEV RNA-positive healthy blood donors [30].
HEV3 and HEV4 can persist in immunocompromised patients, including solid organ transplant (SOT) patients [31,32,33] and those co-infected with the human immunodeficiency virus (HIV) with a T CD4+ count < 200/mm3 [34,35,36]. There have also been reports of chronic HEV infections in patients with hematological disease receiving chemotherapy [37,38,39,40], those given stem cell transplants [41] or patients with rheumatic disorders on heavy immunosuppression immunotherapy [42,43]. Only one case of a chronic HEV1 infection has been reported to date [44], but the finding has been debated because the HEV RNA was not quantified [45]. A patient who had consumed camel meat and milk is also believed to have developed a chronic HEV7 infection [16]. Chronic HEV infection is defined as HEV replication that persists for more than 3 months [46]. A chronic HEV infection can lead to chronic hepatitis and progress rapidly to cirrhosis in 10% of the chronically infected patients [31,32,47,48]. Some of these patients may die from decompensated cirrhosis 2–3 years after the diagnosis. There have been no reports of HEV-infected transplant recipients developing fulminant hepatitis.
4. Extra-Hepatic Manifestations
Extra-hepatic manifestations can occur both in patients with acute or chronic HEV infections. They include a range of neurological symptoms and impaired kidney function associated with cryoglobulinemia. Little is known about the mechanisms leading to these extra-hepatic manifestations, although direct viral effects due to HEV replication in affected tissues and indirect effects mediated by the immune system could be involved.
4.1. Neurological Manifestations
Neurological disorders have frequently been reported in patients with acute or chronic HEV infections and approximately 150 cases of neurological injury in HEV1-infected Asians and HEV3-infected Europeans have been described to date [49]. These include neuralgic amyotrophy (NA), Guillain–Barré syndrome, Bell’s palsy, and polyradiculopathy [50]. The several cohorts and case-studies of HEV infection in patients with NA have almost all been done on HEV3-infected Europeans. A European study covering four centers in France, UK and the Netherlands prospectively tested over 450 consecutive patients with acute-onset non-traumatic neurological injury. It found that 2.4% of them showed evidence of an HEV infection [50]. The three cases of NA were all HEV-associated. Similarly, an Anglo/Dutch cohort study found that 5/47 (10.6%) patients with NA had evidence of an HEV infection at the onset of their illness [51]. The multi-centric European study of 118 patients with NA showed that patients with HEV-associated disease have a distinct clinical phenotype, compared to patients with NA, without evidence of HEV infection. Patients with HEV-associated NA were significantly more likely to have bilateral involvement of, and more extensive damage to, the brachial plexus. Neurological damage outside the brachial plexus, particularly phrenic nerve involvement, was also more likely in these patients [52]. Lastly, a recent report described finding HEV RNA and intrathecal synthesis of anti-HEV immunoglobulin M (IgM) in a patient with NA, indicating a neurotropic HEV infection [53].
The association between HEV infection and Guillain–Barré syndrome is supported by three case–control studies from Bangladesh [54], Japan [55] and the Netherlands [56]. They found evidence of a recent HEV infection in 5%–11% of patients with Guillain–Barré syndrome, which was a significantly greater frequency than in the healthy control group. Lastly, 6/73 (8%) Belgian patients with Guillain–Barré syndrome had an HEV infection [57].
Other manifestations, like neuropathic pain, painless sensory disorders, encephalitis/myelitis, mononeuritis multiplex, vestibular neuritis, myositis or peripheral neuropathy, have also been described in HEV-infected patients [49,58]. Almost all of these patients with neurological manifestations had normal or modestly abnormal liver function, indicating that the neurological symptoms and signs dominate the clinical picture in these patients.
The pathophysiology of HEV-associated neurological injury remains uncertain, although the immune response triggered by the virus may play a role. A recent study showed that neurological injuries were more frequent in immunocompetent patients (22.6%) than in immunocompromised ones (3.2%, p < 0.001) [58], suggesting that Guillain–Barré syndrome and NA are immune-mediated, due to molecular mimicry. This hypothesis fits well with the current thinking about Guillain–Barré syndrome pathophysiology and NA. Another possibility is direct virus neurotropism: pyramidal syndrome was found in a chronically HEV-infected kidney recipient. The variants characterized in the cerebrospinal fluid of this patient differed from those in the serum at the same time, suggesting the emergence of neurotropic variants [59], and possibly HEV replication in the central nervous system.
Using a gerbil model infected intraperitoneally with a 6.57 × 107 genome equivalent of an HEV4 strain isolated from a swine liver sample, negative strands of HEV RNA and ORF2 protein were detected by polymerase chain reaction (PCR) and immunohistochemical staining respectively, in the brains and spinal cords of the animals, suggesting that HEV4 damages the blood–brain barrier and replicates in these tissues. Pathological changes, including neuron degeneration and necrosis, microglia nodules, Purkinje cells necrosis and infiltration by inflammatory cells, were reported [60]. These features parallel the neurological manifestations observed in humans. Immunohistochemical studies on rabbits inoculated with 6.63 × 108 copies via intraperitoneal injection also found the ORF2 antigen in the brains and spinal cords of these animals [61]. Intravenous inoculations of the HEV4 KM01 strain isolated from stools of a swine in mouse 1 × 105 copies/mL and in rhesus macaques (2 × 104 copies) lead to the detection of the ORF2 protein by immunohistochemistry analysis in the cerebellum of the mouse brain tissue and in the granule layer of cerebellum of the rhesus macaque brain [62]. HEV3 Kernow-C1 p6 clone can directly infect human neural cells, neuroblastoma SH-SH5Y, neuroepithelioma SK-N-MC and glioblastoma U87 and U343 cells in vitro [62]. Another study demonstrated that M03.13 oligodendrocytic cells are invaded and support the lifecycle of the HEV3 Kernow-C1 p6 clone when a monolayer of 5 × 105 cells were inoculated with 3 × 107 RNA copies [63]. Human mesodermal and neuroprogenitor cells derived from pluripotent stem cells support HEV replication only when transfected with subgenomic replicon derived from HEV3 Kernow-C1 p6 clone [64]. Lastly, a recent in vitro study showed changes in the tight junction proteins (including Claudin 5, Occludin and Zonula occludens-1) of primary cultures of human brain microvascular cells (HBMVCs) inoculated with swine HEV4 strain for 48 h at a multiplicity of infection of 300 [61]. This observation could explain how HEV crosses the blood–brain barrier to access the central nervous system. However, another group fails to show susceptibility of mice to HEV1, HEV3 or HEV4 [65]. This could be due to the difference of viral isolates used for the different experiments. Whether only specific HEV strains are able to infect brain tissue remains to be determined.
4.2. Renal Manifestations
Both acute and chronic HEV infections can lead to kidney injuries and impaired renal function [66,67], but little is known about the underlying mechanisms. Renal biopsies from patients infected with HEV1 or HEV3 show signs of glomerular disease [67], including membranoproliferative glomerulonephritis (MPGN) with or without cryoglobulinemia and membranous glomerulonephritis [67,68,69]. HEV may also trigger a flare-up of a pre-existing IgA nephropathy [67,70]. HEV RNA has been detected in the cryoprecipitate from the serum of an immunocompetent patient with an acute HEV infection who presented with cryoglobulinemic MPGN [69], and HEV infection was found to be an independent predictive factor for cryoglobulinemia in SOT recipients [71]. Renal function improves after HEV clearance, and proteinuria is reduced in most patients [67,69,70].
Patients with a hepatitis C virus (HCV) infection and kidney MPGN disease can develop deposits of immune complexes formed from the HCV antigen, anti-HCV IgG antibodies and a rheumatoid factor in their glomeruli [72]. Perhaps a similar mechanism is at work in HEV infections. Both HEV antigen and RNA have been detected in the urine of patients with chronic HEV4 [73] or HEV3 infections [74]. High concentrations of HEV antigen were found in the urine of immunocompromised patients, independently of the detection of HEV RNA. High molecular weight ORF2 molecules, like HEV virions, should not freely cross the glomerular filtration barrier [5,75,76], but lower molecular weight by-products of ORF2s could be secreted into the urine and be detected by the assay used. It is also possible that the HEV antigen could be secreted into the urine by kidney epithelial cells. However, there is still no evidence that HEV is directly nephrotoxic or that it can replicate in human renal cells. HEV-positive and negative strand RNA and ORF2 protein have also been detected in the kidneys of infected animals, especially swine [77], gerbils [78], monkeys [73] and rabbits [79]. Histopathological analyses of kidney biopsies identified tubule-interstitial with interstitial inflammatory cell infiltrates [73,78,79]. Immunohistochemistry also detected ORF3 protein in the kidneys of infected rabbits [80]. This suggests that the kidneys or the urinary tract could be an HEV reservoir, although little is known of the pathogenicity of HEV for the human kidney.
HEV can also have hematological manifestations such as anemia and severe thrombocytopenia. Hemolytic anemia was reported in HEV-infected patients with a glucose 6-phosphate dehydrogenase (G6PD) [81,82,83,84]. A Pakistani male patient developed aplastic anemia following severe acute hepatitis E [85], while Woolson et al. reported that 12/106 (11%) HEV-infected patients had low platelet counts [86]. Just how HEV induces thrombocytopenia is unknown. It could be immune-mediated as in other virus infections or be linked to the development of fibrosis with splenomegaly. Lastly, several cases of HEV-induced acute pancreatitis have been reported [87,88,89,90,91]. A single-center study from India found that 2.1% (16/790) of patients with acute pancreatitis had serological evidence of a recent HEV infection with no other discernible cause of pancreatitis [92]. The authors suggested that this was due to edema developing in the ambulla of Vater, which then obstructs pancreatic fluid flow.
5. Pathogenesis
The pathogenesis of hepatitis E remains poorly understood. It is unclear how, and in what form, the virus particles reach the liver since it is transmitted by the fecal oral route. Recent data showed that primary cultures of intestinal cells support HEV1 and HEV3 replication, and HEV RNA and ORF2 antigen have been detected in the intestinal crypts of a chronically infected patient [93]. These elements suggest that HEV first replicates in the intestinal tract before reaching the liver via the blood in a quasi-enveloped form. It could then replicate in the cytoplasm of hepatocytes and be released as lipid-associated particles into the blood and bile. Capelli et al. used polarized hepatocytes in vitro to show that most HEV particles are released at the apical membrane, that is, the bile side [94]. Bile salts then strip the lipids from the virus shed in the stool. Since HEV is not cytopathic, the liver damage induced by an HEV infection may be immune-mediated by cytotoxic T cells and natural killer cells [95].
5.1. Innate Immune Response
The innate immune response during an acute or chronic HEV infection is poorly understood and has not been studied intensively. Microarray analyses of the intrahepatic transcriptome in serial liver biopsies obtained from chimpanzees infected with HEV1 or HCV suggest that the innate immunity mediated by interferon (IFN)-α restricts the replication of HEV more efficiently than that of HCV [96]. Comparison of the results of this study with those for hepatitis A virus (HAV)-infected chimpanzees [97] indicates that HEV triggers a stronger IFN response than does HAV. Analysis of the rhesus macaque liver gene expression showed that the profile differs depending on the genotype (HEV1 or HEV3) of the strain used for infection [98]. HEV1 and HEV3 infections may trigger different host mechanisms to control viral infection: 25% of the interferon-responsive genes were down-regulated during early viremia following an HEV1 infection, including interferon regulatory factor (IRF)3 and IRF7, or interferon-stimulated gene (ISG)15. These same genes were up-regulated during HEV3 infection. The origin of the cytokines, especially type 1 IFN, remains to be determined, since liver biopsies contain both infiltrating leukocytes and infected hepatocytes.
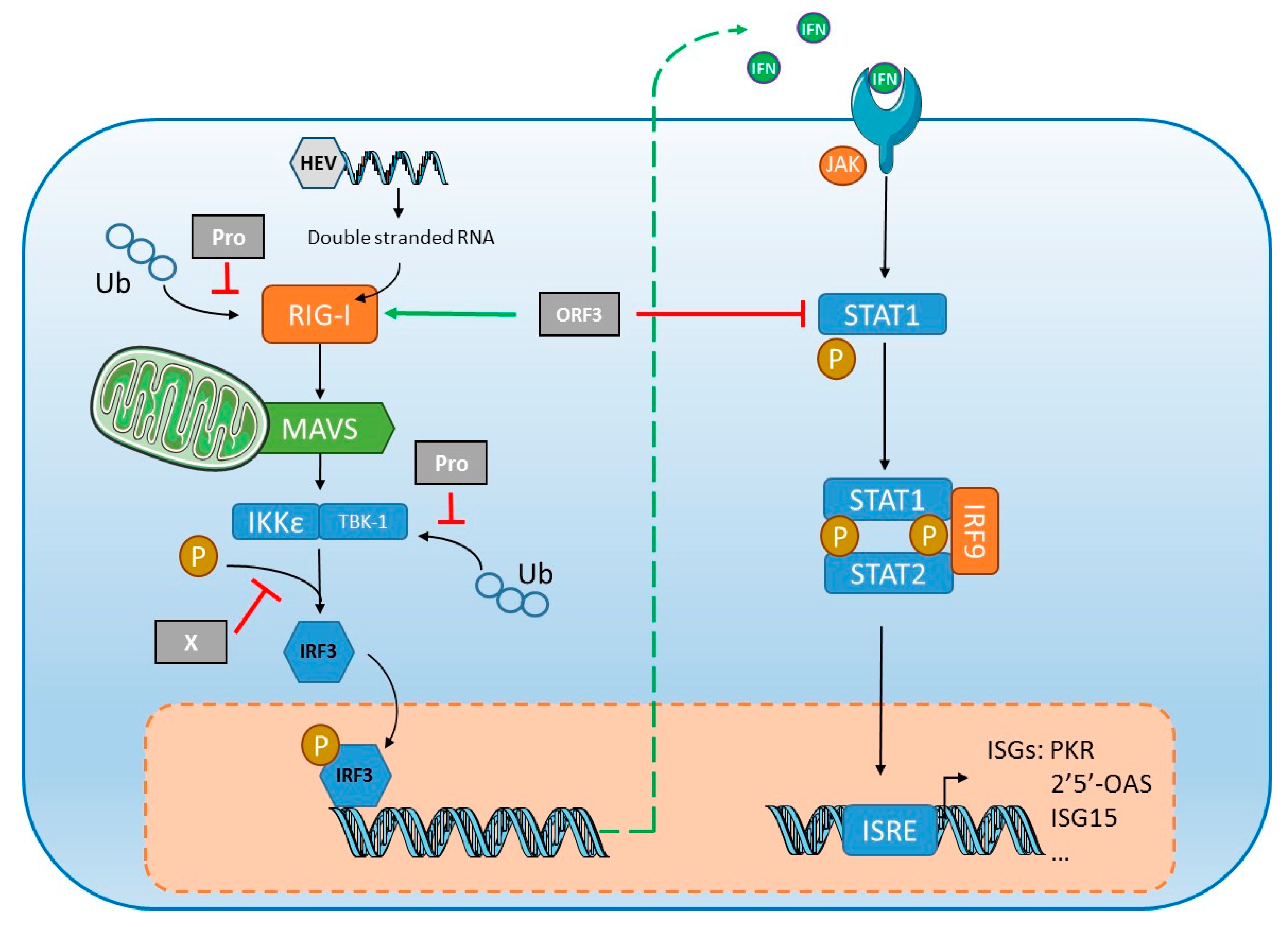
However, HEV has developed mechanisms to suppress IFN-α signaling (Figure 3). In vitro studies on human lung epithelial A549 cells [99] and hepatocarcinoma Huh7 cells [100] indicate that interferon-induced phosphorylation of the signal transducer and activator of transcription STAT1 are inhibited by the ORF3 protein, blocking the synthesis of two key antiviral proteins, double stranded (ds) RNA-activated protein kinase (PKR) and 2′,5′-oligoadenylate synthetase (2′5′-OAS) (Figure 3). A group showed that ORF3 protein enhanced type I IFN production by HEK293T cells by interacting directly with the pattern recognition receptor (PRR) retinoic acid-inducible gene I (RIG-I) [101]. This group used the same in vitro system to show that ORF1 protein inhibits RIG-I signaling and prevents IFN-β induction by de-ubiquitination of RIG-I and tank binding kinase 1 (TBK1) [102]. The inhibitory effect is rather small, contributing in a minor way to HEV’s resistance to IFN, but silencing the key component gene of the Janus Kinase (JAK)-STAT cascade of IFN signaling, including JAK1, STAT1 and IRF9, stimulates HEV infection and replication, indicating that the IFN cascade can restrict HEV infection [100]. Analyses of gene/protein expression in A549 cells infected with HEV showed the robust induction of inflammatory cytokines/chemokines including TNF-α, IL-6, IL-8 and RANTES (regulated on activation, normal T cell expressed and secreted). HEV infection also led to activation of both NF-κB and IRF3, two transcription factors activated in innate immune signaling pathways [103]. These results obtained with different cell lines need further confirmation in other systems, like primary hepatocytes and/or in vivo animal models. Using primary human hepatocytes, it was shown that HEV was able to persist despite production of type III IFNs [104], suggesting that HEV may more efficiently block the signaling pathway of IFNs rather than their production in a more physiological system. Elevated ISG expression was detected in the livers of chimeric mice engrafted with human hepatocytes without adaptive immunity [105]. Conversely, another study found no intrahepatic ISG induction in humanized uPA/NOG mice infected with HEV1 or HEV3 [106], but the earliest tests were done two weeks post-inoculation, which would have missed an early, transient ISG induction. Lastly, high concentrations of IFN-λ3 were found in the serum of patients at the early phase of acute HEV infection [107]. In vitro experiments with A549 or human epithelial colorectal adenocarcinoma Caco-2 cells also showed that IFN-λ3 inhibited HEV replication in a dose-dependent manner [107]. These results need further confirmation using hepatocarcinoma cell lines like HepG2. Interestingly, even though HEV can replicate in the presence of IFNs, HEV can be cleared when infected HepG2 cells were treated with high doses of IFNs for a prolonged period [104].
Natural killer (NK) and natural killer T (NKT) cells constitute a major fraction of the lymphocytes in the liver, where they are important for the pathogenesis of viral hepatitis. Consequently, these cells could also play a major role together with infected hepatocytes in the innate immune response to HEV. The peripheral blood of acutely infected patients contains a higher proportion of CD4+ cells than in the blood of uninfected controls, but that of CD8+ cells is unchanged [108]. However, this increase in CD4+ cells is not associated with an expansion of HEV ORF2-specific CD4+ CD69+ cells producing helper T cell type 1 (IFN-γ and TNF-α) cytokines or helper T cell type 2 (IL-4) cytokines. The authors suggest that the expansion of CD4+ cells could reflect an increase in NKT cells, which can be either CD3+ CD4+ or CD3+ CD4− CD8− [108]. Another study showed that there are significantly fewer NK (CD3− CD56+) and NKT (CD3+ CD56+) cells in peripheral blood mononuclear cells (PBMCs) during acute hepatitis E than in uninfected controls [109]. However, the blood of patients with acute hepatitis E has a much greater proportion of activated NK cells than does the blood of uninfected controls. The apparent depletion of total NK and NKT cell fractions in PBMCs could reflect the increased migration of these cells to the liver of infected patients or their apoptosis following activation [109]. An immune-histological analysis of liver biopsies from HEV-infected acute liver failure patients showed that their cell counts of CD56+ were significantly higher than in biopsies from patients infected with HAV, HBV or HCV [110]. The number and the degree of activation of mast cells in Mongolian gerbils experimentally infected with HEV4 was also increased in both the liver and small intestine, suggesting that they are implicated in infection [111].
5.2. Humoral and Cell-Mediated Immune Responses
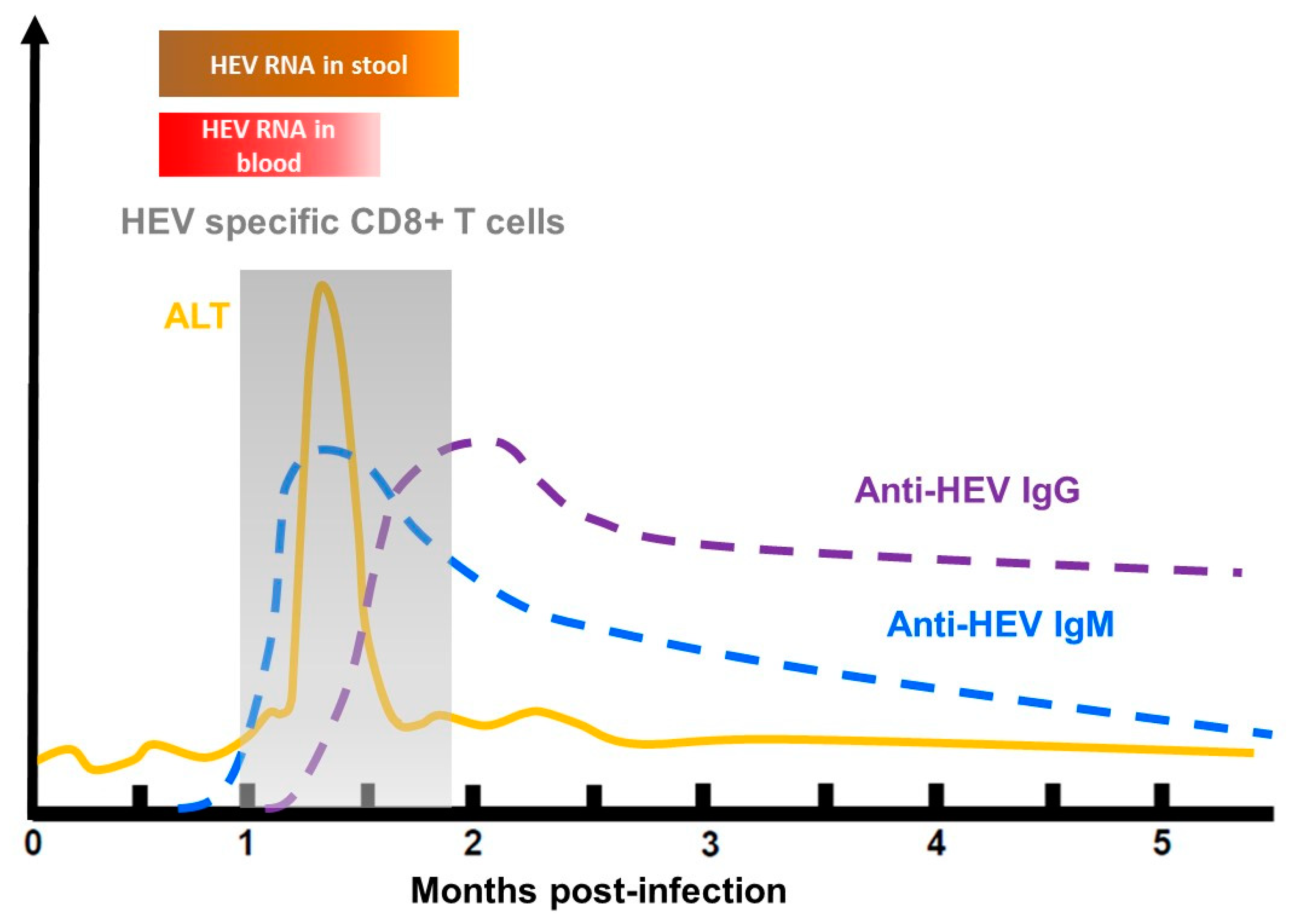
HEV-infected patients generally produce a serological anti-HEV response at the onset of illness. Anti-HEV IgMs are present in the early phase of clinical illness, and they can persist for several months. IgM are usually used to diagnose an HEV infection [112]. Anti-HEV IgG appears shortly after the IgM response and can last up to 14 years [113] (Figure 4). The capsid protein contains several neutralizing epitopes and is thus the main target of anti-HEV neutralizing antibodies [114]. Vaccination can also induce anti-HEV antibodies. Hecolin®, the only licensed vaccine, is composed of a truncated HEV capsid protein, p239, and confers protection against hepatitis E for up to 4.5 years [115,116]. To date, it is only available in China.
The risk of HEV reinfection is unclear. Cross-protection is possible as there is only one serotype [117]. Studies on non-human primates indicate that anti-HEV antibodies induced by passive or active immunization protect animals against infection. Antibodies also protect humans during outbreaks and non-outbreaks [118,119,120]. Although the minimum protective concentration of antibodies remains to be determined, an antibody concentration of 2.5 World Health Organization (WHO) units/mL was found to be protective in a vaccine study [116,121]. SOT recipients can be re-infected when the antibody concentration is below 7 WHO units/mL [122]. Lastly, the role of ORF2 during natural HEV infection remains to be determined. In vitro studies indicate that ORF2 is not essential for the HEV life cycle, but it reduces antibody-mediated neutralization [5]. Therefore, ORF2s may favor evasion of humoral immunity during HEV infection. This is intriguing since HEV particles in the blood are insensitive to neutralizing antibodies because of their associated lipid [123].
Studies have shown that effector T cells are activated during acute hepatitis E, with more CD8+ cells in infected patients than uninfected controls [124,125]. Husain et al. used an enzyme-linked immunospot (ELISPOT) assay to show that the proportions of PBMCs producing IFN-γ in response to stimulation with ORF2/ORF3 peptides are higher in infected patients than in uninfected controls [124], while the immunohistochemistry studies of Prabhu on liver biopsies from patients with HEV-induced acute liver failure indicate that their liver were infiltrated with activated CD8+ T cells containing granzymes [110].
An increased expression of CD11a integrin in naïve CD45RA+ T cells, as well as overexpression of CCR5 and CCR9, two chemokine receptors that play important roles in cell trafficking and homing, was also reported in peripheral blood of acutely infected patients. The expanded CD45RA+ CD11a high subpopulations present during the early phase of acute infection suggests the recruitment of these cells from the periphery to the liver, thus contributing to the pathogenesis of the infection [125]. Patients with acute HEV infection have a higher proportion of CD4+ CD25+ forkhead box P3+ (FoxP3+) and CD4+ CD25− FoxP3+ regulatory T cells and a rise in IL-10, a signature cytokine of regulatory T cells, than uninfected controls [126]. This suggests that the immunosuppressive immune response is involved in the acute phase of the infection, but its exact role remains to be clarified.
Several studies have reported a broad HEV-specific T cell response to both non-structural and structural proteins [127,128]. HEV-specific T cells are also present in the blood of convalescent patients [127,128] but their frequencies decline rapidly after resolution of the infection [127]. Lastly, specific T cell immunity also seems to confer cross-protection against HEV1 and HEV3 [129], which could protect patients previously exposed to HEV3 when travelling to areas where HEV1 is endemic.
6. Pathogenesis of Severe Hepatitis
Fulminant hepatitis is more frequent in men and women suffering from chronic liver disease [25,26,130] and in pregnant women [22].
6.1. Severe Hepatitis E in the General Population
The reasons why a HEV infection becomes severe or fulminant are still obscure. It was reported that stimulating both type Th1 and Th2 immune responses could play a role in liver failure. Patients with fulminant hepatic failure (FHF) have higher anti-HEV IgM and IgG concentrations than those with self-limiting infections [131]. Similarly, PBMCs from patients with FHF produce higher IFN-γ, TNF-α, IL-2 and IL-10 concentrations after stimulation with ORF2 peptides than do PBMCs from healthy controls [131]. In contrast, another study reported that the antiviral cellular immune responses and heightened humoral antiviral responses in patients with fulminant hepatitis E were less marked than those of patients with uncomplicated infection or healthy controls [132]. The heightened humoral response was associated with a more severe HEV disease in both studies. The situation in peripheral blood may not reflect that at the site of infection. CD4+ T cells are more frequent in the livers of patients with FHF due to HEV [110] and CD8+ T cells have been shown to infiltrate the livers of patients with fulminant hepatitis E [110,133]. Thus, cytotoxic CD8+ T cells could be particularly important in the pathogenesis of fulminant hepatitis. It was shown, recently, that liver damage during an HAV infection is mediated by the innate-like cytotoxicity of IL-15-activated bystander CD8+ T cells, i.e., stimulation of unrelated (heterologous) T cells by cytokines during an antigen-specific T-cell response [134]. Whether the liver damage is mediated in this way during HEV infection remains to be determined.
Viral factors may also be important. For example, HEV4 infections tend to be more severe than those due to other genotypes [135]. A Belgian study also found that the risk of HEV3c-infected patients being hospitalized was lower than that of patients infected with HEV3f [136]. The severity of an infection could be linked to the genotype and/or the subgenotype, perhaps due to specific mutations in the polyprotein. The HEV1 strains from six Indian patients with FHF contained six mutations in the ORF1 polyprotein (F179S, A317T, T735I, L1110F, V1120I and FG1439Y) which were not found in the strains from patients with uncomplicated acute hepatitis E [137]. However, Smith et al. re-examined the association between FHF and HEV genotypes and concluded that host factors are responsible for the development of fulminant hepatitis rather than virus genotype, variants or specific aa substitutions [138]. Despite this analysis, one recent study found two mutations (C1483W and N1530T) in the HEV1 polymerases from all 25 patients with acute liver failure but in none of the patients with acute hepatitis E [139]. The same group also identified three mutations in the methyltransferase (V27A, D29N, H105R) of the HEV1 from 16 patients with acute liver failure, which were not detected in patients with uncomplicated acute hepatitis E [140]. Consequently, the real impact of virus polymorphism on disease severity still needs to be clarified.
6.2. Severe Hepatitis in Pregnant Women
The cause of elevated maternal mortality (30%, with most deaths occurring in the third trimester) of pregnant women infected with HEV1 living in developing countries has been the subject of many studies. HEV genotype could explain the poorer outcome in pregnant women, at least in part, since HEV3 is not particularly deadly for pregnant women [141,142,143]. There are no data available for HEV4 infection during pregnancy. A recent study demonstrated that HEV1 replicates more efficiently than HEV3 ex vivo in tissue explants of decidua basalis and fetal placenta, and also in stromal cells [144]. HEV1 is associated with increased apoptosis and necrosis at the maternal–fetal interface with alterations in the architecture of the placental barrier. HEV1 also produces more infectious virions and triggers the production of a panel of pro-inflammatory cytokines like IL-6 and chemokines. These changes in the cytokine microenvironment correlate with virus load and contribute to tissue damage [144]. Such findings are consistent with data correlating the increase in several pro-inflammatory factors (TNF-α, IL-6, IFN-γ and TGF-β1) in the peripheral blood of HEV1-infected women with adverse pregnancy outcomes [145].
However, exactly how severe liver injury occurs during pregnancy remains unclear. Pregnancy is associated with changes in the sex hormone profile and the immune system to protect the fetus from the maternal immune system. A shift from a Th1-dominated immune response to a Th2-dominated one, “Th2 bias”, may help protect the fetus by suppressing macrophage activation [146]. Although the existence of a Th2 bias in HEV-infected pregnant women was confirmed, its implication for the severity of the infection is unknown [147]. Women with acute liver failure have a reduced expression of toll-like receptor (TLR) 3/TLR7/TLR9, the key pattern recognition receptor in innate immunity, and their phagocytic macrophages are weaker than those of women with acute viral hepatitis E [148]. However, the phagocytic capacities of monocytes from the two groups are essentially the same. Impaired monocyte-macrophage function in pregnant women with acute liver failure could contribute to an inadequate innate immune response, and hence to the development and severity of acute liver failure.
Other host factors, such as nutritional status, including micronutrient or folate deficiencies, or differences in major histocompatibility complex, may also influence the immune response of pregnant women to HEV [149]. This may be why HEV infections are benign in pregnant women in Egypt, although they are caused by HEV1 [150]. Lastly, pregnancy-associated hormones can also contribute to a poor outcome. HEV-positive pregnant women who develop FHF have higher concentrations of estrogen, progesterone and β-human chorionic gonadotrophin (β-HCG) than HEV-negative pregnant women with FHF or healthy controls [151]. An in vitro study showed that serum from pregnant women, especially those in the third trimester, enhanced the replication of HEV by inhibiting estrogen receptors and the synthesis of type I IFNs [152]. While some studies have found that the high HEV RNA concentrations in HEV-infected pregnant women were associated with a poor outcome [153,154,155], another study has not confirmed these results, with HEV RNA detected only in 1/7 pregnant women [131].
7. Pathogenesis of Chronic Infection in Immunocompromised Patients
Most studies on the pathogenesis of chronic HEV installation have focused on SOT recipients. The incidence of HEV infection in these patients varies from 0.9% to 3.5%, based on detecting HEV RNA, and acute infections become chronic in ~60% of them [48]. Other studies that were interested in the seroconversion rate among transplant recipients have reported rates of progression to chronicity ranging from 21% to 50% [156,157,158].
Whether an acute HEV infection resolves without sequelae or progresses to chronic hepatitis seems to be related primarily to the host’s immune response. The ISGs response of renal transplant recipients who did not clear their HEV infection was higher than that of patients who cleared their HEV [159]. This suggests that activation of the IFN signaling pathway does not always lead to spontaneous HEV clearance. The increased expression of ISGs in patients with a chronic HEV infection seems to favor virus persistence by causing the IFN signaling pathway to become refractory [159]. HEV can persist in vitro in human hepatoma cells and primary human hepatocytes despite the continuous production of type III IFNs [104]. In addition, the persistent activation of the JAK/STAT signaling rendered infected cells refractory to exogenous IFN [104]. Low concentrations of IL-1Ra and sIL-2R, together with high concentrations of chemokines, during the acute phase, are also associated with HEV persistence [160]. An HEV infection is likely to become chronic in profoundly immunosuppressed patients. The numbers of CD2+, CD3+ and CD4+ T cells are significantly lower in these patients than in those who spontaneously eliminate the virus [33]. Chronic HEV infections are more frequent in patients who are also infected with HIV, especially those with a low CD4+ T cell count [34,35,36]. In addition, the HEV-specific T cell proliferative responses of SOT patients, especially those with a chronic infection, are weaker. The development of HEV-specific IFN-γ-producing cells seems to be associated with a favorable outcome in SOT patients [161,162]. Another study has shown that the γδ cells of SOT patients are mobilized during the acute phase of infection [163]. Immunocompetent patients do not produce such an immune response, suggesting that SOT patients mobilize a larger fraction of their immunity due to immunosuppressive drug treatment. The antiviral role of these cells at the acute phase of infection needs further investigation.
The immunosuppressive regimen can also influence the development of a chronic infection. Pigs given cyclosporin, azathioprine and prednisolone and infected with HEV3 developed chronic infections [164]. In humans, HEV is more likely to be persistent in SOT patients treated with tacrolimus rather than cyclosporin [48]. Cyclosporin and tacrolimus are both immunosuppressive drugs that inhibit the calcineurin phosphatase in lymphocytes, but tacrolimus impairs the specific T cell response more efficiently than does cyclosporin [165]. In vitro studies have shown that both of these calcineurin inhibitors promote HEV replication by inhibiting cyclophilins A and B, while mycophenolic acid, an inhibitor of inosine 5′ monophosphate dehydrogenase, inhibits HEV replication [166]. Rapamycin and everolimus also promote HEV replication in vitro by inhibiting the mechanistic target of rapamycin (mTOR), showing that the PI3K-PKB-mTOR pathway acts as a cell restriction factor [167]. In line with this observation, patients given mTOR inhibitors have higher HEV RNA concentrations in the blood, while those given mycophenolic acid do not have lower HEV RNA concentrations [168].
Lastly, virus factors may favor the persistence of HEV. Greater quasi-species heterogeneity in the ORF1 and ORF2 regions during the acute phase of infection is associated with HEV persistence [160,169]. Chronic infection was found to be rare in a large cohort of Japanese liver transplant recipients, suggesting that there are differences in HEV subtype, strains, or host genetic factors that influence persistence [170,171]. Fulminant hepatitis has not been reported in HEV-infected transplant recipients.
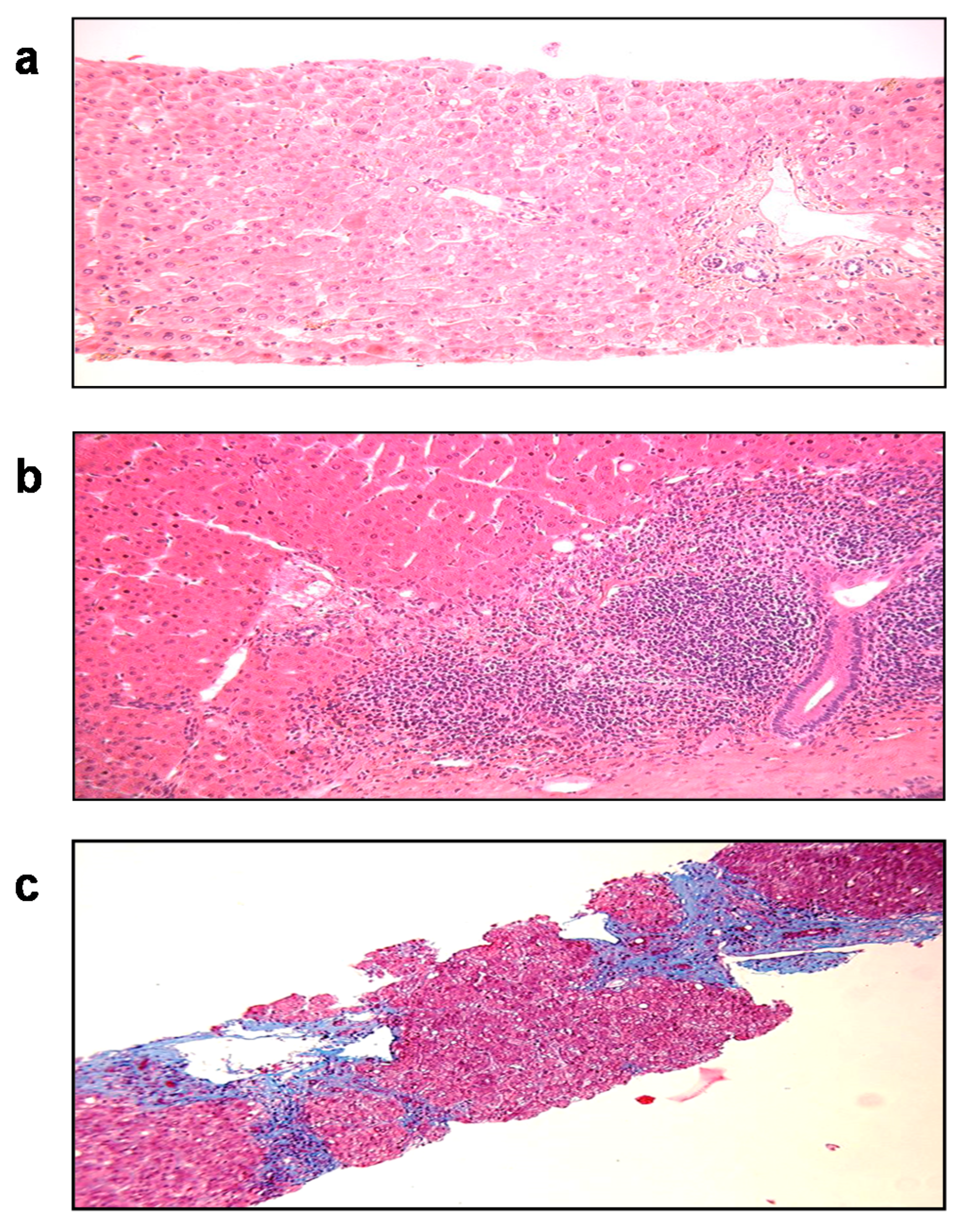
Nearly 10% of SOT patients with HEV develop cirrhosis within 3 to 5 years after the primary infection (Figure 5). Progression to liver fibrosis seems to be associated with the slow diversification of the P domain of the capsid [160]. More aggressive variants may be selected in fibroses, although this needs confirmation in a larger cohort.
Lastly, chronically infected patients have recently been found to harbor recombinant HEV-host variants [172,173,174]. The PPR regions of these recombinant variants included fragments of human genes of varying origin (inter alpha trypsin inhibitor (ITI-H2), ribosomal genes S17 or S19 and tyrosine aminotransferase). Variants harboring the S17, S19 or ITI fragment had a replicative advantage in vitro, while the impact of TAT was not studied. Duplications and insertions of the HEV genome were also described [173,175]. Their influence on HEV infection is unknown and needs further investigation.
8. Treatment
There is no recommended treatment for acute HEV infections. They are usually self-limiting with spontaneous HEV clearance. Ribavirin has been suggested for some cases of immunocompetent patients with severe hepatitis [176], but as there was no control group, it is difficult to claim that ribavirin really improved the course of the infection.
Most studies of the treatment of chronic HEV infection have involved SOT patients. Since the intensity of the immune host response favors HEV persistence, reducing immunosuppressive drugs, particularly those targeting T cells, is the initial option. It leads to HEV clearance in nearly 1/3 of chronically HEV-infected SOT recipients [177]. Antiviral therapy is required for the remaining patients. Pegylated-interferon-α has been successfully used to treat some liver transplant recipients and a hemodialysis patient who cleared HEV after a three-month course of therapy [178,179,180]. However, interferon is generally contraindicated in kidney-, pancreas-, heart- and lung-transplant recipients because it increases the risk of acute rejection by stimulating the immune system.
Ribavirin monotherapy has been extensively studied for treating chronic HEV infections in SOT recipients [158,181,182]. The sustained virological response (SVR) of 59 SOT recipients was 78%, after treatment with ribavirin (median dose: 600 mg/day; range: 29–1200) for three (range, 1–18) months as part of a multicenter retrospective study. Relapse patients who were retreated with ribavirin for a longer period (six months) cleared the virus and achieved SVR, increasing the overall SVR to 90% [183]. The optimal duration of treatment has still to be determined. Treatment should be continued for a further three months if HEV RNA is detected in the stool at the end of the scheduled duration [184]. Thus, HEV RNA in the stools should be monitored before stopping therapy. Ribavirin may block HEV production, as the active metabolite ribavirin monophosphate is a competitive inhibitor of cellular enzyme inosine monophosphate dehydrogenase (IMPDH). In vitro experiments suggest that ribavirin activity results in depletion of the intracellular guanosine triphosphate (GTP) pools, which impedes RNA replication by inhibiting IMPDH [185]. Ribavirin may also act by inducing mutagenesis of the HEV genome [186], therefore disrupting virus replication due to error catastrophe [187]. Mutations in the polymerase, especially the 1634R mutation, have been found in two patients with ribavirin failure [188], but its detection at baseline does not preclude treatment failure [189]. Other mutations in the HEV polymerase have been described: K1383N, D1384G, K1398R, V1479I and Y1587F [186,190], but their presence before treatment does not influence SVR [191].
No other antiviral therapy is presently known to be effective against a chronic HEV infection. Sofosbuvir was shown to have antiviral activity against HEV in vitro [192] but had limited efficacy in HEV-infected patients [193,194,195,196,197,198,199,200]. A recent pilot study of nine patients (seven for whom ribavirin had previously failed) given sofosbuvir (400 mg/day) for 24 weeks found that their HEV viremia decreased, but was not cleared. This suggests that sofosbuvir alone is not a cure for HEV [201]. Zinc salts can block HEV replication in vitro by inhibiting RNA polymerase [202]. However, two female transplant-recipients with chronic HEV infections did not respond to ribavirin despite having elevated intra-erythrocyte zinc concentrations [203]. Clearly, further work with a larger cohort is needed. The natural compound silvestrol blocks HEV replication in vitro [204], and the HEV RNA concentrations in the feces of treated mice rapidly decreased. Whether this compound has a similar action in humans remains to be determined. Screening to identify new anti-HEV drugs found two novel HEV antiviral candidates: NITD008, a broad-spectrum chain-terminating adenosine nucleoside analogue initially developed to treat dengue virus, and GPC-N114, which binds to the RNA channels of picornavirus polymerases [205,206]. These compounds are promising HEV antiviral candidates. Lastly, T cell therapy may be an alternative to drugs [207].
9. Conclusions
The pathogenesis of HEV infection involves a complex interplay between the virus and the host immune response. While the clinical phenotype of an HEV infection is primarily hepatological, the range and incidence of HEV-associated clinical symptoms is much broader, including neurological and renal disorders. Ribavirin is currently the treatment of choice for chronically infected patients but there is an urgent need to find other compounds with which to treat patients who fail to clear HEV after ribavirin therapy.
Author Contributions
All the authors helped draft the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
The English text was edited by Owen Parkes.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kamar, N.; Izopet, J.; Pavio, N.; Aggarwal, R.; Labrique, A.; Wedemeyer, H.; Dalton, H.R. Hepatitis E virus infection. Nat. Rev. Dis. Primers 2017, 3, 17086. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.J. Hepatitis E Virus Genome Structure and Replication Strategy. Cold Spring Harb. Perspect. Med. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Karlin, D.G. Sequence analysis reveals a conserved extension in the capping enzyme of the alphavirus supergroup, and a homologous domain in nodaviruses. Biol. Direct 2015, 10, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar, B.N.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ying, D.; Lhomme, S.; Tang, Z.; Walker, C.M.; Xia, N.; Zheng, Z.; Feng, Z. Origin, antigenicity, and function of a secreted form of ORF2 in hepatitis E virus infection. Proc. Natl. Acad. Sci. USA 2018, 115, 4773–4778. [Google Scholar] [CrossRef] [Green Version]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked Viruses that aren’t Always Naked: Quasi-Enveloped Agents of Acute Hepatitis. Annu. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef]
- Feng, Z.; Lemon, S.M. Peek-a-boo: Membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 2014, 22, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Smith, D.B.; Simmonds, P. Classification and Genomic Diversity of Enterically Transmitted Hepatitis Viruses. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Nicot, F.; Jeanne, N.; Roulet, A.; Lefebvre, C.; Carcenac, R.; Manno, M.; Dubois, M.; Kamar, N.; Lhomme, S.; Abravanel, F.; et al. Diversity of hepatitis E virus genotype 3. Rev. Med. Virol. 2018, 28, e1987. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Izopet, J.; Oliveira-Filho, E.F.; Ulrich, R.G.; Johne, R.; Koenig, M.; Jameel, S.; Harrison, T.J.; Meng, X.J.; et al. Proposed reference sequences for hepatitis E virus subtypes. J. Gen. Virol. 2016, 97, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.P. The Current Host Range of Hepatitis E Viruses. Viruses 2019, 11, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Izopet, J.; Dubois, M.; Bertagnoli, S.; Lhomme, S.; Marchandeau, S.; Boucher, S.; Kamar, N.; Abravanel, F.; Guerin, J.L. Hepatitis E virus strains in rabbits and evidence of a closely related strain in humans, France. Emerg. Infect. Dis. 2012, 18, 1274–1281. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Lhomme, S.; El Costa, H.; Schvartz, B.; Peron, J.M.; Kamar, N.; Izopet, J. Rabbit Hepatitis E Virus Infections in Humans, France. Emerg. Infect. Dis. 2017, 23, 1191–1193. [Google Scholar] [CrossRef] [Green Version]
- Sahli, R.; Fraga, M.; Semela, D.; Moradpour, D.; Gouttenoire, J. Rabbit HEV in immunosuppressed patients with hepatitis E acquired in Switzerland. J. Hepatol. 2019, 70, 1023–1025. [Google Scholar] [CrossRef] [Green Version]
- Lee, G.H.; Tan, B.H.; Teo, E.C.; Lim, S.G.; Dan, Y.Y.; Wee, A.; Aw, P.P.; Zhu, Y.; Hibberd, M.L.; Tan, C.K.; et al. Chronic Infection With Camelid Hepatitis E Virus in a Liver Transplant Recipient Who Regularly Consumes Camel Meat and Milk. Gastroenterology 2016, 150, 355–357. [Google Scholar] [CrossRef] [Green Version]
- Li, T.C.; Bai, H.; Yoshizaki, S.; Ami, Y.; Suzaki, Y.; Doan, Y.H.; Takahashi, K.; Mishiro, S.; Takeda, N.; Wakita, T. Genotype 5 Hepatitis E Virus Produced by a Reverse Genetics System Has the Potential for Zoonotic Infection. Hepatol. Commun. 2019, 3, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Teng, J.L.L.; Lau, S.K.P.; Sridhar, S.; Fu, H.; Gong, W.; Li, M.; Xu, Q.; He, Y.; Zhuang, H.; et al. Transmission of a Novel Genotype of Hepatitis E Virus from Bactrian Camels to Cynomolgus Macaques. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Sridhar, S.; Yip, C.C.Y.; Wu, S.; Cai, J.; Zhang, A.J.; Leung, K.H.; Chung, T.W.H.; Chan, J.F.W.; Chan, W.M.; Teng, J.L.L.; et al. Rat Hepatitis E Virus as Cause of Persistent Hepatitis after Liver Transplant. Emerg. Infect. Dis. 2018, 24, 2241–2250. [Google Scholar] [CrossRef] [Green Version]
- Andonov, A.; Robbins, M.; Borlang, J.; Cao, J.; Hattchete, T.; Stueck, A.; Deschaumbault, Y.; Murnaghan, K.; Varga, J.; Johnston, B. Rat hepatitis E virus linked to severe acute hepatitis in an immunocompetent patient. J. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Wedemeyer, H.; Pischke, S.; Manns, M.P. Pathogenesis and treatment of hepatitis e virus infection. Gastroenterology 2012, 142, 1388–1397. [Google Scholar] [CrossRef] [PubMed]
- Navaneethan, U.; Al Mohajer, M.; Shata, M.T. Hepatitis E and pregnancy: Understanding the pathogenesis. Liver Int. 2008, 28, 1190–1199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khuroo, M.S.; Kamili, S.; Khuroo, M.S. Clinical course and duration of viremia in vertically transmitted hepatitis E virus (HEV) infection in babies born to HEV-infected mothers. J. Viral Hepat. 2009, 16, 519–523. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Singhal, A.; Panda, S.K.; Acharya, S.K. A 20-year single-center experience with acute liver failure during pregnancy: Is the prognosis really worse? Hepatology 2008, 48, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Dalton, H.R.; Hazeldine, S.; Banks, M.; Ijaz, S.; Bendall, R. Locally acquired hepatitis E in chronic liver disease. Lancet 2007, 369, 1260. [Google Scholar] [CrossRef]
- Peron, J.M.; Bureau, C.; Poirson, H.; Mansuy, J.M.; Alric, L.; Selves, J.; Dupuis, E.; Izopet, J.; Vinel, J.P. Fulminant liver failure from acute autochthonous hepatitis E in France: Description of seven patients with acute hepatitis E and encephalopathy. J. Viral Hepat. 2007, 14, 298–303. [Google Scholar] [CrossRef]
- Guillois, Y.; Abravanel, F.; Miura, T.; Pavio, N.; Vaillant, V.; Lhomme, S.; Le Guyader, F.S.; Rose, N.; Le Saux, J.C.; King, L.A.; et al. High Proportion of Asymptomatic Infections in an Outbreak of Hepatitis E Associated With a Spit-Roasted Piglet, France, 2013. Clin. Infect. Dis. 2016, 62, 351–357. [Google Scholar] [CrossRef]
- Said, B.; Ijaz, S.; Kafatos, G.; Booth, L.; Thomas, H.L.; Walsh, A.; Ramsay, M.; Morgan, D.; Hepatitis, E.I.I.T. Hepatitis E outbreak on cruise ship. Emerg. Infect. Dis. 2009, 15, 1738–1744. [Google Scholar] [CrossRef]
- Faber, M.; Willrich, N.; Schemmerer, M.; Rauh, C.; Kuhnert, R.; Stark, K.; Wenzel, J.J. Hepatitis E virus seroprevalence, seroincidence and seroreversion in the German adult population. J. Viral Hepat. 2018, 25, 752–758. [Google Scholar] [CrossRef]
- Lhomme, S.; Gallian, P.; Dimeglio, C.; Assal, A.; Abravanel, F.; Tiberghien, P.; Izopet, J. Viral load and clinical manifestations of hepatitis E virus genotype 3 infections. J. Viral Hepat. 2019, 26, 1139–1142. [Google Scholar] [CrossRef]
- Gerolami, R.; Moal, V.; Colson, P. Chronic hepatitis E with cirrhosis in a kidney-transplant recipient. N. Engl. J. Med. 2008, 358, 859–860. [Google Scholar] [CrossRef] [PubMed]
- Haagsma, E.B.; van den Berg, A.P.; Porte, R.J.; Benne, C.A.; Vennema, H.; Reimerink, J.H.; Koopmans, M.P. Chronic hepatitis E virus infection in liver transplant recipients. Liver Transplant. 2008, 14, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Peron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E virus and chronic hepatitis in organ-transplant recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colson, P.; Kaba, M.; Moreau, J.; Brouqui, P. Hepatitis E in an HIV-infected patient. J. Clin. Virol. 2009, 45, 269–271. [Google Scholar] [CrossRef]
- Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Tedder, R.S.; Ijaz, S. Persistent carriage of hepatitis E virus in patients with HIV infection. N. Engl. J. Med. 2009, 361, 1025–1027. [Google Scholar] [CrossRef]
- Kenfak-Foguena, A.; Schoni-Affolter, F.; Burgisser, P.; Witteck, A.; Darling, K.E.; Kovari, H.; Kaiser, L.; Evison, J.M.; Elzi, L.; Gurter-De La Fuente, V.; et al. Hepatitis E Virus seroprevalence and chronic infections in patients with HIV, Switzerland. Emerg. Infect. Dis. 2011, 17, 1074–1078. [Google Scholar] [CrossRef]
- Tamura, A.; Shimizu, Y.K.; Tanaka, T.; Kuroda, K.; Arakawa, Y.; Takahashi, K.; Mishiro, S.; Shimizu, K.; Moriyama, M. Persistent infection of hepatitis E virus transmitted by blood transfusion in a patient with T-cell lymphoma. Hepatol. Res. 2007, 37, 113–120. [Google Scholar] [CrossRef]
- Ollier, L.; Tieulie, N.; Sanderson, F.; Heudier, P.; Giordanengo, V.; Fuzibet, J.G.; Nicand, E. Chronic hepatitis after hepatitis E virus infection in a patient with non-Hodgkin lymphoma taking rituximab. Ann. Intern. Med. 2009, 150, 430–431. [Google Scholar] [CrossRef]
- Geng, Y.; Zhang, H.; Huang, W.; Harrison, T.J.; Geng, K.; Li, Z.; Wang, Y.. Persistent hepatitis e virus genotype 4 infection in a child with acute lymphoblastic leukemia. Hepat. Mon. 2014, 14, e15618. [Google Scholar] [CrossRef] [Green Version]
- Peron, J.M.; Mansuy, J.M.; Recher, C.; Bureau, C.; Poirson, H.; Alric, L.; Izopet, J.; Vinel, J.P. Prolonged hepatitis E in an immunocompromised patient. J. Gastroenterol. Hepatol. 2006, 21, 1223–1224. [Google Scholar] [CrossRef]
- Versluis, J.; Pas, S.D.; Agteresch, H.J.; de Man, R.A.; Maaskant, J.; Schipper, M.E.; Osterhaus, A.D.; Cornelissen, J.J.; van der Eijk, A.A. Hepatitis E virus: An underestimated opportunistic pathogen in recipients of allogeneic hematopoietic stem cell transplantation. Blood 2013, 122, 1079–1086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, H.; Luxembourger, C.; Gottenberg, J.E.; Fournier, S.; Abravanel, F.; Cantagrel, A.; Chatelus, E.; Claudepierre, P.; Hudry, C.; Izopet, J.; et al. Outcome of hepatitis E virus infection in patients with inflammatory arthritides treated with immunosuppressants: A French retrospective multicenter study. Medicine 2015, 94, e675. [Google Scholar] [CrossRef] [PubMed]
- Pischke, S.; Peron, J.M.; von Wulffen, M.; von Felden, J.; Honer Zu Siederdissen, C.; Fournier, S.; Lutgehetmann, M.; Iking-Konert, C.; Bettinger, D.; Par, G.; et al. Chronic Hepatitis E in Rheumatology and Internal Medicine Patients: A Retrospective Multicenter European Cohort Study. Viruses 2019, 11, 186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robins, A.E.M.; Bowden, D.J.; Gelson, W.T.H. Chronic genotype 1 hepatitis E infection from immunosuppression for ileo-colonic Crohn’s disease. Oxf. Med. Case Rep. 2018, 2018, omy059. [Google Scholar] [CrossRef]
- Ankcorn, M.; Haywood, B.; Tedder, R.; Ijaz, S. Response to: ‘Chronic genotype 1 hepatitis E infection from immunosuppression for ileo-colonic Crohn’s disease’. Oxf. Med. Case Rep. 2019, 2019, omy125. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Legrand-Abravanel, F.; Izopet, J. How should hepatitis E virus infection be defined in organ-transplant recipients? Am. J. Transplant. 2013, 13, 1935–1936. [Google Scholar] [CrossRef]
- Kamar, N.; Mansuy, J.M.; Cointault, O.; Selves, J.; Abravanel, F.; Danjoux, M.; Otal, P.; Esposito, L.; Durand, D.; Izopet, J.; et al. Hepatitis E virus-related cirrhosis in kidney- and kidney-pancreas-transplant recipients. Am. J. Transplant. 2008, 8, 1744–1748. [Google Scholar] [CrossRef]
- Kamar, N.; Garroutste, C.; Haagsma, E.B.; Garrigue, V.; Pischke, S.; Chauvet, C.; Dumortier, J.; Cannesson, A.; Cassuto-Viguier, E.; Thervet, E.; et al. Factors associated with chronic hepatitis in patients with hepatitis E virus infection who have received solid organ transplants. Gastroenterology 2011, 140, 1481–1489. [Google Scholar] [CrossRef]
- Dalton, H.R.; Kamar, N.; van Eijk, J.J.; McLean, B.N.; Cintas, P.; Bendall, R.P.; Jacobs, B.C. Hepatitis E virus and neurological injury. Nat. Rev. Neurol. 2016, 12, 77–85. [Google Scholar] [CrossRef]
- Dalton, H.R.; van Eijk, J.J.J.; Cintas, P.; Madden, R.G.; Jones, C.; Webb, G.W.; Norton, B.; Pique, J.; Lutgens, S.; Devooght-Johnson, N.; et al. Hepatitis E virus infection and acute non-traumatic neurological injury: A prospective multicentre study. J. Hepatol. 2017, 67, 925–932. [Google Scholar] [CrossRef]
- van Eijk, J.J.; Madden, R.G.; van der Eijk, A.A.; Hunter, J.G.; Reimerink, J.H.; Bendall, R.P.; Pas, S.D.; Ellis, V.; van Alfen, N.; Beynon, L.; et al. Neuralgic amyotrophy and hepatitis E virus infection. Neurology 2014, 82, 498–503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Eijk, J.J.J.; Dalton, H.R.; Ripellino, P.; Madden, R.G.; Jones, C.; Fritz, M.; Gobbi, C.; Melli, G.; Pasi, E.; Herrod, J.; et al. Clinical phenotype and outcome of hepatitis E virus-associated neuralgic amyotrophy. Neurology 2017, 89, 909–917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fritz, M.; Berger, B.; Schemmerer, M.; Endres, D.; Wenzel, J.J.; Stich, O.; Panning, M. Pathological Cerebrospinal Fluid Findings in Patients with Neuralgic Amyotrophy and Acute Hepatitis E Virus Infection. J. Infect. Dis. 2018, 217, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Geurtsvankessel, C.H.; Islam, Z.; Mohammad, Q.D.; Jacobs, B.C.; Endtz, H.P.; Osterhaus, A.D. Hepatitis E and Guillain-Barre syndrome. Clin. Infect. Dis. 2013, 57, 1369–1370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukae, J.; Tsugawa, J.; Ouma, S.; Umezu, T.; Kusunoki, S.; Tsuboi, Y. Guillain-Barre and Miller Fisher syndromes in patients with anti-hepatitis E virus antibody: A hospital-based survey in Japan. Neurol. Sci. 2016, 37, 1849–1851. [Google Scholar] [CrossRef] [PubMed]
- van den Berg, B.; van der Eijk, A.A.; Pas, S.D.; Hunter, J.G.; Madden, R.G.; Tio-Gillen, A.P.; Dalton, H.R.; Jacobs, B.C. Guillain-Barre syndrome associated with preceding hepatitis E virus infection. Neurology 2014, 82, 491–497. [Google Scholar] [CrossRef]
- Stevens, O.; Claeys, K.G.; Poesen, K.; Saegeman, V.; Van Damme, P. Diagnostic Challenges and Clinical Characteristics of Hepatitis E Virus-Associated Guillain-Barre Syndrome. JAMA Neurol. 2017, 74, 26–33. [Google Scholar] [CrossRef]
- Abravanel, F.; Pique, J.; Couturier, E.; Nicot, F.; Dimeglio, C.; Lhomme, S.; Chiabrando, J.; Saune, K.; Peron, J.M.; Kamar, N.; et al. Acute hepatitis E in French patients and neurological manifestations. J. Infect. 2018, 77, 220–226. [Google Scholar] [CrossRef]
- Kamar, N.; Izopet, J.; Cintas, P.; Garrouste, C.; Uro-Coste, E.; Cointault, O.; Rostaing, L. Hepatitis E virus-induced neurological symptoms in a kidney-transplant patient with chronic hepatitis. Am. J. Transplant. 2010, 10, 1321–1324. [Google Scholar] [CrossRef]
- Shi, R.; Soomro, M.H.; She, R.; Yang, Y.; Wang, T.; Wu, Q.; Li, H.; Hao, W. Evidence of Hepatitis E virus breaking through the blood-brain barrier and replicating in the central nervous system. J. Viral Hepat. 2016, 23, 930–939. [Google Scholar] [CrossRef]
- Tian, J.; Shi, R.; Liu, T.; She, R.; Wu, Q.; An, J.; Hao, W.; Soomro, M.H. Brain Infection by Hepatitis E Virus Probably via Damage of the Blood-Brain Barrier Due to Alterations of Tight Junction Proteins. Front. Cell. Infect. Microbiol. 2019, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, F.; Xu, L.; Lin, Z.; de Vrij, F.M.S.; Ayo-Martin, A.C.; van der Kroeg, M.; Zhao, M.; Yin, Y.; Wang, W.; et al. Hepatitis E Virus Infects Neurons and Brains. J. Infect. Dis. 2017, 215, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Helsen, N.; Debing, Y.; Paeshuyse, J.; Dallmeier, K.; Boon, R.; Coll, M.; Sancho-Bru, P.; Claes, C.; Neyts, J.; Verfaillie, C.M. Stem cell-derived hepatocytes: A novel model for hepatitis E virus replication. J. Hepatol. 2016, 64, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Li, T.C.; Suzaki, Y.; Ami, Y.; Tsunemitsu, H.; Miyamura, T.; Takeda, N. Mice are not susceptible to hepatitis E virus infection. J. Vet. Med. Sci. 2008, 70, 1359–1362. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Mansuy, J.M.; Esposito, L.; Legrand-Abravanel, F.; Peron, J.M.; Durand, D.; Rostaing, L.; Izopet, J. Acute hepatitis and renal function impairment related to infection by hepatitis E virus in a renal allograft recipient. Am. J. Kidney Dis. 2005, 45, 193–196. [Google Scholar] [CrossRef]
- Kamar, N.; Weclawiak, H.; Guilbeau-Frugier, C.; Legrand-Abravanel, F.; Cointault, O.; Ribes, D.; Esposito, L.; Cardeau-Desangles, I.; Guitard, J.; Sallusto, F.; et al. Hepatitis E virus and the kidney in solid-organ transplant patients. Transplantation 2012, 93, 617–623. [Google Scholar] [CrossRef]
- Taton, B.; Moreau, K.; Lepreux, S.; Bachelet, T.; Trimoulet, P.; De Ledinghen, V.; Pommereau, A.; Ronco, P.; Kamar, N.; Merville, P.; et al. Hepatitis E virus infection as a new probable cause of de novo membranous nephropathy after kidney transplantation. Transpl. Infect. Dis. 2013, 15, E211–E215. [Google Scholar] [CrossRef]
- Guinault, D.; Ribes, D.; Delas, A.; Milongo, D.; Abravanel, F.; Puissant-Lubrano, B.; Izopet, J.; Kamar, N. Hepatitis E Virus-Induced Cryoglobulinemic Glomerulonephritis in a Nonimmunocompromised Person. Am. J. Kidney Dis. 2016, 67, 660–663. [Google Scholar] [CrossRef]
- Del Bello, A.; Guilbeau-Frugier, C.; Josse, A.G.; Rostaing, L.; Izopet, J.; Kamar, N. Successful treatment of hepatitis E virus-associated cryoglobulinemic membranoproliferative glomerulonephritis with ribavirin. Transpl. Infect. Dis. 2015, 17, 279–283. [Google Scholar] [CrossRef]
- Marion, O.; Abravanel, F.; Del Bello, A.; Esposito, L.; Lhomme, S.; Puissant-Lubrano, B.; Alric, L.; Faguer, S.; Izopet, J.; Kamar, N. Hepatitis E virus-associated cryoglobulinemia in solid-organ-transplant recipients. Liver Int. 2018, 38, 2178–2189. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G. Renal involvement in hepatitis C infection: Cryoglobulinemic glomerulonephritis. Kidney Int. 1998, 54, 650–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Zhao, C.; Huang, W.; Harrison, T.J.; Zhang, H.; Geng, K.; Wang, Y. Detection and assessment of infectivity of hepatitis E virus in urine. J. Hepatol. 2016, 64, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Marion, O.; Capelli, N.; Lhomme, S.; Dubois, M.; Pucelle, M.; Abravanel, F.; Kamar, N.; Izopet, J. Hepatitis E virus genotype 3 and capsid protein in the blood and urine of immunocompromised patients. J. Infect. 2019, 78, 232–240. [Google Scholar] [CrossRef]
- Montpellier, C.; Wychowski, C.; Sayed, I.M.; Meunier, J.C.; Saliou, J.M.; Ankavay, M.; Bull, A.; Pillez, A.; Abravanel, F.; Helle, F.; et al. Hepatitis E Virus Lifecycle and Identification of 3 Forms of the ORF2 Capsid Protein. Gastroenterology 2018, 154, 211–223. [Google Scholar] [CrossRef]
- Robinson, R.A.; Burgess, W.H.; Emerson, S.U.; Leibowitz, R.S.; Sosnovtseva, S.A.; Tsarev, S.; Purcell, R.H. Structural characterization of recombinant hepatitis E virus ORF2 proteins in baculovirus-infected insect cells. Protein Expr. Purif. 1998, 12, 75–84. [Google Scholar] [CrossRef]
- Williams, T.P.; Kasorndorkbua, C.; Halbur, P.G.; Haqshenas, G.; Guenette, D.K.; Toth, T.E.; Meng, X.J. Evidence of extrahepatic sites of replication of the hepatitis E virus in a swine model. J. Clin. Microbiol. 2001, 39, 3040–3046. [Google Scholar] [CrossRef] [Green Version]
- Soomro, M.H.; Shi, R.; She, R.; Yang, Y.; Hu, F.; Li, H. Antigen detection and apoptosis in Mongolian gerbil’s kidney experimentally intraperitoneally infected by swine hepatitis E virus. Virus Res. 2016, 213, 343–352. [Google Scholar] [CrossRef]
- Han, J.; Lei, Y.; Liu, L.; Liu, P.; Xia, J.; Zhang, Y.; Zeng, H.; Wang, L.; Wang, L.; Zhuang, H. SPF rabbits infected with rabbit hepatitis E virus isolate experimentally showing the chronicity of hepatitis. PLoS ONE 2014, 9, e99861. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Xia, J.; Wang, L.; Wang, Y. Experimental infection of rabbits with genotype 3 hepatitis E virus produced both chronicity and kidney injury. Gut 2017, 66, 561–562. [Google Scholar] [CrossRef]
- Zamvar, V.; McClean, P.; Odeka, E.; Richards, M.; Davison, S. Hepatitis E virus infection with nonimmune hemolytic anemia. J. Pediatr. Gastroenterol. Nutr. 2005, 40, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Abid, S.; Khan, A.H. Severe hemolysis and renal failure in glucose-6-phosphate dehydrogenase deficient patients with hepatitis E. Am. J. Gastroenterol. 2002, 97, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Thapa, R.; Pramanik, S.; Biswas, B.; Mallick, D. Hepatitis E virus infection in a 7-year-old boy with glucose 6-phosphate dehydrogenase deficiency. J. Pediatr. Hematol. Oncol. 2009, 31, 223–224. [Google Scholar] [CrossRef] [PubMed]
- Monga, A.; Makkar, R.P.; Arora, A.; Mukhopadhyay, S.; Gupta, A.K. Case report: Acute hepatitis E infection with coexistent glucose-6-phosphate dehydrogenase deficiency. Can. J. Infect. Dis. 2003, 14, 230–231. [Google Scholar] [CrossRef] [Green Version]
- Shah, S.A.; Lal, A.; Idrees, M.; Hussain, A.; Jeet, C.; Malik, F.A.; Iqbal, Z.; Rehman, H. Hepatitis E virus-associated aplastic anaemia: The first case of its kind. J. Clin. Virol. 2012, 54, 96–97. [Google Scholar] [CrossRef]
- Woolson, K.L.; Forbes, A.; Vine, L.; Beynon, L.; McElhinney, L.; Panayi, V.; Hunter, J.G.; Madden, R.G.; Glasgow, T.; Kotecha, A.; et al. Extra-hepatic manifestations of autochthonous hepatitis E infection. Aliment. Pharmacol. Ther. 2014, 40, 1282–1291. [Google Scholar] [CrossRef]
- Jaroszewicz, J.; Flisiak, R.; Kalinowska, A.; Wierzbicka, I.; Prokopowicz, D. Acute hepatitis E complicated by acute pancreatitis: A case report and literature review. Pancreas 2005, 30, 382–384. [Google Scholar] [CrossRef]
- Makharia, G.K.; Garg, P.K.; Tandon, R.K. Acute pancreatitis associated with acute hepatitis E infection. Trop. Gastroenterol. 2003, 24, 200–201. [Google Scholar]
- Bhagat, S.; Wadhawan, M.; Sud, R.; Arora, A. Hepatitis viruses causing pancreatitis and hepatitis: A case series and review of literature. Pancreas 2008, 36, 424–427. [Google Scholar] [CrossRef]
- Nayak, H.K.; Kamble, N.L.; Raizada, N.; Garg, S.; Daga, M.K. Acute pancreatitis complicating acute hepatitis e virus infection: A case report and review. Case Rep. Hepatol. 2013, 2013, 531235. [Google Scholar] [CrossRef] [Green Version]
- Deniel, C.; Coton, T.; Brardjanian, S.; Guisset, M.; Nicand, E.; Simon, F. Acute pancreatitis: A rare complication of acute hepatitis E. J. Clin. Virol. 2011, 51, 202–204. [Google Scholar] [CrossRef] [PubMed]
- Raj, M.; Kumar, K.; Ghoshal, U.C.; Saraswat, V.A.; Aggarwal, R.; Mohindra, S. Acute Hepatitis E-Associated Acute Pancreatitis: A Single Center Experience and Literature Review. Pancreas 2015, 44, 1320–1322. [Google Scholar] [CrossRef] [PubMed]
- Marion, O.; Lhomme, S.; Nayrac, M.; Dubois, M.; Pucelle, M.; Requena, M.; Migueres, M.; Abravanel, F.; Peron, J.M.; Carrere, N.; et al. Hepatitis E virus replication in human intestinal cells. Gut 2019. [Google Scholar] [CrossRef] [PubMed]
- Capelli, N.; Marion, O.; Dubois, M.; Allart, S.; Bertrand-Michel, J.; Lhomme, S.; Abravanel, F.; Izopet, J.; Chapuy-Regaud, S. Vectorial Release of Hepatitis E Virus in Polarized Human Hepatocytes. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, S.U.; Purcell, R.H. Fields Virology 2013, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 2242–2258. [Google Scholar]
- Yu, C.; Boon, D.; McDonald, S.L.; Myers, T.G.; Tomioka, K.; Nguyen, H.; Engle, R.E.; Govindarajan, S.; Emerson, S.U.; Purcell, R.H. Pathogenesis of hepatitis E virus and hepatitis C virus in chimpanzees: Similarities and differences. J. Virol. 2010, 84, 11264–11278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanford, R.E.; Feng, Z.; Chavez, D.; Guerra, B.; Brasky, K.M.; Zhou, Y.; Yamane, D.; Perelson, A.S.; Walker, C.M.; Lemon, S.M. Acute hepatitis A virus infection is associated with a limited type I interferon response and persistence of intrahepatic viral RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 11223–11228. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.H.; Zhang, X.; Tran, C.; Skinner, B. Expression profiles of host immune response-related genes against HEV genotype 3 and genotype 1 infections in rhesus macaques. J. Viral Hepat. 2018. [Google Scholar] [CrossRef]
- Dong, C.; Zafrullah, M.; Mixson-Hayden, T.; Dai, X.; Liang, J.; Meng, J.; Kamili, S. Suppression of interferon-alpha signaling by hepatitis E virus. Hepatology 2012, 55, 1324–1332. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, L.; Wang, W.; Watashi, K.; Wang, Y.; Sprengers, D.; de Ruiter, P.E.; van der Laan, L.J.; Metselaar, H.J.; Kamar, N.; et al. Disparity of basal and therapeutically activated interferon signalling in constraining hepatitis E virus infection. J. Viral Hepat. 2016, 23, 294–304. [Google Scholar] [CrossRef]
- Nan, Y.; Ma, Z.; Wang, R.; Yu, Y.; Kannan, H.; Fredericksen, B.; Zhang, Y.J. Enhancement of interferon induction by ORF3 product of hepatitis E virus. J. Virol. 2014, 88, 8696–8705. [Google Scholar] [CrossRef] [Green Version]
- Nan, Y.; Yu, Y.; Ma, Z.; Khattar, S.K.; Fredericksen, B.; Zhang, Y.J. Hepatitis E virus inhibits type I interferon induction by ORF1 products. J. Virol. 2014, 88, 11924–11932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devhare, P.B.; Chatterjee, S.N.; Arankalle, V.A.; Lole, K.S. Analysis of antiviral response in human epithelial cells infected with hepatitis E virus. PLoS ONE 2013, 8, e63793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Li, X.; Ambardekar, C.; Hu, Z.; Lhomme, S.; Feng, Z. Hepatitis E virus persists in the presence of a type III interferon response. PLoS Pathog. 2017, 13, e1006417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, I.M.; Verhoye, L.; Cocquerel, L.; Abravanel, F.; Foquet, L.; Montpellier, C.; Debing, Y.; Farhoudi, A.; Wychowski, C.; Dubuisson, J.; et al. Study of hepatitis E virus infection of genotype 1 and 3 in mice with humanised liver. Gut 2017, 66, 920–929. [Google Scholar] [CrossRef] [PubMed]
- van de Garde, M.D.; Pas, S.D.; van der Net, G.; de Man, R.A.; Osterhaus, A.D.; Haagmans, B.L.; Boonstra, A.; Vanwolleghem, T. Hepatitis E Virus (HEV) Genotype 3 Infection of Human Liver Chimeric Mice as a Model for Chronic HEV Infection. J. Virol. 2016, 90, 4394–4401. [Google Scholar] [CrossRef] [Green Version]
- Murata, K.; Kang, J.H.; Nagashima, S.; Matsui, T.; Karino, Y.; Yamamoto, Y.; Atarashi, T.; Oohara, M.; Uebayashi, M.; Sakata, H.; et al. IFN-lambda3 as a host immune response in acute hepatitis E virus infection. Cytokine 2019, 125, 154816. [Google Scholar] [CrossRef]
- Srivastava, R.; Aggarwal, R.; Jameel, S.; Puri, P.; Gupta, V.K.; Ramesh, V.S.; Bhatia, S.; Naik, S. Cellular immune responses in acute hepatitis E virus infection to the viral open reading frame 2 protein. Viral Immunol. 2007, 20, 56–65. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, R.; Aggarwal, R.; Bhagat, M.R.; Chowdhury, A.; Naik, S. Alterations in natural killer cells and natural killer T cells during acute viral hepatitis E. J. Viral Hepat. 2008, 15, 910–916. [Google Scholar] [CrossRef]
- Prabhu, S.B.; Gupta, P.; Durgapal, H.; Rath, S.; Gupta, S.D.; Acharya, S.K.; Panda, S.K. Study of cellular immune response against Hepatitis E virus (HEV). J. Viral Hepat. 2011, 18, 587–594. [Google Scholar] [CrossRef]
- Liu, T.; Xiao, P.; Li, R.; She, R.; Tian, J.; Wang, J.; Mao, J.; Yin, J.; Shi, R. Increased Mast Cell Activation in Mongolian Gerbils Infected by Hepatitis E Virus. Front. Microbiol. 2018, 9, 2226. [Google Scholar] [CrossRef]
- Lhomme, S.; Legrand-Abravanel, F.; Kamar, N.; Izopet, J. Screening, diagnosis and risks associated with Hepatitis E virus infection. Expert Rev. Anti Infect. Ther. 2019, 17, 403–418. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S.; Kamili, S.; Dar, M.Y.; Moecklii, R.; Jameel, S. Hepatitis E and long-term antibody status. Lancet 1993, 341, 1355. [Google Scholar] [PubMed]
- Tang, X.; Yang, C.; Gu, Y.; Song, C.; Zhang, X.; Wang, Y.; Zhang, J.; Hew, C.L.; Li, S.; Xia, N.; et al. Structural basis for the neutralization and genotype specificity of hepatitis E virus. Proc. Natl. Acad. Sci. USA 2011, 108, 10266–10271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, F.C.; Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Wang, H.; Yang, C.L.; Jiang, H.M.; Cai, J.P.; et al. Efficacy and safety of a recombinant hepatitis E vaccine in healthy adults: A large-scale, randomised, double-blind placebo-controlled, phase 3 trial. Lancet 2010, 376, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shih, J.W.; Xia, N.S. Long-term efficacy of a hepatitis E vaccine. N. Engl. J. Med. 2015, 372, 2265–2266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emerson, S.U.; Purcell, R.H. Recombinant vaccines for hepatitis E. Trends Mol. Med. 2001, 7, 462–466. [Google Scholar] [CrossRef]
- Bryan, J.P.; Tsarev, S.A.; Iqbal, M.; Ticehurst, J.; Emerson, S.; Ahmed, A.; Duncan, J.; Rafiqui, A.R.; Malik, I.A.; Purcell, R.H.; et al. Epidemic hepatitis E in Pakistan: Patterns of serologic response and evidence that antibody to hepatitis E virus protects against disease. J. Infect. Dis. 1994, 170, 517–521. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Yao, X.; Liang, Z.L.; Wu, T.; Li, J.X.; Yan, Q.; et al. Protection against hepatitis E virus infection by naturally acquired and vaccine-induced immunity. Clin. Microbiol. Infect. 2014, 20, O397–O405. [Google Scholar] [CrossRef] [Green Version]
- Shata, M.T.; Daef, E.A.; Zaki, M.E.; Abdelwahab, S.F.; Marzuuk, N.M.; Sobhy, M.; Rafaat, M.; Abdelbaki, L.; Nafeh, M.A.; Hashem, M.; et al. Protective role of humoral immune responses during an outbreak of hepatitis E in Egypt. Trans. R. Soc. Trop. Med. Hyg. 2012, 106, 613–618. [Google Scholar] [CrossRef]
- Shrestha, M.P.; Scott, R.M.; Joshi, D.M.; Mammen, M.P., Jr.; Thapa, G.B.; Thapa, N.; Myint, K.S.; Fourneau, M.; Kuschner, R.A.; Shrestha, S.K.; et al. Safety and efficacy of a recombinant hepatitis E vaccine. N. Engl. J. Med. 2007, 356, 895–903. [Google Scholar] [CrossRef]
- Abravanel, F.; Lhomme, S.; Chapuy-Regaud, S.; Mansuy, J.M.; Muscari, F.; Sallusto, F.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus reinfections in solid-organ-transplant recipients can evolve into chronic infections. J. Infect. Dis. 2014, 209, 1900–1906. [Google Scholar] [CrossRef]
- Chapuy-Regaud, S.; Dubois, M.; Plisson-Chastang, C.; Bonnefois, T.; Lhomme, S.; Bertrand-Michel, J.; You, B.; Simoneau, S.; Gleizes, P.E.; Flan, B.; et al. Characterization of the lipid envelope of exosome encapsulated HEV particles protected from the immune response. Biochimie 2017, 141, 70–79. [Google Scholar] [CrossRef]
- Husain, M.M.; Aggarwal, R.; Kumar, D.; Jameel, S.; Naik, S. Effector T cells immune reactivity among patients with acute hepatitis E. J. Viral Hepat. 2011, 18, e603–e608. [Google Scholar] [CrossRef]
- TrehanPati, N.; Sukriti, S.; Geffers, R.; Hissar, S.; Riese, P.; Toepfer, T.; Guzman, C.A.; Sarin, S.K. Gene expression profiles of T cells from hepatitis E virus infected patients in acute and resolving phase. J. Clin. Immunol. 2011, 31, 498–508. [Google Scholar] [CrossRef] [Green Version]
- Tripathy, A.S.; Das, R.; Rathod, S.B.; Arankalle, V.A. Cytokine profiles, CTL response and T cell frequencies in the peripheral blood of acute patients and individuals recovered from hepatitis E infection. PLoS ONE 2012, 7, e31822. [Google Scholar] [CrossRef] [Green Version]
- Brown, A.; Halliday, J.S.; Swadling, L.; Madden, R.G.; Bendall, R.; Hunter, J.G.; Maggs, J.; Simmonds, P.; Smith, D.B.; Vine, L.; et al. Characterization of the Specificity, Functionality, and Durability of Host T-Cell Responses Against the Full-Length Hepatitis E Virus. Hepatology 2016, 64, 1934–1950. [Google Scholar] [CrossRef]
- Al-Ayoubi, J.; Behrendt, P.; Bremer, B.; Suneetha, P.V.; Gisa, A.; Rinker, F.; Manns, M.P.; Cornberg, M.; Wedemeyer, H.; Kraft, A.R.M. Hepatitis E virus ORF 1 induces proliferative and functional T-cell responses in patients with ongoing and resolved hepatitis E. Liver Int. 2018, 38, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Gisa, A.; Suneetha, P.V.; Behrendt, P.; Pischke, S.; Bremer, B.; Falk, C.S.; Manns, M.P.; Cornberg, M.; Wedemeyer, H.; Kraft, A.R. Cross-genotype-specific T-cell responses in acute hepatitis E virus (HEV) infection. J. Viral Hepat. 2016, 23, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Aggarwal, R.; Naik, S.R.; Saraswat, V.; Ghoshal, U.C.; Naik, S. Hepatitis E virus is responsible for decompensation of chronic liver disease in an endemic region. Indian J. Gastroenterol. 2004, 23, 59–62. [Google Scholar] [PubMed]
- Saravanabalaji, S.; Tripathy, A.S.; Dhoot, R.R.; Chadha, M.S.; Kakrani, A.L.; Arankalle, V.A. Viral load, antibody titers and recombinant open reading frame 2 protein-induced TH1/TH2 cytokines and cellular immune responses in self-limiting and fulminant hepatitis e. Intervirology 2009, 52, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Aggarwal, R.; Sachdeva, S.; Alam, M.I.; Jameel, S.; Naik, S. Adaptive immune responses during acute uncomplicated and fulminant hepatitis E. J. Gastroenterol. Hepatol. 2011, 26, 306–311. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, V.; Goel, A.; Rawat, A.; Naik, S.; Aggarwal, R. Histological and immunohistochemical features in fatal acute fulminant hepatitis E. Indian J. Pathol. Microbiol. 2012, 55, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Chang, D.Y.; Lee, H.W.; Lee, H.; Kim, J.H.; Sung, P.S.; Kim, K.H.; Hong, S.H.; Kang, W.; Lee, J.; et al. Innate-like Cytotoxic Function of Bystander-Activated CD8(+) T Cells Is Associated with Liver Injury in Acute Hepatitis, A. Immunity 2018, 48, 161–173.e165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeblaoui, A.; Haim-Boukobza, S.; Marchadier, E.; Mokhtari, C.; Roque-Afonso, A.M. Genotype 4 hepatitis e virus in france: An autochthonous infection with a more severe presentation. Clin. Infect. Dis. 2013, 57, e122–e126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subissi, L.; Peeters, M.; Lamoral, S.; Klamer, S.; Suin, V.; Van Gucht, S. Subtype-specific differences in the risk of hospitalisation among patients infected with hepatitis E virus genotype 3 in Belgium, 2010–2018. Epidemiol. Infect. 2019, 147, e224. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Walimbe, A.M.; Arankalle, V.A. Hepatitis E virus from India exhibits significant amino acid mutations in fulminant hepatic failure patients. Virus Genes 2013, 46, 47–53. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P. Hepatitis E virus and fulminant hepatitis-A virus or host-specific pathology? Liver Int. 2015, 35, 1334–1340. [Google Scholar] [CrossRef] [Green Version]
- Borkakoti, J.; Ahmed, G.; Kar, P. Report of a novel C1483W mutation in the hepatitis E virus polymerase in patients with acute liver failure. Infect. Genet. Evol. 2016, 44, 51–54. [Google Scholar] [CrossRef]
- Borkakoti, J.; Ahmed, G.; Rai, A.; Kar, P. Report of novel H105R, D29N, V27A mutations in the methyltransferase region of the HEV genome in patients with acute liver failure. J. Clin. Virol. 2017, 91, 1–4. [Google Scholar] [CrossRef]
- Anty, R.; Ollier, L.; Peron, J.M.; Nicand, E.; Cannavo, I.; Bongain, A.; Giordanengo, V.; Tran, A. First case report of an acute genotype 3 hepatitis E infected pregnant woman living in South-Eastern France. J. Clin. Virol. 2012, 54, 76–78. [Google Scholar] [CrossRef]
- Tabatabai, J.; Wenzel, J.J.; Soboletzki, M.; Flux, C.; Navid, M.H.; Schnitzler, P. First case report of an acute hepatitis E subgenotype 3c infection during pregnancy in Germany. J. Clin. Virol. 2014, 61, 170–172. [Google Scholar] [CrossRef] [PubMed]
- Bouthry, E.; Benachi, A.; Vivanti, A.J.; Letamendia, E.; Vauloup-Fellous, C.; Roque-Afonso, A.M. Autochthonous Hepatitis E during Pregnancy, France. Emerg. Infect. Dis. 2018, 24, 1586–1587. [Google Scholar] [CrossRef] [PubMed]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat. Commun. 2018, 9, 4748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Devi, S.G.; Kar, P.; Agarwal, S.; Husain, S.A.; Gupta, R.K.; Sharma, S. Association of cytokines in hepatitis E with pregnancy outcome. Cytokine 2014, 65, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, S. The Th1/Th2 paradigm. Immunol. Today 1997, 18, 263–266. [Google Scholar] [CrossRef]
- Pal, R.; Aggarwal, R.; Naik, S.R.; Das, V.; Das, S.; Naik, S. Immunological alterations in pregnant women with acute hepatitis E. J. Gastroenterol. Hepatol. 2005, 20, 1094–1101. [Google Scholar] [CrossRef]
- Sehgal, R.; Patra, S.; David, P.; Vyas, A.; Khanam, A.; Hissar, S.; Gupta, E.; Kumar, G.; Kottilil, S.; Maiwall, R.; et al. Impaired monocyte-macrophage functions and defective Toll-like receptor signaling in hepatitis E virus-infected pregnant women with acute liver failure. Hepatology 2015, 62, 1683–1696. [Google Scholar] [CrossRef]
- Kmush, B.L.; Labrique, A.; Li, W.; Klein, S.L.; Schulze, K.; Shaikh, S.; Ali, H.; Engle, R.E.; Wu, L.; Purcell, R.H.; et al. The Association of Cytokines and Micronutrients with Hepatitis E Virus Infection During Pregnancy and the Postpartum Period in Rural Bangladesh. Am. J. Trop. Med. Hyg. 2016, 94, 203–211. [Google Scholar] [CrossRef]
- Stoszek, S.K.; Abdel-Hamid, M.; Saleh, D.A.; El Kafrawy, S.; Narooz, S.; Hawash, Y.; Shebl, F.M.; El Daly, M.; Said, A.; Kassem, E.; et al. High prevalence of hepatitis E antibodies in pregnant Egyptian women. Trans R. Soc. Trop. Med. Hyg. 2006, 100, 95–101. [Google Scholar] [CrossRef]
- Jilani, N.; Das, B.C.; Husain, S.A.; Baweja, U.K.; Chattopadhya, D.; Gupta, R.K.; Sardana, S.; Kar, P. Hepatitis E virus infection and fulminant hepatic failure during pregnancy. J. Gastroenterol. Hepatol. 2007, 22, 676–682. [Google Scholar] [CrossRef]
- Bi, Y.; Yang, C.; Yu, W.; Zhao, X.; Zhao, C.; He, Z.; Jing, S.; Wang, H.; Huang, F. Pregnancy serum facilitates hepatitis E virus replication in vitro. J. Gen. Virol. 2015, 96, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Kar, P.; Jilani, N.; Husain, S.A.; Pasha, S.T.; Anand, R.; Rai, A.; Das, B.C. Does hepatitis E viral load and genotypes influence the final outcome of acute liver failure during pregnancy? Am. J. Gastroenterol. 2008, 103, 2495–2501. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.D.; Das, B.C.; Kumar, A.; Gondal, R.; Kumar, D.; Kar, P. High viral load and deregulation of the progesterone receptor signaling pathway: Association with hepatitis E-related poor pregnancy outcome. J. Hepatol. 2011, 54, 1107–1113. [Google Scholar] [CrossRef] [PubMed]
- Borkakoti, J.; Hazam, R.K.; Mohammad, A.; Kumar, A.; Kar, P. Does high viral load of hepatitis E virus influence the severity and prognosis of acute liver failure during pregnancy? J. Med. Virol. 2013, 85, 620–626. [Google Scholar] [CrossRef] [PubMed]
- Legrand-Abravanel, F.; Kamar, N.; Sandres-Saune, K.; Lhomme, S.; Mansuy, J.M.; Muscari, F.; Sallusto, F.; Rostaing, L.; Izopet, J. Hepatitis E virus infection without reactivation in solid-organ transplant recipients, France. Emerg. Infect. Dis. 2011, 17, 30–37. [Google Scholar] [CrossRef]
- Pischke, S.; Stiefel, P.; Franz, B.; Bremer, B.; Suneetha, P.V.; Heim, A.; Ganzenmueller, T.; Schlue, J.; Horn-Wichmann, R.; Raupach, R.; et al. Chronic hepatitis E in heart transplant recipients. Am. J. Transplant. 2012, 12, 3128–3133. [Google Scholar] [CrossRef]
- Pischke, S.; Hardtke, S.; Bode, U.; Birkner, S.; Chatzikyrkou, C.; Kauffmann, W.; Bara, C.L.; Gottlieb, J.; Wenzel, J.; Manns, M.P.; et al. Ribavirin treatment of acute and chronic hepatitis E: A single-centre experience. Liver Int. 2013, 33, 722–726. [Google Scholar] [CrossRef]
- Moal, V.; Textoris, J.; Ben Amara, A.; Mehraj, V.; Berland, Y.; Colson, P.; Mege, J.L. Chronic hepatitis E virus infection is specifically associated with an interferon-related transcriptional program. J. Infect. Dis. 2013, 207, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Rostaing, L.; Kamar, N.; Izopet, J. Hepatitis E virus quasispecies and the outcome of acute hepatitis E in solid-organ transplant patients. J. Virol. 2012, 86, 10006–10014. [Google Scholar] [CrossRef] [Green Version]
- Suneetha, P.V.; Pischke, S.; Schlaphoff, V.; Grabowski, J.; Fytili, P.; Gronert, A.; Bremer, B.; Markova, A.; Jaroszewicz, J.; Bara, C.; et al. Hepatitis E virus (HEV)-specific T-cell responses are associated with control of HEV infection. Hepatology 2012, 55, 695–708. [Google Scholar] [CrossRef]
- Kamar, N.; Legrand-Abravanel, F.; Dalton, H.R.; Izopet, J. Hepatitis E virus-specific T-cell response after transplantation. Hepatology 2012, 55, 1643. [Google Scholar] [CrossRef] [PubMed]
- Abravanel, F.; Barrague, H.; Dorr, G.; Saune, K.; Peron, J.M.; Alric, L.; Kamar, N.; Izopet, J.; Champagne, E. Conventional and innate lymphocytes response at the acute phase of HEV infection in transplanted patients. J. Infect. 2016, 72, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Cao, Q.M.; Subramaniam, S.; Yugo, D.M.; Heffron, C.L.; Rogers, A.J.; Kenney, S.P.; Tian, D.; Matzinger, S.R.; Overend, C.; et al. Pig model mimicking chronic hepatitis E virus infection in immunocompromised patients to assess immune correlates during chronicity. Proc. Natl. Acad. Sci. USA 2017, 114, 6914–6923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McAlister, V.C.; Haddad, E.; Renouf, E.; Malthaner, R.A.; Kjaer, M.S.; Gluud, L.L. Cyclosporin versus tacrolimus as primary immunosuppressant after liver transplantation: A meta-analysis. Am. J. Transplant. 2006, 6, 1578–1585. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, X.; Debing, Y.; Chen, K.; Van Der Laan, L.J.; Neyts, J.; Janssen, H.L.; Metselaar, H.J.; Peppelenbosch, M.P.; Pan, Q. Calcineurin inhibitors stimulate and mycophenolic acid inhibits replication of hepatitis E virus. Gastroenterology 2014, 146, 1775–1783. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Y.; Metselaar, H.J.; Janssen, H.L.; Peppelenbosch, M.P.; Pan, Q. Rapamycin and everolimus facilitate hepatitis E virus replication: Revealing a basal defense mechanism of PI3K-PKB-mTOR pathway. J. Hepatol. 2014, 61, 746–754. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Lhomme, S.; Abravanel, F.; Cointault, O.; Esposito, L.; Cardeau-Desangles, I.; Del Bello, A.; Dorr, G.; Lavayssiere, L.; Nogier, M.B.; et al. An Early Viral Response Predicts the Virological Response to Ribavirin in Hepatitis E Virus Organ Transplant Patients. Transplantation 2015, 99, 2124–2131. [Google Scholar] [CrossRef]
- Lhomme, S.; Garrouste, C.; Kamar, N.; Saune, K.; Abravanel, F.; Mansuy, J.M.; Dubois, M.; Rostaing, L.; Izopet, J. Influence of polyproline region and macro domain genetic heterogeneity on HEV persistence in immunocompromised patients. J. Infect. Dis. 2014, 209, 300–303. [Google Scholar] [CrossRef] [Green Version]
- Inagaki, Y.; Oshiro, Y.; Tanaka, T.; Yoshizumi, T.; Okajima, H.; Ishiyama, K.; Nakanishi, C.; Hidaka, M.; Wada, H.; Hibi, T.; et al. A Nationwide Survey of Hepatitis E Virus Infection and Chronic Hepatitis E in Liver Transplant Recipients in Japan. EBioMedicine 2015, 2, 1607–1612. [Google Scholar] [CrossRef] [Green Version]
- Pischke, S.; Wedemeyer, H. Hepatitis E In Transplant Recipients: Why Is This Not A Problem In Japan? EBioMedicine 2015, 2, 1564–1565. [Google Scholar] [CrossRef] [Green Version]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the polyproline region of the hepatitis E virus in immunocompromised patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenroder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef]
- Peron, J.M.; Dalton, H.; Izopet, J.; Kamar, N. Acute autochthonous hepatitis E in western patients with underlying chronic liver disease: A role for ribavirin? J. Hepatol. 2011, 54, 1323–1324. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Abravanel, F.; Selves, J.; Garrouste, C.; Esposito, L.; Lavayssiere, L.; Cointault, O.; Ribes, D.; Cardeau, I.; Nogier, M.B.; et al. Influence of immunosuppressive therapy on the natural history of genotype 3 hepatitis-E virus infection after organ transplantation. Transplantation 2010, 89, 353–360. [Google Scholar] [CrossRef]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Esposito, L.; Cardeau-Desangles, I.; Mansuy, J.M.; Selves, J.; Peron, J.M.; Otal, P.; et al. Pegylated interferon-alpha for treating chronic hepatitis E virus infection after liver transplantation. Clin. Infect. Dis. 2010, 50, e30–e33. [Google Scholar] [CrossRef] [Green Version]
- Haagsma, E.B.; Riezebos-Brilman, A.; van den Berg, A.P.; Porte, R.J.; Niesters, H.G. Treatment of chronic hepatitis E in liver transplant recipients with pegylated interferon alpha-2b. Liver Transplant. 2010, 16, 474–477. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Garrouste, C.; Cardeau-Desangles, I.; Mansuy, J.M.; Weclawiak, H.; Izopet, J.; Rostaing, L. Three-month pegylated interferon-alpha-2a therapy for chronic hepatitis E virus infection in a haemodialysis patient. Nephrol. Dial. Transplant. 2010, 25, 2792–2795. [Google Scholar] [CrossRef] [Green Version]
- Mallet, V.; Nicand, E.; Sultanik, P.; Chakvetadze, C.; Tesse, S.; Thervet, E.; Mouthon, L.; Sogni, P.; Pol, S. Brief communication: Case reports of ribavirin treatment for chronic hepatitis E. Ann. Intern. Med. 2010, 153, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Lhomme, S.; Esposito, L.; Basse, G.; Cointault, O.; Ribes, D.; Nogier, M.B.; et al. Ribavirin therapy inhibits viral replication on patients with chronic hepatitis e virus infection. Gastroenterology 2010, 139, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Tripon, S.; Bismuth, M.; Hillaire, S.; Dumortier, J.; Radenne, S.; Coilly, A.; Garrigue, V.; D’Alteroche, L.; et al. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N. Engl. J. Med. 2014, 370, 1111–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, O.; Lhomme, S.; Del Bello, A.; Abravanel, F.; Esposito, L.; Hebral, A.L.; Lavayssiere, L.; Cointault, O.; Ribes, D.; Izopet, J.; et al. Monitoring hepatitis E virus fecal shedding to optimize ribavirin treatment duration in chronically infected transplant patients. J. Hepatol. 2019, 70, 206–209. [Google Scholar] [CrossRef] [Green Version]
- Debing, Y.; Emerson, S.U.; Wang, Y.; Pan, Q.; Balzarini, J.; Dallmeier, K.; Neyts, J. Ribavirin inhibits in vitro hepatitis E virus replication through depletion of cellular GTP pools and is moderately synergistic with alpha interferon. Antimicrob. Agents Chemother. 2014, 58, 267–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.; Manns, M.P.; et al. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotty, S.; Cameron, C.E.; Andino, R. RNA virus error catastrophe: Direct molecular test by using ribavirin. Proc. Natl. Acad. Sci. USA 2001, 98, 6895–6900. [Google Scholar] [CrossRef] [Green Version]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar] [CrossRef]
- Lhomme, S.; Kamar, N.; Nicot, F.; Ducos, J.; Bismuth, M.; Garrigue, V.; Petitjean-Lecherbonnier, J.; Ollivier, I.; Alessandri-Gradt, E.; Goria, O.; et al. Mutation in the Hepatitis E Virus Polymerase and Outcome of Ribavirin Therapy. Antimicrob. Agents Chemother. 2015, 60, 1608–1614. [Google Scholar] [CrossRef] [Green Version]
- Debing, Y.; Ramiere, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.A.; Lebosse, F.; Scholtes, C.; Roche, M.; Legras-Lachuer, C.; de Lamballerie, X.; et al. Hepatitis E virus mutations associated with ribavirin treatment failure result in altered viral fitness and ribavirin sensitivity. J. Hepatol. 2016, 65, 499–508. [Google Scholar] [CrossRef]
- Kamar, N.; Abravanel, F.; Behrendt, P.; Hofmann, J.; Pageaux, G.P.; Barbet, C.; Moal, V.; Couzi, L.; Horvatits, T.; De Man, R.A.; et al. Ribavirin for Hepatitis E Virus Infection After Organ Transplantation: A Large European Retrospective Multicenter Study. Clin. Infect. Dis. 2019. [Google Scholar] [CrossRef]
- Dao Thi, V.L.; Debing, Y.; Wu, X.; Rice, C.M.; Neyts, J.; Moradpour, D.; Gouttenoire, J. Sofosbuvir Inhibits Hepatitis E Virus Replication In Vitro and Results in an Additive Effect When Combined With Ribavirin. Gastroenterology 2016, 150, 82–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Wezel, E.M.; de Bruijne, J.; Damman, K.; Bijmolen, M.; van den Berg, A.P.; Verschuuren, E.A.M.; Ruigrok, G.A.; Riezebos-Brilman, A.; Knoester, M. Sofosbuvir Add-on to Ribavirin Treatment for Chronic Hepatitis E Virus Infection in Solid Organ Transplant Recipients Does Not Result in Sustained Virological Response. Open Forum Infect. Dis. 2019, 6. [Google Scholar] [CrossRef] [PubMed]
- Drinane, M.; Jing Wang, X.; Watt, K. Sofosbuvir and Ribavirin Eradication of Refractory Hepatitis E in an Immunosuppressed Kidney Transplant Recipient. Hepatology 2019, 69, 2297–2299. [Google Scholar] [CrossRef] [PubMed]
- van der Valk, M.; Zaaijer, H.L.; Kater, A.P.; Schinkel, J. Sofosbuvir shows antiviral activity in a patient with chronic hepatitis E virus infection. J. Hepatol. 2017, 66, 242–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todesco, E.; Demeret, S.; Calin, R.; Roque-Afonso, A.M.; Thibault, V.; Mallet, V.; Akhavan, S.; Jaspard, M.; Peytavin, G.; Poynard, T.; et al. Chronic hepatitis E in HIV/HBV coinfected patient: Lack of power of sofosbuvir-ribavirin. AIDS 2017, 31, 1346–1348. [Google Scholar] [CrossRef] [PubMed]
- Todesco, E.; Mazzola, A.; Akhavan, S.; Abravanel, F.; Poynard, T.; Roque-Afonso, A.M.; Peytavin, G.; Marcelin, A.G.; Calmus, Y.; Lecuyer, L.; et al. Chronic hepatitis E in a heart transplant patient: Sofosbuvir and ribavirin regimen not fully effective. Antivir. Ther. 2018. [Google Scholar] [CrossRef]
- Donnelly, M.C.; Imlach, S.N.; Abravanel, F.; Ramalingam, S.; Johannessen, I.; Petrik, J.; Fraser, A.R.; Campbell, J.D.; Bramley, P.; Dalton, H.R.; et al. Sofosbuvir and Daclatasvir Anti-Viral Therapy Fails to Clear HEV Viremia and Restore Reactive T Cells in a HEV/HCV Co-Infected Liver Transplant Recipient. Gastroenterology 2017, 152, 300–301. [Google Scholar] [CrossRef] [Green Version]
- De Martin, E.; Antonini, T.M.; Coilly, A.; Pittau, G.; Vibert, E.; Duclos-Vallee, J.C.; Samuel, D.; Roque-Afonso, A.M. HCV and HEV recurrence after liver transplantation: One antiviral therapy for two viruses. Transpl. Int. 2017, 30, 318–319. [Google Scholar] [CrossRef] [Green Version]
- Kamar, N.; Pan, Q. No Clear Evidence for an Effect of Sofosbuvir against Hepatitis E Virus in Organ Transplant Patients. Hepatology 2019, 69, 1846–1847. [Google Scholar] [CrossRef]
- Cornberg, M.; Pischke, S.; Müller, T.; Behrendt, P.; Piecha, F.; Benckert, J.; Smith, A.; Koch, A.; Lohse, A.; Hardtke, S.; et al. LBO-04; Efficacy and safety of sofosbuvir monotherapy in patients with chronic hepatitis E-The HepNet SofE pilot study. J. Hepatol. 2019, 70, e129–e130. [Google Scholar] [CrossRef]
- Kaushik, N.; Subramani, C.; Anang, S.; Muthumohan, R.; Shalimar, B.N.; Ranjith-Kumar, C.T.; Surjit, M. Zinc Salts Block Hepatitis E Virus Replication by Inhibiting the Activity of Viral RNA-Dependent RNA Polymerase. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marion, O.; Abravanel, F.; Izopet, J.; Kamar, N. Failure to respond to ribavirin despite elevated intra-erythrocyte zinc level in transplant-patients with chronic hepatitis E virus infection. Transpl. Infect. Dis. 2019, 21, e13050. [Google Scholar] [CrossRef]
- Todt, D.; Moeller, N.; Praditya, D.; Kinast, V.; Friesland, M.; Engelmann, M.; Verhoye, L.; Sayed, I.M.; Behrendt, P.; Dao Thi, V.L.; et al. The natural compound silvestrol inhibits hepatitis E virus (HEV) replication in vitro and in vivo. Antivir. Res. 2018, 157, 151–158. [Google Scholar] [CrossRef]
- Netzler, N.E.; Enosi Tuipulotu, D.; Vasudevan, S.G.; Mackenzie, J.M.; White, P.A. Antiviral Candidates for Treating Hepatitis E Virus Infection. Antimicrob. Agents Chemother. 2019, 63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishiyama, T.; Kobayashi, T.; Jirintai, S.; Kii, I.; Nagashima, S.; Prathiwi Primadharsini, P.; Nishizawa, T.; Okamoto, H. Screening of novel drugs for inhibiting hepatitis E virus replication. J. Virol. Methods 2019, 270, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Soon, C.F.; Behrendt, P.; Todt, D.; Manns, M.P.; Wedemeyer, H.; Sallberg Chen, M.; Cornberg, M. Defining virus-specific CD8+ TCR repertoires for therapeutic regeneration of T cells against chronic hepatitis E. J. Hepatol. 2019, 71, 673–684. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hepatitis E virus (HEV) genome. Open reading frames (ORF1) (blue box) encodes nonstructural proteins. ORF4 has been found only in HEV1. 7 mG: 7-methylguanosine; Hel: helicase; MT: methyl transferase; polyA: polyadenylated tail; PPR: polyproline region; Pro: cysteine protease; RdRp: RNA polymerase; X: X domain; Y: Y domain.
Figure 1.
Hepatitis E virus (HEV) genome. Open reading frames (ORF1) (blue box) encodes nonstructural proteins. ORF4 has been found only in HEV1. 7 mG: 7-methylguanosine; Hel: helicase; MT: methyl transferase; polyA: polyadenylated tail; PPR: polyproline region; Pro: cysteine protease; RdRp: RNA polymerase; X: X domain; Y: Y domain.

Figure 2.
Phylogenetic tree based on full-length sequences of HEV strains. Sequence alignment was performed using ClustalW (MEGA5) and BioEdit, version 7.0. The neighbor-joining method was used to create the phylogenetic tree, with a bootstrap of 100 replicates.
Figure 2.
Phylogenetic tree based on full-length sequences of HEV strains. Sequence alignment was performed using ClustalW (MEGA5) and BioEdit, version 7.0. The neighbor-joining method was used to create the phylogenetic tree, with a bootstrap of 100 replicates.

Figure 3.
Interference of HEV with the innate antiviral response. HEV RNA is detected in the cytoplasm by the retinoic acid-inducible gene I (RIG-I), leading to type I and type III interferon (IFN) production. The protease domain (Pro) of the ORF1 protein inhibits signaling via RIG-I and prevents IFN induction by de-ubiquitining RIG-I and TANK binding kinase 1 (TBK-1). The X domain (X) inhibits the phosphorylation (P) of IFN regulatory protein 3 (IRF3). Conversely, the ORF3 protein stimulates the direct interaction of type I IFN with RIG-I, but ORF3 also binds to STAT1 to restrict its phosphorylation and activation of the downstream cascade, thus inhibiting the expression of the IFN-stimulated genes (ISGs), including the double stranded (ds)RNA-activated protein kinase (PKR) and 2′,5′-oligoadenylate synthetase (2′5′-OAS) or ISG15. IKKε: IκB-Kinase-epsilon; IRF9: IFN regulatory protein 9; ISRE: interferon stimulated response element; MAVS: mitochondrial antiviral-signaling protein; Ub: ubiquitin.
Figure 3.
Interference of HEV with the innate antiviral response. HEV RNA is detected in the cytoplasm by the retinoic acid-inducible gene I (RIG-I), leading to type I and type III interferon (IFN) production. The protease domain (Pro) of the ORF1 protein inhibits signaling via RIG-I and prevents IFN induction by de-ubiquitining RIG-I and TANK binding kinase 1 (TBK-1). The X domain (X) inhibits the phosphorylation (P) of IFN regulatory protein 3 (IRF3). Conversely, the ORF3 protein stimulates the direct interaction of type I IFN with RIG-I, but ORF3 also binds to STAT1 to restrict its phosphorylation and activation of the downstream cascade, thus inhibiting the expression of the IFN-stimulated genes (ISGs), including the double stranded (ds)RNA-activated protein kinase (PKR) and 2′,5′-oligoadenylate synthetase (2′5′-OAS) or ISG15. IKKε: IκB-Kinase-epsilon; IRF9: IFN regulatory protein 9; ISRE: interferon stimulated response element; MAVS: mitochondrial antiviral-signaling protein; Ub: ubiquitin.

Figure 4.
Course of an acute HEV infection. ALT: alanine aminotransferase activity.

Figure 5.
Histology of liver biopsies from a chronically HEV-infected patient. (a) Initial liver biopsy, (b) inflammation after 15 months of infection, and (c) Cirrhosis after 38 months of infection (Masson’s trichrome, magnification 100×).
Figure 5.
Histology of liver biopsies from a chronically HEV-infected patient. (a) Initial liver biopsy, (b) inflammation after 15 months of infection, and (c) Cirrhosis after 38 months of infection (Masson’s trichrome, magnification 100×).

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lhomme, S.; Marion, O.; Abravanel, F.; Izopet, J.; Kamar, N. Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections. J. Clin. Med. 2020, 9, 331. https://doi.org/10.3390/jcm9020331
AMA Style
Lhomme S, Marion O, Abravanel F, Izopet J, Kamar N. Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections. Journal of Clinical Medicine. 2020; 9(2):331. https://doi.org/10.3390/jcm9020331
Chicago/Turabian StyleLhomme, Sébastien, Olivier Marion, Florence Abravanel, Jacques Izopet, and Nassim Kamar. 2020. "Clinical Manifestations, Pathogenesis and Treatment of Hepatitis E Virus Infections" Journal of Clinical Medicine 9, no. 2: 331. https://doi.org/10.3390/jcm9020331
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.