Sex Differences in Urate Handling


Department of Physiology, University of Maryland School of Medicine, Baltimore, MD 21201, USA
*
Authors to whom correspondence should be addressed.
Int. J. Mol. Sci. 2020, 21(12), 4269; https://doi.org/10.3390/ijms21124269
Submission received: 27 May 2020
/
Revised: 11 June 2020
/
Accepted: 12 June 2020
/
Published: 16 June 2020
(This article belongs to the Special Issue Membrane Proteins of Urinary System in Physiology, Pathobiology and Medicine)
Abstract
:Hyperuricemia, or elevated serum urate, causes urate kidney stones and gout and also increases the incidence of many other conditions including renal disease, cardiovascular disease, and metabolic syndrome. As we gain mechanistic insight into how urate contributes to human disease, a clear sex difference has emerged in the physiological regulation of urate homeostasis. This review summarizes our current understanding of urate as a disease risk factor and how being of the female sex appears protective. Further, we review the mechanisms of renal handling of urate and the significant contributions from powerful genome-wide association studies of serum urate. We also explore the role of sex in the regulation of specific renal urate transporters and the power of new animal models of hyperuricemia to inform on the role of sex and hyperuricemia in disease pathogenesis. Finally, we advocate the use of sex differences in urate handling as a potent tool in gaining a further understanding of physiological regulation of urate homeostasis and for presenting new avenues for treating the constellation of urate related pathologies.
1. Introduction
Understanding the mechanisms of common heritable diseases has long proven to be challenging [1]. One such common human disease is gout, the most common inflammatory arthritis [2]. Gout is caused by the deposition of sodium urate (UA) crystals in the synovial fluid of joints, a process that leads to a cascade of inflammatory responses and extreme discomfort [3]. Precipitation of the weakly soluble UA occurs as the UA levels in the blood and other body fluids rise, termed hyperuricemia. Gout, or podagra, is one of the most well documented human diseases, recognized as early as 2460 BCE by the ancient Egyptians [4]. A staple throughout the development of the civilized world, cases were also noted by Hippocrates and Galen in ancient Greece, with a surge in cases in the 17th and 18th centuries [4] during the industrial revolution, commiserate with rising wealth across the western world. Today, the prevalence of gout is roughly 4% of the population of the United States, Europe, and Southeast Asia [5]. Interestingly, recent research has found that gout is only one of many potential diseases caused or contributed to by hyperuricemia (HUA); HUA is also an independent risk factor for additional pathologies including renal diseases, cardiovascular disease, hypertension, and metabolic syndrome [5,6,7] (Figure 1). As this list of diseases continues to grow, so does the critical need for a deeper understanding of not only how we regulate UA homeostasis, but also the causes of disruptions of these processes.
A mechanistic picture of the effectors of UA homeostasis has emerged, yet we lack an understanding of how these proteins are regulated. New tools are needed to examine this problem to provide relief for the large and growing population of hyperuricemic individuals [1]. Additionally, new work has reinforced the importance of an ancient observation [4], the male sex is a significant risk factor for hyperuricemia and gout [8,9], where men are up to four times more likely to be affected than women [5]. This observation has now been coupled to investigations of differences in UA handling in the kidney, as well as differential effects of pathogenic variants in UA transporter genes, presenting the potential to use sex differences in UA handling as a powerful comparative analysis revealing how these systems are regulated. Here, we review sex differences in UA handling to explore how the female sex may be protective against the development of UA related diseases and to illustrate the power of using sex differences as a tool.
2. Urate as a Risk Factor in Disease
HUA is clinically defined as elevated serum UA (SUA, >6mg/dL), which increases the risk of the precipitation of weakly soluble UA. Increased UA leads to precipitation of monosodium UA crystals, which can cause UA kidney stones and gout. The number of affected individuals continues to climb with current estimates being approximately 47.2 million HUA individuals in the United States; roughly 27.9 million individuals have severe HUA (>7mg/dL), with men being affected five times more often than women [6]. Similar results have been observed in China, with an overall HUA prevalence of 13.3%, with striking differences in prevalence by sex, with 19.4% in men and only 7.9% in women [10]. However, the risk of the development of HUA increases roughly four-fold for women after menopause, and postmenopausal hormone replacement therapy reduces this risk [11,12,13], providing evidence that female hormones may contribute to the protection from HUA.
HUA is just one of many conditions that have observable sex differences, as differing pathologies based on sex have been observed in a variety of fields [14,15,16,17] including cardiovascular [18,19,20,21], neurological [22,23,24,25], immunological [26,27,28], and renal diseases [29,30,31,32,33]. The architecture of the female kidney is likely distinct from that of the male kidney [34], given women have a lower blood pressure than men [35], women are less likely to develop acute kidney injury than men [36,37], women demonstrate improved tolerance to renal ischemia [38,39], and women are protected from renal and cardiovascular disease before menopause as compared to men [40,41]. A recent study determined that females with chronic kidney disease (CKD) had a slower decline in glomerular filtration rate (GFR), lower risk of progression to end-stage renal disease (ERSD), and a lower risk of death compared to age-matched men with similar mild-to-moderate CKD [42]. In diabetic kidney disease, men tended to demonstrate renal complications roughly 10 years earlier than women [40], while sex differences in obesity-related kidney disease [43,44,45] demonstrated that female sex hormones may safeguard against worsening pathology [45]. Thus, being female may have a protective effect on the kidney, preventing women from developing the more severe phenotypes observed in men. All these studies illustrate the need for greater emphasis on the idea of sex as a biological variable [46,47,48], going beyond merely looking at sex differences, and instead examining the influence of sex on various physiological and pathophysiological pathways [49] to help elucidate underlying mechanisms.
Although the overall kidney function seems to be improved in females when compared to males, once female kidney function does begin to decline, the resulting pathologies can be more extreme than in males, in part due to the fact that this decline is likely to occur later in life in women. As kidney function declines, SUA levels tend to increase, and increases have been reported to be associated with all-cause and cardiovascular mortality in a dose-dependent manner. Thus, as UA levels increase, so does the risk of developing other conditions, including CKD, hypertension, and diabetes mellitus [50,51,52,53,54,55], especially in women [12,56]. Table 1 summarizes data that compares the comorbidities of individuals who have low SUA with individuals who have high SUA, stratified by sex. HUA increases the risk of developing CKD for both men and women and increases the likelihood of progression to ESRD in both sexes, yet this risk is much higher in HUA females than HUA males [50]. Similarly, the incidence of CKD increases two-fold to six-fold higher in HUA females than HUA males [51,52], demonstrating that once females have lost the benefit of low SUA, renal health is more likely to decline. Males and females with both HUA and CKD had a higher incidence of left ventricular hypertrophy and hypertension, however, this association was only significant in females [53]. HUA also increases the overall risk for hypertension for both sexes, but once again females with HUA are more likely to develop hypertension than HUA males compared to non-HUA controls [51,54,57]. Finally, females have a higher incidence of type 2 diabetes mellitus than males [52], yet HUA females are even more likely to develop diabetes than HUA males [54], further emphasizing that an increase in SUA can be more detrimental to females than males. However, since HUA is only one of several pathologies to affect people later in life, the role of UA in the biogenesis of the underlying pathology is difficult to tease out, demonstrating the critical need for additional studies.
Taken together, this evidence implies that both biological sex and SUA level affect the kidney, in that the interaction of these two variables may influence renal function. The female sex must somehow affect the balance of UA levels, leading to a decrease in SUA when compared to males. Unraveling the mechanism behind this effect could lead to a better understanding of the underlying physiology and improved treatment for HUA. However, UA handling in the kidney is an incredibly complicated process, the details of which have yet to be fully elucidated.
3. Urate Homeostasis
Renal UA handling involves multiple transporter proteins. UA is the protonated form of uric acid and enters the circulation as the terminal metabolite of purine metabolism in humans and the other great apes. It is produced from the degradation of purine nucleotides and amino acids, mediated by xanthine oxidase. Higher-order primates have lost the activity of the enzyme uricase, which further metabolizes UA to the much more soluble allantoin [58]. Loss of uricase gene function in the ape lineage supports the idea of a possible selective advantage of increased SUA [59,60]. Contributing to this complicated process is the fact that the physiological concentrations of UA occupy a wide range, which is higher in men (3.5 to 7.2 mg/dL) than in pre-menopausal women (2.6 to 6.0 mg/dL) [61]. Interestingly, concentrations in post-menopausal women increase to levels observed in men [62]. Since UA is weakly soluble, high concentrations (>6 mg/dL) can lead to precipitation and crystal formation resulting in UA kidney stones. These crystals can also precipitate in joints and synovial fluid causing gout [6].
Of the UA excretion, 70% is mediated by the kidney and 30% by extrarenal pathways including the gut and the liver [63,64,65]. A delicate balance between secretion and reabsorption exists in the kidney to maintain UA homeostasis. UA is freely filtered at the renal glomerulus, then reabsorbed, actively secreted, and reabsorbed again in the proximal tubule [66,67,68,69]. Several proteins have been identified as UA transporters based primarily on in vitro studies demonstrating UA affinity. Based on in vivo renal tubule expression, the initial reabsorption of 95% of the initial filtered UA load may occur via organic anion transporter (OAT) family proteins, including OAT1 (encoded by SLC22A6), OAT3 (encoded by SLC22A8), OAT4 (encoded by SLC22A11), and OAT10 (encoded by SLC22A13), as well as SLC2A9 (also called GLUT9) [70,71,72,73,74]. Approximately 50% of the initial filtered load is then actively secreted back into the tubular lumen, primarily where ABCG2, NPT1 (encoded by SLC17A1), and NPT4 (encoded by SLC17A3) are expressed [75,76,77,78,79,80]. Reabsorption of another 40% of the filtered UA occurs downstream of the secretion in the S3 segment of the proximal tubule, where expression of URAT1 (encoded by SLC22A12) and SLC2A9 have been reported [71,81], resulting in a fractional excretion (FEUA) of 4%–8% of the initial filtered load. This somewhat controversial [69] and complicated mechanism of filtration, reabsorption, secretion, and a second round of reabsorption demonstrates that the kidney spends an exorbitant amount of energy fueling these transporters in an effort to carefully regulate UA excretion from the kidney for reasons yet to be fully understood.
Functional data of these and other [82,83,84] transporters demonstrate that these proteins can transport UA. However, whether or not transport of UA occurs in the renal tubules by these particular transporters remains unclear. The next logical step would be to explore genetic perturbations in these transporters to determine if those alterations affect SUA levels. Thus, exploring human genetics and genetic variations in the form of genome-wide association studies (GWAS) can be a powerful tool for understanding which genes are most important to UA handling. GWAS use common single nucleotide polymorphisms (SNPs) that exist in a given population as signposts for genomic space that correlate with a given condition [1], including SUA levels. The larger the study population, the higher the genomic resolution to identify regions of interest that associate with altered SUA. Once these genomic regions have been identified, additional analyses can then be performed to identify those genes most likely to underlie the associated SNP, in some cases identifying novel causal variants that contribute to the development of HUA.
4. Genetics of Hyperuricemia
SUA levels display a strong heritable component, estimated between 40%–70% [85,86,87]. The first gene associated with UA transport in humans was SLC22A12 [81]. A labor-intensive comparative and candidate based cloning approach identified the locus of SLC22A12 as potentially harboring the key gene. The resulting protein product, URAT1, proved to be a kidney-specific UA transporter, and variants were found in a Japanese family with significant hypouricemia [81]. SLC22A12 variants causing hypouricemia have been reported at high frequencies in Japanese [88], Korean [89], and European Roma [90] populations, reinforcing the importance of URAT1 function on SUA levels. Sex differences, however, have not been reported in the context of hypouricemia. Subsequent work proved that other members of the SLC22A gene family (the OATs) proved to have UA affinity [70,91], but their physiological relevance proved difficult to substantiate [69]. Progress was only made in identifying other key UA related genes with the advent of GWAS, allowing researchers to use an unbiased approach to discover genes and gene products that are involved in specific phenotypes like SUA levels or gout risk [85,86,87,92]. Because of the ease of measurement and the availability of serum samples from large cohorts of individuals, GWAS for SUA have played a driving role in demonstrating the incredible power of the tool to find gene networks or even specific transporter genes playing a significant role in UA homeostasis in humans [92,93,94]. The GWAS published on UA over the past decade [94,95,96,97,98,99,100,101,102] have been progressively increasing genomic resolution by increasing the number of individuals used, thus genes with smaller and smaller contributions to the SUA phenotype were able to be resolved. A recent effort, the largest yet, performed by Tin et al. [101], explored variants in 457,690 individuals of various ethnic backgrounds and found 147 new SNPs and replicated 36 previously reported SNPs that associate with SUA, all of which were further replicated in an independent cohort of 334,880 individuals. As part of their analysis, Tin et al. also attempted to identify common variants causal for alterations in UA levels. Emphasis was placed on missense variants with the most significant combined annotation dependent depletion Phred score, revealing six potential causal variants. These potential causal variants included SNPs in the well-established UA transporter gene ABCG2 [76], as well as in the transcription factor genes HNF1A and HNF4A [101]. Investigators demonstrated the causative nature of the HNF4A variant by showing that it altered the ability of its protein product HNF4α to directly regulate trans-activation of ABCG2 [101].
Only 6 of the 183 loci in that same study demonstrated sex-specific effect differences, including SLC2A9, ABCG2, CAPN1, GCKR, IDH2, and SLC22A12 [101]. However, of these six, only SLC2A9 and ABCG2 were found to have significant associations in the more stringent genome-wide test for SNP effects on UA levels based on sex, confirming previously established findings [92,93,94,103,104]. Table 2 displays a current list of SNPs that have reported sex differences in genes that associate with SUA levels confirmed by multiple GWAS. Initial reports demonstrate that SLC2A9 had a stronger association with lower SUA levels in females, whereas ABCG2 had a stronger association with higher SUA levels in males, demonstrating that regulation and/or activity of these transporters may be influenced by factors that contribute to biological sex [92]. Significant association with HUA associated gout risk has been reported in males, but not females, for several other transporter genes (SLC16A9, SLC17A1, SLC22A11, and SLC22A12), as well as UA transporter “keystone” scaffold protein encoded by PDZK1 [100]. Additional sex differences in these genes are detailed in Table 2, as well as some of the UA associated transcription factors HNF4A and HNF4G [105,106,107]. Understanding the interactions between these genes will lead to a more accurate model of renal UA handling, revealing those genes with the most significant contributions to UA levels.
The picture of the genetic architecture emerging is one of two tiers, first a base composed of a large number of genes that provide a significant but small contribution to the variance in SUA, and then a top tier with the three critical UA transporter genes: ABCG2, SLC2A9, and SLC22A12. As mentioned above, variants in ABCG2 and SLC2A9 have been demonstrated to exert sex differences in effect size, but not SLC22A12. This may speak to one of the limitations of GWAS, such that these studies can only use common variants present in high enough frequencies to be detected, leaving rare but potential associated and causal variants to remain undetected in these studies. To address this, recent work that used whole-exome sequencing (WES) to probe for rare variants that contribute to SUA variance found numerous rare variants including two in the SLC22A12 gene (rs150255373 encoding R325W and rs147647315 encoding R434H) that had large effect sizes [108]. The lack of common variants in SLC22A12 may have contributed to its under-reporting of sex effect. Additionally, SLC22A12 expression was found to be higher in males over the age of 50 compared to females of a similar age, yet SLC22A12 expression was the highest in females <50 as reported in a smaller transporter targeted study [109]. Increased SLC22A12 expression could indicate an increase in UA reabsorption, yet females under the age of 50 have some of the lowest levels of SUA of all populations sampled, implying that the reabsorption of UA by URAT1 may not be the primary contributor to SUA in females. This primary contribution is much more likely due to commiserate changes in other UA transporters.
4.1. SLC2A9
Not coincidentally, the two genes variants with the largest sex-specific effect differences are also the two with the common variants contributing the most to SUA variability (SLC2A9) and gout risk (ABCG2) for both adult [125] and pediatric-onset [126]. SLC2A9 is a high affinity UA transporter and one of two transporters that are primarily responsible for the reabsorption of UA [71,104]. A recent WES association study revealed dozens of variants in SLC2A9, including 90 rare variants that associated with SUA, and an additional number of common variants that map onto important functional regions of the protein [108]. Interestingly, SNPs of SLC2A9 are associated with both decreased SUA [92,94,127,128] and increased SUA [97], dependent upon the SNP. Of identified SLC2A9 SNPs associated with a decrease in SUA, this decrease was much greater in women than in men (rs7442295 yielded −0.503 mg/dL in women, while only −0.202 mg/dL in men), with a higher heritability component in females than in males in European populations [104]. These results were further confirmed in a case-control study, where the partial loss of function variant of SLC2A9 contributed to a lowering of UA levels between 0.30–0.35 mg/dL or 5%–6% of all populations sampled, with a much stronger association in women [129]. These results support the idea that SLC2A9 mediated reabsorption is an important mechanism of SUA regulation in females.
4.2. ABCG2
ABCG2, the other major transporter, transports UA out of the cell [76], and is expressed in the kidney, liver, and small intestine, as well as the blood-brain and blood-placental barriers, and in mammary epithelial cells [125]. It is expressed in the renal proximal tubule in both humans and mice [130] where it plays an important role in UA secretion. Several variants in ABCG2 have been associated with SUA levels, the most well-characterized of which is rs2231142, which encodes the missense mutation Q141K. This risk allele is common among several populations ranging from 3% in populations of African ancestry, 11% in those of European ancestry, and up to 31% in East Asian populations [131]. The Q141K variant confers a partial loss of function, characterized by a loss of transporter activity and reduced stability of the N-terminal nucleotide-binding domain leading to decreased protein abundance at the cell surface via increased endoplasmic-reticulum-associated-degradation [94]. However, in humans, possessing the Q141K variant did not translate to the predicted significant changes in FEUA [94,132,133,134].
A recent study attempted to better understand the role of ABCG2 in the kidney and the impact of the Q141K variant on kidney function in a relatively normal young population [130]. The participants were challenged with oral inosine to acutely increase SUA levels. Participants with the Q141K variant had significantly higher SUA levels at baseline and throughout, yet a comparison of the FEUA between genotypes revealed no differences. These findings suggest that changes in FEUA did not contribute significantly to increases in SUA [130] in the Q141K individuals. Instead, the Q141K individuals lost a significant portion of the extra-renal contribution to SUA variance, implicating a strong role for ABCG2 in intestinal UA secretion, but unfortunately not clarifying the role, if any, of ABCG2 in the kidney. Interestingly, that same study examined non-risk allele carriers stratified by sex and found females to have a lower SUA in response to inosine, with higher FEUA and higher urinary UA excretion (Figure 2). These data implied that the female kidney responded differently to the higher UA load, but also better compensated for this higher UA load by increasing excretion.
5. Regulation of UA Transporters
5.1. Transcription Factors
The effectors of UA homeostasis, the transporters, are an obvious mechanism for explaining sex differences but they exist downstream of other differentially regulated pathways, such as transcription factors. While several transcription factors have been implicated in the regulation of UA transporters, for example, HIF-1α and Nrf2 can regulate ABCG2 expression [135,136,137], these transcription factors do not associate with changes in SUA levels in reported GWAS. A more appealing and probable target would be a transcription factor that does associate with SUA levels in humans. Such a target was recently revealed, where transcription factor genes HNF1A (encoding HNF1α) and HNF4A (encoding HNF4α) had significant associations with SUA [101]. Targeted studies have shown that the transcription of UA transporters can be directly regulated by these HNF family transcription factors [138]. Specifically, HNF1α can regulate expression of SLC22A12 [139] and important scaffold gene PDZK1 [140], while HNF4α has been reported to trans-activate SLC2A9 [122] and ABCG2 [101], and also facilitates the proper expression of PDZK1 [141,142]. Index SNPs in HNF1A demonstrated a borderline significant increase in SUA and a decrease in FEUA in women, but not men [94]. Both HNF1A and HNF4A may be influenced by sex hormones [105]. The HNF4A gene has been reported to have estrogen receptor (ER) enhancer elements [143], and the HNF4α protein may interact with ER-α to affect the transcription of target genes [144]. In humans, pancreatic islets demonstrate differential methylation patters of the HNF4A locus between males and females [106], supporting evidence that HNF4α may be associated with islet function [123]. Relatedly, cultured primary hepatocytes isolated from females have reported roughly two-fold higher non-sex hormone-stimulated activation of HNF4α than those isolated from males [107], demonstrating that HNF4α activity may be increased in females when compared to males. HNF4A has been further characterized in mice, in which male HNF4α-null mice demonstrated 1000 more affected genes than females [145]. Thus, since HNF1α and HNF4α affect UA handling machinery and may be affected by sex hormones, these transcription factors are promising candidates for further study.
5.2. Estrogen
To explore the possible role of sex hormones in regulating UA transporters and more generally UA levels, an early study examined the effects of oral administration of synthetic sex hormones on both women and men [146]. The study demonstrated an inverse relationship between SUA and administered exogenous estrogen, as well as an increase in urinary UA. Similar effects were observed when post-menopausal women received progesterone. Additional studies demonstrated changes in SUA during ovulation in pre-menopausal women [147], with peak SUA levels occurring during the follicular phase [148] when estrogen levels are lowest. Furthermore, SUA was positively correlated with follicle stimulating hormone and inversely associated with estradiol and progesterone [148]. Another study examined the effects of hormone therapy on transsexual participants, demonstrating a decrease in SUA in male-to-female (MTF) participants, and an increase in SUA in female-to-male (FTM) participants after one year of cross-sex hormone administration. Furthermore, baseline FEUA was higher in FTM subjects and significantly decreased after the loss of estrogen, while MTF subjects demonstrated an increase in FEUA with the addition of estrogen [149]. These studies provide strong evidence for the role of female sex hormones in regulating FEUA, which can strongly contribute to SUA. Finally, as female sex hormone levels decrease after menopause, SUA increases, but this increase in SUA can be mitigated by hormone replacement therapy [12]. This evidence further reinforces that the female sex hormones may be regulating UA handling machinery, at either the transcriptional or post-transcriptional levels. Estrogen could explain the observed sex differences in UA associated UA transporters. Therefore, understanding the role of estrogen in regulating UA transporters is critical in unraveling the complex mechanisms of UA handling.
Estrogen has been reported to have direct effects on sex-associated UA transporters. For example, ER binding sites have been identified in the promoter region of ABCG2 [150], implying ABCG2 can be transcriptionally regulated by estrogen. Additional reports have demonstrated that ABCG2 protein expression is down-regulated by estrogen [151], possibly through proteasome activation via ER-β induced PTEN/PI3K/AKT signaling [152]. Based on these studies, increased estrogen could decrease ABCG2 expression, which would decrease secretion, leading to an increase in SUA. This seems to be in conflict with the fact that females have lower SUA levels, demonstrating that in vitro models may have limited utility regarding ABCG2 regulation. Combinatorial signaling through ER-α and ER-β may also induce internalization of ABCG2 protein at the plasma membrane, decreasing ABCG2 activity, as observed in the blood-brain-barrier of mice [152]. This study provides some evidence of estrogen-mediated post-transcriptional regulation of ABCG2, yet further studies are required to elucidate the role of estrogen regulation of ABCG2 in the kidney. Similarly, estrogen has also been shown to downregulate SLC2A9 at the post-transcriptional level through ER-β induced proteasomal activation [153]. Decreased SUA levels in females could be explained as ER-β signaling causing a greater decrease in SLC2A9 than ABCG2, resulting in a greater decrease in the reabsorption of UA, leading to a higher FEUA, as seen in females, however, this requires additional study.
6. Modeling HUA Associated Sex Differences
6.1. Established Mouse Models
The complexity of human physiology and the heterogeneous nature of human populations make mechanistic studies difficult. Thus, effective model systems are needed to further progress our understanding of UA homeostasis and the role of sex in its regulation. Animal models are widely used to provide insights into human disease, but the specific differences in human and rodent UA physiology present additional hurdles [154,155]. One hurdle that needs to be potentially overcome when using mice to model HUA is that mice have a functional Uox gene, resulting in the production of the uricase enzyme, which then breaks down UA into allantoin. To remove this hurdle, a mouse with genetic ablation of the Uox gene was first described in 1994 [156], resulting in increased SUA levels in both males and females. Unfortunately, as much as 40% of the animals died within the first five weeks of life, with even the survivors displaying significantly increased incidence of UA crystal deposition in the kidney and other tissues, limiting the utility as a model of hyperuricemia. Therefore, additional mouse models that develop HUA by slightly different mechanisms are required for further study.
Other key UA transporter genes have been targeted in attempts to raise the SUA levels in mice, including URAT1, SLC2A9, and ABCG2. SLC22A12 has been knocked-out on both a mixed [157] and a pure C57Bl/6J background [70]. Both SLC22A12 knock-out models are grossly physiologically normal, with either an increased urinary UA to creatinine ration along with higher FEUA [158] or increased urinary UA excretion with no apparent differences in SUA [70]. Whole-body knock-outs of SLC2A9 [159] demonstrated HUA, as well as nephropathy with UA crystals, hyperuricosuria, polyuria, and increased water intake. Interestingly, sex differences in FEUA were observed in these animals, corroborating the predicted sex differences from the genetic studies in humans. Sex differences in FEUA were lost in a liver-specific SLC2A9 knock-out and there were no sex differences reported in the intestinal enterocyte-specific knock-out of SLC2A9 [160]. Therefore, like the SLC22A12 knock-out mice, SLC2A9 knock-out mice have limited use when exploring sex differences in HUA.
6.2. ABCG2-Q140K Mouse Model
In contrast, a new HUA mouse model produced by inserting a human ABCG2 variant (Q140K; orthologous to the human Q141K), not only accurately recapitulates renal and gut UA transport defects observed in humans possessing the Q141K variant, but also results in profound sex differences in how the deleterious ABCG2 variant effects function. The mouse model was created in an effort to better understand if ABCG2 played a significant role in kidney UA excretion and the deleterious effects of the Q140K variant. The model demonstrated that ABCG2 is a key secretory transporter in the kidney and small intestines, however, while the Q140K variant was extremely deleterious in the intestines, it surprisingly had only a small effect on ABCG2 abundance and function in the kidney. This discrepancy in phenotypes appeared to explain why humans with the Q141K variant are at high risk for HUA: the mutant protein has little effect on transporter function in the kidney, thus differences in observed FEUA are small or are absent, yet the Q141K variant produces a profound loss of intestinal secretion of UA, driving increases in SUA [130].
A second startling finding from the ABCG2-Q140K mouse model was that the female Q140K mice did not show any phenotypes (Figure 3). The female Q140K mice had unaltered SUA, FEUA, and even unaltered renal ABCG2 protein abundance [130], suggesting these mice were representative of the sex differences observed in humans associated with HUA, and therefore could prove to be the most useful model to date. These sex differences may be due, at least in part, to the fact that ABCG2 can be regulated by estrogen [150,151,152], as hypothesized in humans. Comparing transcriptional profiles and post-translational modifications in the HUA male Q140K mice against the non-HUA Q140K female mice may reveal genes or signaling cascades critical for the regulation of UA handling in the kidney. Luckily, these further studies are now possible.
With the right in vivo models, a number of important questions are possible to address. Since increases in SUA are associated with a myriad of other pathologies, including CKD, hypertension, and diabetes, it is difficult to determine which is causal and which is the co-morbidity. In the case of CKD, can HUA promote CKD, and will amelioration of SUA levels improve the prognosis for CKD? How does biological sex potentially contribute to this relationship? There is emerging evidence that treatment of SUA levels in patients with CKD can in fact improve CKD progression in patients receiving UA lowering therapy [161,162]. In order to better answer these questions, improved models are required. An ideal model would most closely mimic the human condition, with a slower onset of increased SUA as the animals age, and distinct differences in phenotypes between male and female animals, in which females demonstrate milder symptoms. The humanized ABCG2-Q140K mouse model [130] serves as an excellent jumping-off point to begin comparisons, as these mice recapitulate the human pathophysiology and the sex differences phenotypes.
7. Conclusions
Female sex hormones, specifically estrogen, may play a role in the regulation of expression or activity of UA transporters, specifically ABCG2 and SLC2A9. This is emphasized by the fact that females may have differences in renal UA transporter expression, localization, or activity. Estrogen could mediate either direct transcriptional regulation of the transporter genes, or activate transporter specific transcription factors, including HNF4α. Observable differences in how transporter transcription and post-translational modification are regulated between the sexes may reveal significant information about these regulatory pathways and provide targets for therapy. Thus, using sex as a biological variable may provide key insights into understanding UA handling, elucidating underlying physiological regulatory mechanisms, and underscoring a critical need for future studies.
Author Contributions
V.L.H.K. wrote the manuscript. O.M.W. edited the manuscript. Both authors revised and approved the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the National Institute of Diabetes and Digestive and Kidney Diseases, R01DK114091 (O.M.W.) and F32DK124960 (V.L.H.K.).
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| CKD | Chronic Kidney Disease |
| ER | Estrogen Receptor |
| ESRD | End Stand Renal Disease |
| FEUA | Fractional Excretion of Urate |
| FTM | Female-to-Male |
| GFR | Glomerular Filtration Rate |
| GWAS | Genome-Wide Association Studies |
| HUA | Hyperuricemia |
| MTF | Male-to-Female |
| OAT | Organic Anion Transporter |
| SNP | Single Nucleotide Polymorphism |
| SUA | Serum Urate |
| UA | Urate |
| UUE | Urinary Excretion of Urate |
| WES | Whole Exome Sequencing |
References
- Kottgen, A. Genome-wide association studies in nephrology research. Am. J. Kidney Dis. 2010, 56, 743–758. [Google Scholar] [CrossRef]
- Nakagawa, T.; Hu, H.; Zharikov, S.; Tuttle, K.R.; Short, R.A.; Glushakova, O.; Ouyang, X.; Feig, D.I.; Block, E.R.; Herrera-Acosta, J.; et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2006, 290, F625–F631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Major, T.J.; Dalbeth, N.; Stahl, E.A.; Merriman, T.R. An update on the genetics of hyperuricaemia and gout. Nat. Rev. Rheumatol. 2018, 14, 341–353. [Google Scholar] [CrossRef]
- Nuki, G.; Simkin, P.A. A concise history of gout and hyperuricemia and their treatment. Arthritis Res. 2006, 8 (Suppl. 1), S1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, C.F.; Grainge, M.J.; Zhang, W.; Doherty, M. Global epidemiology of gout: Prevalence, incidence and risk factors. Nat. Rev. Rheumatol. 2015, 11, 649–662. [Google Scholar] [CrossRef] [PubMed]
- Chen-Xu, M.; Yokose, C.; Rai, S.K.; Pillinger, M.H.; Choi, H.K. Contemporary Prevalence of Gout and Hyperuricemia in the United States and Decadal Trends: The National Health and Nutrition Examination Survey 2007–2016. Arthritis Rheumatol. 2019. [Google Scholar] [CrossRef]
- Mazzali, M.; Kanbay, M.; Segal, M.S.; Shafiu, M.; Jalal, D.; Feig, D.I.; Johnson, R.J. Uric acid and hypertension: Cause or effect? Curr. Rheumatol. Rep. 2010, 12, 108–117. [Google Scholar] [CrossRef]
- Harrold, L.R.; Etzel, C.J.; Gibofsky, A.; Kremer, J.M.; Pillinger, M.H.; Saag, K.G.; Schlesinger, N.; Terkeltaub, R.; Cox, V.; Greenberg, J.D. Sex differences in gout characteristics: Tailoring care for women and men. BMC Musculoskelet Disord. 2017, 18, 108. [Google Scholar] [CrossRef] [Green Version]
- Harrold, L.R.; Yood, R.A.; Mikuls, T.R.; Andrade, S.E.; Davis, J.; Fuller, J.; Chan, K.A.; Roblin, D.; Raebel, M.A.; Von Worley, A.; et al. Sex differences in gout epidemiology: Evaluation and treatment. Ann. Rheum. Dis. 2006, 65, 1368–1372. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Han, C.; Wu, D.; Xia, X.; Gu, J.; Guan, H.; Shan, Z.; Teng, W. Prevalence of Hyperuricemia and Gout in Mainland China from 2000 to 2014: A Systematic Review and Meta-Analysis. Biomed. Res. Int. 2015, 2015, 762820. [Google Scholar] [CrossRef] [Green Version]
- Hak, A.E.; Curhan, G.C.; Grodstein, F.; Choi, H.K. Menopause, postmenopausal hormone use and risk of incident gout. Ann. Rheum. Dis. 2010, 69, 1305–1309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ioannou, G.N.; Boyko, E.J. Effects of menopause and hormone replacement therapy on the associations of hyperuricemia with mortality. Atherosclerosis 2013, 226, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumino, H.; Ichikawa, S.; Kanda, T.; Nakamura, T.; Sakamaki, T. Reduction of serum uric acid by hormone replacement therapy in postmenopausal women with hyperuricaemia. Lancet 1999, 354, 650. [Google Scholar] [CrossRef]
- Regitz-Zagrosek, V. Sex and gender differences in health. Embo. Rep. 2012, 13, 596–603. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.Y.; Schaub, M.A.; Sirota, M.; Butte, A.J. Sex differences in disease risk from reported genome-wide association study findings. Hum. Genet. 2012, 131, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Ruau, D.; Liu, L.Y.; Clark, J.D.; Angst, M.S.; Butte, A.J. Sex differences in reported pain across 11,000 patients captured in electronic medical records. J. Pain 2012, 13, 228–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shansky, R.M. Sex differences in mechanisms of disease. Genes Brain Behav. 2020, 19, e12646. [Google Scholar] [CrossRef]
- Humphries, K.H.; Izadnegahdar, M.; Sedlak, T.; Saw, J.; Johnston, N.; Schenck-Gustafsson, K.; Shah, R.U.; Regitz-Zagrosek, V.; Grewal, J.; Vaccarino, V.; et al. Sex differences in cardiovascular disease—Impact on care and outcomes. Front. Neuroendocr. 2017, 46, 46–70. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Dworatzek, E.; Seeland, U.; Kararigas, G.; Arnal, J.F.; Brunelleschi, S.; Carpenter, T.C.; Erdmann, J.; Franconi, F.; Giannetta, E.; et al. Sex in basic research: Concepts in the cardiovascular field. Cardiovasc. Res. 2017, 113, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yu, M.; Yuan, J.; Hu, F.; Liu, S.; Yang, Z.; Cui, J.; Qiao, S. Sex-Related Differences in the Impact of Systemic Hypertension on Left Ventricular Remodeling in Patients with Hypertrophic Obstructive Cardiomyopathy. Cardiology 2020, 145, 203–214. [Google Scholar] [CrossRef]
- Agarwala, A.; Michos, E.D.; Samad, Z.; Ballantyne, C.M.; Virani, S.S. The Use of Sex-Specific Factors in the Assessment of Women’s Cardiovascular Risk. Circulation 2020, 141, 592–599. [Google Scholar] [CrossRef]
- Kerr, N.; Dietrich, D.W.; Bramlett, H.M.; Raval, A.P. Sexually dimorphic microglia and ischemic stroke. CNS Neurosci. Ther. 2019, 25, 1308–1317. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shields, G.S.; Wu, Q.; Liu, Y.; Guo, C. Obesity is associated with poor working memory in women, not men: Findings from a nationally representative dataset of U.S. adults. Eat. Behav. 2019, 35, 101338. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Shi, W.; Jiang, W.; Rao, S.; Huang, B.; Yan, H.; Gao, X. Sex-specific association of metabolic risk factors with brain ischemic lesions by severity and location. Biol. Sex Differ. 2019, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Meoni, S.; Macerollo, A.; Moro, E. Sex differences in movement disorders. Nat. Rev. Neurol. 2020, 16, 84–96. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Becerra-Diaz, M.; Strickland, A.B.; Keselman, A.; Heller, N.M. Androgen and Androgen Receptor as Enhancers of M2 Macrophage Polarization in Allergic Lung Inflammation. J. Immunol. 2018, 201, 2923–2933. [Google Scholar] [CrossRef] [Green Version]
- Dolsen, M.R.; Crosswell, A.D.; Prather, A.A. Links Between Stress, Sleep, and Inflammation: Are there Sex Differences? Curr. Psychiatry Rep. 2019, 21, 8. [Google Scholar] [CrossRef]
- Costacou, T.; Fried, L.; Ellis, D.; Orchard, T.J. Sex differences in the development of kidney disease in individuals with type 1 diabetes mellitus: A contemporary analysis. Am. J. Kidney Dis. 2011, 58, 565–573. [Google Scholar] [CrossRef] [Green Version]
- Bjornstad, P.; Cherney, D.Z. Renal Hyperfiltration in Adolescents with Type 2 Diabetes: Physiology, Sex Differences, and Implications for Diabetic Kidney Disease. Curr. Diab. Rep. 2018, 18, 22. [Google Scholar] [CrossRef]
- Antlanger, M.; Noordzij, M.; Van de Luijtgaarden, M.; Carrero, J.J.; Palsson, R.; Finne, P.; Hemmelder, M.H.; Areste-Fosalba, N.; Reisaeter, A.V.; Cases, A.; et al. Sex Differences in Kidney Replacement Therapy Initiation and Maintenance. Clin. J. Am. Soc. Nephrol. 2019, 14, 1616–1625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, B.D. Sex Differences in Diabetes and Kidney Disease: Mechanisms and Consequences. Am. J. Physiol. Ren. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Layton, A.T.; Sullivan, J.C. Recent advances in sex differences in kidney function. Am. J. Physiol. Ren. Physiol. 2019, 316, F328–F331. [Google Scholar] [CrossRef] [PubMed]
- Veiras, L.C.; Girardi, A.C.C.; Curry, J.; Pei, L.; Ralph, D.L.; Tran, A.; Castelo-Branco, R.C.; Pastor-Soler, N.; Arranz, C.T.; Yu, A.S.L.; et al. Sexual Dimorphic Pattern of Renal Transporters and Electrolyte Homeostasis. J. Am. Soc. Nephrol. 2017, 28, 3504–3517. [Google Scholar] [CrossRef]
- Writing Group, M.; Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; De Ferranti, S.; Despres, J.P.; et al. Executive Summary: Heart Disease and Stroke Statistics—2016 Update: A Report From the American Heart Association. Circulation 2016, 133, 447–454. [Google Scholar] [CrossRef]
- Neugarten, J.; Golestaneh, L. Female sex reduces the risk of hospital-associated acute kidney injury: A meta-analysis. BMC Nephrol. 2018, 19, 314. [Google Scholar] [CrossRef]
- Neugarten, J.; Golestaneh, L.; Kolhe, N.V. Sex differences in acute kidney injury requiring dialysis. BMC Nephrol. 2018, 19, 131. [Google Scholar] [CrossRef] [PubMed]
- Aufhauser, D.D., Jr.; Wang, Z.; Murken, D.R.; Bhatti, T.R.; Wang, Y.; Ge, G.; Redfield, R.R., III; Abt, P.L.; Wang, L.; Svoronos, N.; et al. Improved renal ischemia tolerance in females influences kidney transplantation outcomes. J. Clin. Investig. 2016, 126, 1968–1977. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, R.; Yazawa, M.; Morikawa, Y.; Tsutsui, H.; Ohkita, M.; Yukimura, T.; Matsumura, Y. Sex differences in ischaemia/reperfusion-induced acute kidney injury depends on the degradation of noradrenaline by monoamine oxidase. Clin. Exp. Pharm. Physiol. 2017, 44, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Cobo, G.; Hecking, M.; Port, F.K.; Exner, I.; Lindholm, B.; Stenvinkel, P.; Carrero, J.J. Sex and gender differences in chronic kidney disease: Progression to end-stage renal disease and haemodialysis. Clin. Sci. (Lond.) 2016, 130, 1147–1163. [Google Scholar] [CrossRef] [PubMed]
- Reckelhoff, J.F. Gender differences in the regulation of blood pressure. Hypertension 2001, 37, 1199–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricardo, A.C.; Yang, W.; Sha, D.; Appel, L.J.; Chen, J.; Krousel-Wood, M.; Manoharan, A.; Steigerwalt, S.; Wright, J.; Rahman, M.; et al. Sex-Related Disparities in CKD Progression. J. Am. Soc. Nephrol. 2019, 30, 137–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiwara, A.; Kita, A.; Saruwatari, J.; Miyazaki, H.; Kawata, Y.; Morita, K.; Oniki, K.; Yoshida, A.; Jinnouchi, H.; Nakagawa, K. Sex Differences in the Renal Function Decline of Patients with Type 2 Diabetes. J. Diabetes Res. 2016, 2016, 4626382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakurai, M.; Kobayashi, J.; Takeda, Y.; Nagasawa, S.Y.; Yamakawa, J.; Moriya, J.; Mabuchi, H.; Nakagawa, H. Sex Differences in Associations Among Obesity, Metabolic Abnormalities, and Chronic Kidney Disease in Japanese Men and Women. J. Epidemiol. 2016, 26, 440–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, L.E.; Sullivan, J.C. Sex Differences in Obesity-Induced Hypertension and Vascular Dysfunction: A Protective Role for Estrogen in Adipose Tissue Inflammation? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016. [Google Scholar] [CrossRef] [Green Version]
- Clayton, J.A. Studying both sexes: A guiding principle for biomedicine. FASEB J. 2016, 30, 519–524. [Google Scholar] [CrossRef] [Green Version]
- Clayton, J.A.; Collins, F.S. Policy: NIH to balance sex in cell and animal studies. Nature 2014, 509, 282–283. [Google Scholar] [CrossRef]
- Tannenbaum, C.; Schwarz, J.M.; Clayton, J.A.; De Vries, G.J.; Sullivan, C. Evaluating sex as a biological variable in preclinical research: The devil in the details. Biol. Sex Differ. 2016, 7, 13. [Google Scholar] [CrossRef]
- Clayton, J.A. Applying the new SABV (sex as a biological variable) policy to research and clinical care. Physiol. Behav. 2018, 187, 2–5. [Google Scholar] [CrossRef]
- Chang, H.Y.; Tung, C.W.; Lee, P.H.; Lei, C.C.; Hsu, Y.C.; Chang, H.H.; Yang, H.F.; Lu, L.C.; Jong, M.C.; Chen, C.Y.; et al. Hyperuricemia as an independent risk factor of chronic kidney disease in middle-aged and elderly population. Am. J. Med. Sci. 2010, 339, 509–515. [Google Scholar] [CrossRef]
- Kuwabara, M.; Niwa, K.; Hisatome, I.; Nakagawa, T.; Roncal-Jimenez, C.A.; Andres-Hernando, A.; Bjornstad, P.; Jensen, T.; Sato, Y.; Milagres, T.; et al. Asymptomatic Hyperuricemia Without Comorbidities Predicts Cardiometabolic Diseases: Five-Year Japanese Cohort Study. Hypertension 2017, 69, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Redon, P.; Maloberti, A.; Facchetti, R.; Redon, J.; Lurbe, E.; Bombelli, M.; Mancia, G.; Grassi, G. Gender-related differences in serum uric acid in treated hypertensive patients from central and east European countries: Findings from the Blood Pressure control rate and CArdiovascular Risk profilE study. J. Hypertens. 2019, 37, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Yoshitomi, R.; Fukui, A.; Nakayama, M.; Ura, Y.; Ikeda, H.; Oniki, H.; Tsuchihashi, T.; Tsuruya, K.; Kitazono, T. Sex differences in the association between serum uric acid levels and cardiac hypertrophy in patients with chronic kidney disease. Hypertens. Res. 2014, 37, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Fukatsu, M.; Suzuki, S.; Wada, T.; Joh, T. Elevated serum uric acid predicts impaired fasting glucose and type 2 diabetes only among Japanese women undergoing health checkups. Diabetes Metab. 2011, 37, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Dehghan, A.; Van Hoek, M.; Sijbrands, E.J.; Hofman, A.; Witteman, J.C. High serum uric acid as a novel risk factor for type 2 diabetes. Diabetes Care 2008, 31, 361–362. [Google Scholar] [CrossRef] [Green Version]
- Strasak, A.M.; Kelleher, C.C.; Brant, L.J.; Rapp, K.; Ruttmann, E.; Concin, H.; Diem, G.; Pfeiffer, K.P.; Ulmer, H.; Vhm&PP Study Group. Serum uric acid is an independent predictor for all major forms of cardiovascular death in 28,613 elderly women: A prospective 21-year follow-up study. Int. J. Cardiol. 2008, 125, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.K.; Lin, Y.P.; Lee, J.T.; Lin, C.S.; Wu, T.J.; Tsai, K.Z.; Su, F.Y.; Kwon, Y.; Hoshide, S.; Lin, G.M. Sex-specific association of hyperuricemia with cardiometabolic abnormalities in a military cohort: The CHIEF study. Medicine (Baltimore) 2020, 99, e19535. [Google Scholar] [CrossRef] [PubMed]
- Gutman, A.B. Significance of uric acid as a nitrogenous waste in vertebrate evolution. Arthritis Rheum. 1965, 8, 614–626. [Google Scholar] [CrossRef] [PubMed]
- Kratzer, J.T.; Lanaspa, M.A.; Murphy, M.N.; Cicerchi, C.; Graves, C.L.; Tipton, P.A.; Ortlund, E.A.; Johnson, R.J.; Gaucher, E.A. Evolutionary history and metabolic insights of ancient mammalian uricases. Proc. Natl. Acad. Sci. USA 2014, 111, 3763–3768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, M.; Satta, Y.; Takenaka, O.; Takahata, N. Loss of urate oxidase activity in hominoids and its evolutionary implications. Mol. Biol. Evol. 2002, 19, 640–653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desideri, G.; Castaldo, G.; Lombardi, A.; Mussap, M.; Testa, A.; Pontremoli, R.; Punzi, L.; Borghi, C. Is it time to revise the normal range of serum uric acid levels? Eur. Rev. Med. Pharm. Sci. 2014, 18, 1295–1306. [Google Scholar]
- Mikkelsen, W.M.; Dodge, H.J.; Valkenburg, H. The Distribution of Serum Uric Acid Values in a Population Unselected as to Gout or Hyperuricemia: Tecumseh, Michigan 1959–1960. Am. J. Med. 1965, 39, 242–251. [Google Scholar] [CrossRef]
- Esparza Martin, N.; Garcia Nieto, V. Hypouricemia and tubular transport of uric acid. Nefrologia 2011, 31, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Bardin, T. Gout. Lancet 2010, 375, 318–328. [Google Scholar] [CrossRef]
- Roch-Ramel, F.; Werner, D.; Guisan, B. Urate transport in brush-border membrane of human kidney. Am. J. Physiol. 1994, 266, F797–F805. [Google Scholar] [CrossRef] [PubMed]
- Gutman, A.B.; Yu, T.F. A three-component system for regulation of renal excretion of uric acid in man. Trans. Assoc. Am. Physicians 1961, 74, 353–365. [Google Scholar]
- Gutman, A.B. Renal excretion of uric acid in normal and gouty man. Arthritis Rheum. 1965, 8, 665–670. [Google Scholar] [CrossRef]
- Gutman, A.B.; Yu, T.F. Renal mechanisms for regulation of uric acid excretion, with special reference to normal and gouty man. Semin. Arthritis Rheum. 1972, 2, 1–46. [Google Scholar] [CrossRef]
- Bobulescu, I.A.; Moe, O.W. Renal transport of uric acid: Evolving concepts and uncertainties. Adv. Chronic. Kidney Dis. 2012, 19, 358–371. [Google Scholar] [CrossRef] [Green Version]
- Eraly, S.A.; Vallon, V.; Rieg, T.; Gangoiti, J.A.; Wikoff, W.R.; Siuzdak, G.; Barshop, B.A.; Nigam, S.K. Multiple organic anion transporters contribute to net renal excretion of uric acid. Physiol. Genom. 2008, 33, 180–192. [Google Scholar] [CrossRef] [Green Version]
- Caulfield, M.J.; Munroe, P.B.; O’Neill, D.; Witkowska, K.; Charchar, F.J.; Doblado, M.; Evans, S.; Eyheramendy, S.; Onipinla, A.; Howard, P.; et al. SLC2A9 is a high-capacity urate transporter in humans. PLoS Med. 2008, 5, e197. [Google Scholar] [CrossRef] [PubMed]
- Ichida, K.; Hosoyamada, M.; Kimura, H.; Takeda, M.; Utsunomiya, Y.; Hosoya, T.; Endou, H. Urate transport via human PAH transporter hOAT1 and its gene structure. Kidney Int. 2003, 63, 143–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagos, Y.; Stein, D.; Ugele, B.; Burckhardt, G.; Bahn, A. Human renal organic anion transporter 4 operates as an asymmetric urate transporter. J. Am. Soc. Nephrol. 2007, 18, 430–439. [Google Scholar] [CrossRef] [PubMed]
- So, A.; Thorens, B. Uric acid transport and disease. J. Clin. Investig. 2010, 120, 1791–1799. [Google Scholar] [CrossRef] [Green Version]
- Huls, M.; Brown, C.D.; Windass, A.S.; Sayer, R.; Van den Heuvel, J.J.; Heemskerk, S.; Russel, F.G.; Masereeuw, R. The breast cancer resistance protein transporter ABCG2 is expressed in the human kidney proximal tubule apical membrane. Kidney Int. 2008, 73, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Woodward, O.M.; Kottgen, A.; Coresh, J.; Boerwinkle, E.; Guggino, W.B.; Kottgen, M. Identification of a urate transporter, ABCG2, with a common functional polymorphism causing gout. Proc. Natl. Acad. Sci. USA 2009, 106, 10338–10342. [Google Scholar] [CrossRef] [Green Version]
- Iharada, M.; Miyaji, T.; Fujimoto, T.; Hiasa, M.; Anzai, N.; Omote, H.; Moriyama, Y. Type 1 sodium-dependent phosphate transporter (SLC17A1 Protein) is a Cl(-)-dependent urate exporter. J. Biol. Chem. 2010, 285, 26107–26113. [Google Scholar] [CrossRef] [Green Version]
- Chiba, T.; Matsuo, H.; Kawamura, Y.; Nagamori, S.; Nishiyama, T.; Wei, L.; Nakayama, A.; Nakamura, T.; Sakiyama, M.; Takada, T.; et al. NPT1/SLC17A1 Is a Renal Urate Exporter in Humans and Its Common Gain-of-Function Variant Decreases the Risk of Renal Underexcretion Gout. Arthritis Rheumatol. 2015, 67, 281–287. [Google Scholar] [CrossRef]
- Van Aubel, R.A.; Smeets, P.H.; Van den Heuvel, J.J.; Russel, F.G. Human organic anion transporter MRP4 (ABCC4) is an efflux pump for the purine end metabolite urate with multiple allosteric substrate binding sites. Am. J. Physiol. Ren. Physiol. 2005, 288, F327–F333. [Google Scholar] [CrossRef]
- Jutabha, P.; Anzai, N.; Kitamura, K.; Taniguchi, A.; Kaneko, S.; Yan, K.; Yamada, H.; Shimada, H.; Kimura, T.; Katada, T.; et al. Human sodium phosphate transporter 4 (hNPT4/SLC17A3) as a common renal secretory pathway for drugs and urate. J. Biol. Chem. 2010, 285, 35123–35132. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, A.; Kimura, H.; Chairoungdua, A.; Shigeta, Y.; Jutabha, P.; Cha, S.H.; Hosoyamada, M.; Takeda, M.; Sekine, T.; Igarashi, T.; et al. Molecular identification of a renal urate anion exchanger that regulates blood urate levels. Nature 2002, 417, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Boocock, J.; Leask, M.; Okada, Y.; Asian Genetic Epidemiology Network Consortium; Matsuo, H.; Kawamura, Y.; Shi, Y.; Li, C.; Mount, D.B.; Mandal, A.K.; et al. Genomic dissection of 43 serum urate-associated loci provides multiple insights into molecular mechanisms of urate control. Hum. Mol. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sakiyama, M.; Matsuo, H.; Shimizu, S.; Nakashima, H.; Nakayama, A.; Chiba, T.; Naito, M.; Takada, T.; Suzuki, H.; Hamajima, N.; et al. A common variant of organic anion transporter 4 (OAT4/SLC22A11) gene is associated with renal underexcretion type gout. Drug Metab. Pharm. 2014, 29, 208–210. [Google Scholar] [CrossRef] [PubMed]
- Tanner, C.; Boocock, J.; Stahl, E.A.; Dobbyn, A.; Mandal, A.K.; Cadzow, M.; Phipps-Green, A.J.; Topless, R.K.; Hindmarsh, J.H.; Stamp, L.K.; et al. Population-Specific Resequencing Associates the ATP-Binding Cassette Subfamily C Member 4 Gene With Gout in New Zealand Maori and Pacific Men. Arthritis Rheumatol. 2017, 69, 1461–1469. [Google Scholar] [CrossRef]
- Wilk, J.B.; Djousse, L.; Borecki, I.; Atwood, L.D.; Hunt, S.C.; Rich, S.S.; Eckfeldt, J.H.; Arnett, D.K.; Rao, D.C.; Myers, R.H. Segregation analysis of serum uric acid in the NHLBI Family Heart Study. Hum. Genet. 2000, 106, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Guo, C.Y.; Cupples, L.A.; Levy, D.; Wilson, P.W.; Fox, C.S. Genome-wide search for genes affecting serum uric acid levels: The Framingham Heart Study. Metabolism 2005, 54, 1435–1441. [Google Scholar] [CrossRef] [PubMed]
- Nath, S.D.; Voruganti, V.S.; Arar, N.H.; Thameem, F.; Lopez-Alvarenga, J.C.; Bauer, R.; Blangero, J.; MacCluer, J.W.; Comuzzie, A.G.; Abboud, H.E. Genome scan for determinants of serum uric acid variability. J. Am. Soc. Nephrol. 2007, 18, 3156–3163. [Google Scholar] [CrossRef]
- Wakasugi, M.; Kazama, J.J.; Narita, I.; Konta, T.; Fujimoto, S.; Iseki, K.; Moriyama, T.; Yamagata, K.; Tsuruya, K.; Asahi, K.; et al. Association between hypouricemia and reduced kidney function: A cross-sectional population-based study in Japan. Am. J. Nephrol. 2015, 41, 138–146. [Google Scholar] [CrossRef]
- Cheong, H.I.; Kang, J.H.; Lee, J.H.; Ha, I.S.; Kim, S.; Komoda, F.; Sekine, T.; Igarashi, T.; Choi, Y. Mutational analysis of idiopathic renal hypouricemia in Korea. Pediatr. Nephrol. 2005, 20, 886–890. [Google Scholar] [CrossRef]
- Stiburkova, B.; Gabrikova, D.; Cepek, P.; Simek, P.; Kristian, P.; Cordoba-Lanus, E.; Claverie-Martin, F. Prevalence of URAT1 allelic variants in the Roma population. Nucleosides Nucleotides Nucleic Acids 2016, 35, 529–535. [Google Scholar] [CrossRef]
- Xu, L.; Shi, Y.; Zhuang, S.; Liu, N. Recent advances on uric acid transporters. Oncotarget 2017, 8, 100852–100862. [Google Scholar] [CrossRef] [Green Version]
- Dehghan, A.; Kottgen, A.; Yang, Q.; Hwang, S.J.; Kao, W.L.; Rivadeneira, F.; Boerwinkle, E.; Levy, D.; Hofman, A.; Astor, B.C.; et al. Association of three genetic loci with uric acid concentration and risk of gout: A genome-wide association study. Lancet 2008, 372, 1953–1961. [Google Scholar] [CrossRef] [Green Version]
- Kolz, M.; Johnson, T.; Sanna, S.; Teumer, A.; Vitart, V.; Perola, M.; Mangino, M.; Albrecht, E.; Wallace, C.; Farrall, M.; et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009, 5, e1000504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kottgen, A.; Albrecht, E.; Teumer, A.; Vitart, V.; Krumsiek, J.; Hundertmark, C.; Pistis, G.; Ruggiero, D.; O’Seaghdha, C.M.; Haller, T.; et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat. Genet. 2013, 45, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, B.; Mo, Z.; Wu, C.; Yang, H.; Yang, X.; He, Y.; Gui, L.; Zhou, L.; Guo, H.; Zhang, X.; et al. A genome-wide association study identifies common variants influencing serum uric acid concentrations in a Chinese population. BMC Med. Genom. 2014, 7, 10. [Google Scholar] [CrossRef] [Green Version]
- Dong, Z.; Zhou, J.; Jiang, S.; Li, Y.; Zhao, D.; Yang, C.; Ma, Y.; Wang, Y.; He, H.; Ji, H.; et al. Effects of multiple genetic loci on the pathogenesis from serum urate to gout. Sci. Rep. 2017, 7, 43614. [Google Scholar] [CrossRef] [Green Version]
- Nakayama, A.; Nakaoka, H.; Yamamoto, K.; Sakiyama, M.; Shaukat, A.; Toyoda, Y.; Okada, Y.; Kamatani, Y.; Nakamura, T.; Takada, T.; et al. GWAS of clinically defined gout and subtypes identifies multiple susceptibility loci that include urate transporter genes. Ann. Rheum. Dis. 2017, 76, 869–877. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.J.; Tseng, C.C.; Yen, J.H.; Chang, J.G.; Chou, W.C.; Chu, H.W.; Chang, S.J.; Liao, W.T. ABCG2 contributes to the development of gout and hyperuricemia in a genome-wide association study. Sci. Rep. 2018, 8, 3137. [Google Scholar] [CrossRef]
- Nakatochi, M.; Kanai, M.; Nakayama, A.; Hishida, A.; Kawamura, Y.; Ichihara, S.; Akiyama, M.; Ikezaki, H.; Furusyo, N.; Shimizu, S.; et al. Genome-wide meta-analysis identifies multiple novel loci associated with serum uric acid levels in Japanese individuals. Commun. Biol. 2019, 2, 115. [Google Scholar] [CrossRef]
- Narang, R.K.; Topless, R.; Cadzow, M.; Gamble, G.; Stamp, L.K.; Merriman, T.R.; Dalbeth, N. Interactions between serum urate-associated genetic variants and sex on gout risk: Analysis of the UK Biobank. Arthritis Res. 2019, 21, 13. [Google Scholar] [CrossRef] [Green Version]
- Tin, A.; Marten, J.; Halperin Kuhns, V.L.; Li, Y.; Wuttke, M.; Kirsten, H.; Sieber, K.B.; Qiu, C.; Gorski, M.; Yu, Z.; et al. Target genes, variants, tissues and transcriptional pathways influencing human serum urate levels. Nat. Genet. 2019, 51, 1459–1474. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Shriner, D.; Doumatey, A.P.; Zhou, J.; Bentley, A.R.; Lei, L.; Adeyemo, A.; Rotimi, C.N. Refining genome-wide associated loci for serum uric acid in individuals with African ancestry. Hum. Mol. Genet. 2020, 29, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Doring, A.; Gieger, C.; Mehta, D.; Gohlke, H.; Prokisch, H.; Coassin, S.; Fischer, G.; Henke, K.; Klopp, N.; Kronenberg, F.; et al. SLC2A9 influences uric acid concentrations with pronounced sex-specific effects. Nat. Genet. 2008, 40, 430–436. [Google Scholar] [CrossRef]
- Vitart, V.; Rudan, I.; Hayward, C.; Gray, N.K.; Floyd, J.; Palmer, C.N.; Knott, S.A.; Kolcic, I.; Polasek, O.; Graessler, J.; et al. SLC2A9 is a newly identified urate transporter influencing serum urate concentration, urate excretion and gout. Nat. Genet. 2008, 40, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Selva, D.M.; Hogeveen, K.N.; Innis, S.M.; Hammond, G.L. Monosaccharide-induced lipogenesis regulates the human hepatic sex hormone-binding globulin gene. J. Clin. Investig. 2007, 117, 3979–3987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.; Volkov, P.; Dayeh, T.; Esguerra, J.L.; Salo, S.; Eliasson, L.; Ronn, T.; Bacos, K.; Ling, C. Sex differences in the genome-wide DNA methylation pattern and impact on gene expression, microRNA levels and insulin secretion in human pancreatic islets. Genome Biol. 2014, 15, 522. [Google Scholar] [CrossRef] [Green Version]
- Thangavel, C.; Boopathi, E.; Shapiro, B.H. Inherent sex-dependent regulation of human hepatic CYP3A5. Br. J. Pharm. 2013, 168, 988–1000. [Google Scholar] [CrossRef] [Green Version]
- Tin, A.; Li, Y.; Brody, J.A.; Nutile, T.; Chu, A.Y.; Huffman, J.E.; Yang, Q.; Chen, M.H.; Robinson-Cohen, C.; Mace, A.; et al. Large-scale whole-exome sequencing association studies identify rare functional variants influencing serum urate levels. Nat. Commun. 2018, 9, 4228. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.; Nicolson, T.J.; Hammons, G.; Word, B.; Green-Knox, B.; Lyn-Cook, B. Expression of drug transporters in human kidney: Impact of sex, age, and ethnicity. Biol. Sex Differ. 2015, 6, 4. [Google Scholar] [CrossRef] [Green Version]
- Ichida, K.; Matsuo, H.; Takada, T.; Nakayama, A.; Murakami, K.; Shimizu, T.; Yamanashi, Y.; Kasuga, H.; Nakashima, H.; Nakamura, T.; et al. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nat. Commun. 2012, 3, 764. [Google Scholar] [CrossRef] [Green Version]
- Woodward, O.M. ABCG2: The molecular mechanisms of urate secretion and gout. Am. J. Physiol. Ren. Physiol. 2015, 309, F485–F488. [Google Scholar] [CrossRef]
- Okada, Y.; Sim, X.; Go, M.J.; Wu, J.Y.; Gu, D.; Takeuchi, F.; Takahashi, A.; Maeda, S.; Tsunoda, T.; Chen, P.; et al. Meta-analysis identifies multiple loci associated with kidney function-related traits in east Asian populations. Nat. Genet. 2012, 44, 904–909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le, M.T.; Shafiu, M.; Mu, W.; Johnson, R.J. SLC2A9—A fructose transporter identified as a novel uric acid transporter. Nephrol. Dial. Transpl. 2008, 23, 2746–2749. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Jiang, F.; Zhang, R.; Tang, S.S.; Chen, M.; Peng, D.F.; Yan, J.; Wang, T.; Wang, S.Y.; Bao, Y.Q.; et al. Serum uric acid levels are associated with polymorphisms in the SLC2A9, SF1, and GCKR genes in a Chinese population. Acta Pharm. Sin. 2014, 35, 1421–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futagi, Y.; Narumi, K.; Furugen, A.; Kobayashi, M.; Iseki, K. Molecular characterization of the orphan transporter SLC16A9, an extracellular pH- and Na(+)-sensitive creatine transporter. Biochem. Biophys. Res. Commun. 2020, 522, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, A.; Matsuo, H.; Shimizu, T.; Ogata, H.; Takada, Y.; Nakashima, H.; Nakamura, T.; Shimizu, S.; Chiba, T.; Sakiyama, M.; et al. Common missense variant of monocarboxylate transporter 9 (MCT9/SLC16A9) gene is associated with renal overload gout, but not with all gout susceptibility. Hum. Cell 2013, 26, 133–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koyama, T.; Matsui, D.; Kuriyama, N.; Ozaki, E.; Tanaka, K.; Oze, I.; Hamajima, N.; Wakai, K.; Okada, R.; Arisawa, K.; et al. Genetic variants of SLC17A1 are associated with cholesterol homeostasis and hyperhomocysteinaemia in Japanese men. Sci. Rep. 2015, 5, 15888. [Google Scholar] [CrossRef]
- Breljak, D.; Ljubojevic, M.; Hagos, Y.; Micek, V.; Balen Eror, D.; Vrhovac Madunic, I.; Brzica, H.; Karaica, D.; Radovic, N.; Kraus, O.; et al. Distribution of organic anion transporters NaDC3 and OAT1-3 along the human nephron. Am. J. Physiol. Ren. Physiol. 2016, 311, F227–F238. [Google Scholar] [CrossRef] [Green Version]
- Sato, M.; Mamada, H.; Anzai, N.; Shirasaka, Y.; Nakanishi, T.; Tamai, I. Renal secretion of uric acid by organic anion transporter 2 (OAT2/SLC22A7) in human. Biol. Pharm. Bull. 2010, 33, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Shima, Y.; Teruya, K.; Ohta, H. Association between intronic SNP in urate-anion exchanger gene, SLC22A12, and serum uric acid levels in Japanese. Life Sci. 2006, 79, 2234–2237. [Google Scholar] [CrossRef]
- Ferreira, C.; Prestin, K.; Hussner, J.; Zimmermann, U.; Meyer Zu Schwabedissen, H.E. PDZ domain containing protein 1 (PDZK1), a modulator of membrane proteins, is regulated by the nuclear receptor THRbeta. Mol. Cell Endocrinol. 2018, 461, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Prestin, K.; Wolf, S.; Feldtmann, R.; Hussner, J.; Geissler, I.; Rimmbach, C.; Kroemer, H.K.; Zimmermann, U.; Meyer zu Schwabedissen, H.E. Transcriptional regulation of urate transportosome member SLC2A9 by nuclear receptor HNF4alpha. Am. J. Physiol. Ren. Physiol. 2014, 307, F1041–F1051. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Maechler, P.; Antinozzi, P.A.; Hagenfeldt, K.A.; Wollheim, C.B. Hepatocyte nuclear factor 4alpha regulates the expression of pancreatic beta-cell genes implicated in glucose metabolism and nutrient-induced insulin secretion. J. Biol. Chem. 2000, 275, 35953–35959. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.P.; Zhang, J.L.; Wang, J.Y.; Cui, M.X.; Jia, J.L.; Liu, X.H.; Liang, Q.D. MiR-1246 Promotes LPS-Induced Inflammatory Injury in Chondrogenic Cells ATDC5 by Targeting HNF4gamma. Cell Physiol. Biochem. 2017, 43, 2010–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cleophas, M.C.; Joosten, L.A.; Stamp, L.K.; Dalbeth, N.; Woodward, O.M.; Merriman, T.R. ABCG2 polymorphisms in gout: Insights into disease susceptibility and treatment approaches. Pharmgenom. Pers. Med. 2017, 10, 129–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiburkova, B.; Pavelcova, K.; Pavlikova, M.; Jesina, P.; Pavelka, K. The impact of dysfunctional variants of ABCG2 on hyperuricemia and gout in pediatric-onset patients. Arthritis Res. 2019, 21, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, H.; Chiba, T.; Nagamori, S.; Nakayama, A.; Domoto, H.; Phetdee, K.; Wiriyasermkul, P.; Kikuchi, Y.; Oda, T.; Nishiyama, J.; et al. Mutations in glucose transporter 9 gene SLC2A9 cause renal hypouricemia. Am. J. Hum. Genet. 2008, 83, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Dinour, D.; Gray, N.K.; Campbell, S.; Shu, X.; Sawyer, L.; Richardson, W.; Rechavi, G.; Amariglio, N.; Ganon, L.; Sela, B.A.; et al. Homozygous SLC2A9 mutations cause severe renal hypouricemia. J. Am. Soc. Nephrol. 2010, 21, 64–72. [Google Scholar] [CrossRef] [Green Version]
- Brandstatter, A.; Kiechl, S.; Kollerits, B.; Hunt, S.C.; Heid, I.M.; Coassin, S.; Willeit, J.; Adams, T.D.; Illig, T.; Hopkins, P.N.; et al. Sex-specific association of the putative fructose transporter SLC2A9 variants with uric acid levels is modified by BMI. Diabetes Care 2008, 31, 1662–1667. [Google Scholar] [CrossRef] [Green Version]
- Hoque, K.M.; Dixon, E.E.; Lewis, R.M.; Allan, J.; Gamble, G.D.; Phipps-Green, A.J.; Halperin Kuhns, V.L.; Horne, A.M.; Stamp, L.K.; Merriman, T.R.; et al. The ABCG2 Q141K hyperuricemia and gout associated variant illuminates the physiology of human urate excretion. Nat. Commun. 2020, 11, 2767. [Google Scholar] [CrossRef] [PubMed]
- Woodward, O.M.; Kottgen, A.; Kottgen, M. ABCG transporters and disease. FEBS J. 2011, 278, 3215–3225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dalbeth, N.; House, M.E.; Gamble, G.D.; Pool, B.; Horne, A.; Purvis, L.; Stewart, A.; Merriman, M.; Cadzow, M.; Phipps-Green, A.; et al. Influence of the ABCG2 gout risk 141 K allele on urate metabolism during a fructose challenge. Arthritis Res. 2014, 16, R34. [Google Scholar] [CrossRef] [Green Version]
- Matsuo, H.; Tsunoda, T.; Ooyama, K.; Sakiyama, M.; Sogo, T.; Takada, T.; Nakashima, A.; Nakayama, A.; Kawaguchi, M.; Higashino, T.; et al. Hyperuricemia in acute gastroenteritis is caused by decreased urate excretion via ABCG2. Sci. Rep. 2016, 6, 31003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kannangara, D.R.; Phipps-Green, A.J.; Dalbeth, N.; Stamp, L.K.; Williams, K.M.; Graham, G.G.; Day, R.O.; Merriman, T.R. Hyperuricaemia: Contributions of urate transporter ABCG2 and the fractional renal clearance of urate. Ann. Rheum. Dis. 2016, 75, 1363–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Wang, J.; Wei, W.; Shi, M.; Xin, B.; Zhang, T.; Shen, X. Hypoxia regulates ABCG2 activity through the activivation of ERK1/2/HIF-1alpha and contributes to chemoresistance in pancreatic cancer cells. Cancer Biol. 2016, 17, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Jia, Y.; Chen, J.; Zhu, H.; Jia, Z.H.; Cui, M.H. Aberrantly elevated redox sensing factor Nrf2 promotes cancer stem cell survival via enhanced transcriptional regulation of ABCG2 and Bcl-2/Bmi-1 genes. Oncol. Rep. 2015, 34, 2296–2304. [Google Scholar] [CrossRef]
- Krishnamurthy, P.; Ross, D.D.; Nakanishi, T.; Bailey-Dell, K.; Zhou, S.; Mercer, K.E.; Sarkadi, B.; Sorrentino, B.P.; Schuetz, J.D. The stem cell marker Bcrp/ABCG2 enhances hypoxic cell survival through interactions with heme. J. Biol. Chem. 2004, 279, 24218–24225. [Google Scholar] [CrossRef] [Green Version]
- Martovetsky, G.; Tee, J.B.; Nigam, S.K. Hepatocyte nuclear factors 4alpha and 1alpha regulate kidney developmental expression of drug-metabolizing enzymes and drug transporters. Mol. Pharm. 2013, 84, 808–823. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, R.; Kusuhara, H.; Hattori, N.; Kim, I.; Shiota, K.; Gonzalez, F.J.; Sugiyama, Y. Regulation of tissue-specific expression of the human and mouse urate transporter 1 gene by hepatocyte nuclear factor 1 alpha/beta and DNA methylation. Mol. Pharm. 2007, 72, 1619–1625. [Google Scholar] [CrossRef]
- Prestin, K.; Hussner, J.; Ferreira, C.; Seibert, I.; Breitung, V.; Zimmermann, U.; Meyer Zu Schwabedissen, H.E. Regulation of PDZ domain-containing 1 (PDZK1) expression by hepatocyte nuclear factor-1alpha (HNF1alpha) in human kidney. Am. J. Physiol. Ren. Physiol. 2017, 313, F973–F983. [Google Scholar] [CrossRef] [Green Version]
- Gallegos, T.F.; Martovetsky, G.; Kouznetsova, V.; Bush, K.T.; Nigam, S.K. Organic anion and cation SLC22 “drug” transporter (Oat1, Oat3, and Oct1) regulation during development and maturation of the kidney proximal tubule. PLoS ONE 2012, 7, e40796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ketharnathan, S.; Leask, M.; Boocock, J.; Phipps-Green, A.J.; Antony, J.; O’Sullivan, J.M.; Merriman, T.R.; Horsfield, J.A. A non-coding genetic variant maximally associated with serum urate levels is functionally linked to HNF4A-dependent PDZK1 expression. Hum. Mol. Genet. 2018, 27, 3964–3973. [Google Scholar] [CrossRef] [PubMed]
- Weltmeier, F.; Borlak, J. A high resolution genome-wide scan of HNF4alpha recognition sites infers a regulatory gene network in colon cancer. PLoS ONE 2011, 6, e21667. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Yeh, S.H.; Lin, W.H.; Yeh, K.H.; Yuan, Q.; Xia, N.S.; Chen, D.S.; Chen, P.J. Estrogen receptor alpha represses transcription of HBV genes via interaction with hepatocyte nuclear factor 4alpha. Gastroenterology 2012, 142, 989–998.e4. [Google Scholar] [CrossRef] [PubMed]
- Holloway, M.G.; Miles, G.D.; Dombkowski, A.A.; Waxman, D.J. Liver-specific hepatocyte nuclear factor-4alpha deficiency: Greater impact on gene expression in male than in female mouse liver. Mol. Endocrinol. 2008, 22, 1274–1286. [Google Scholar] [CrossRef] [Green Version]
- Adamopoulos, D.; Vlassopoulos, C.; Seitanides, B.; Contoyiannis, P.; Vassilopoulos, P. The relationship of sex steroids to uric acid levels in plasma and urine. Acta. Endocrinol. (Copenh.) 1977, 85, 198–208. [Google Scholar] [CrossRef]
- Taddeo, A.; Renieri, A.; Fioravanti, A.; Morozzi, G.; Parrini, E.; D’Amato, M.S.; Ricci, M.G. Effect of sex steroids in vivo and in vitro on the binding of uric acid to plasma proteins. Boll. Soc. Ital. Biol. Sper. 1984, 60, 1709–1714. [Google Scholar]
- Mumford, S.L.; Dasharathy, S.S.; Pollack, A.Z.; Perkins, N.J.; Mattison, D.R.; Cole, S.R.; Wactawski-Wende, J.; Schisterman, E.F. Serum uric acid in relation to endogenous reproductive hormones during the menstrual cycle: Findings from the BioCycle study. Hum. Reprod. 2013, 28, 1853–1862. [Google Scholar] [CrossRef] [Green Version]
- Yahyaoui, R.; Esteva, I.; Haro-Mora, J.J.; Almaraz, M.C.; Morcillo, S.; Rojo-Martinez, G.; Martinez, J.; Gomez-Zumaquero, J.M.; Gonzalez, I.; Hernando, V.; et al. Effect of long-term administration of cross-sex hormone therapy on serum and urinary uric acid in transsexual persons. J. Clin. Endocrinol. Metab. 2008, 93, 2230–2233. [Google Scholar] [CrossRef] [Green Version]
- Ee, P.L.; Kamalakaran, S.; Tonetti, D.; He, X.; Ross, D.D.; Beck, W.T. Identification of a novel estrogen response element in the breast cancer resistance protein (ABCG2) gene. Cancer Res. 2004, 64, 1247–1251. [Google Scholar] [CrossRef] [Green Version]
- Imai, Y.; Ishikawa, E.; Asada, S.; Sugimoto, Y. Estrogen-mediated post transcriptional down-regulation of breast cancer resistance protein/ABCG2. Cancer Res. 2005, 65, 596–604. [Google Scholar] [PubMed]
- Hartz, A.M.; Madole, E.K.; Miller, D.S.; Bauer, B. Estrogen receptor beta signaling through phosphatase and tensin homolog/phosphoinositide 3-kinase/Akt/glycogen synthase kinase 3 down-regulates blood-brain barrier breast cancer resistance protein. J. Pharm. Exp. 2010, 334, 467–476. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Chen, B.; Qing, Y.; Xie, W.; Dang, W.; Zhao, M.; Zhou, J. Estrogen receptor beta signaling induces autophagy and downregulates Glut9 expression. Nucleosides Nucleotides Nucleic Acids 2014, 33, 455–465. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Dalbeth, N.; Yin, H.; Li, C.; Merriman, T.R.; Wei, W.H. Mouse models for human hyperuricaemia: A critical review. Nat. Rev. Rheumatol. 2019, 15, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Peng, X.; Ling, G. An update on the animal models in hyperuricaemia research. Clin. Exp. Rheumatol. 2017, 35, 860–864. [Google Scholar]
- Wu, X.; Wakamiya, M.; Vaishnav, S.; Geske, R.; Montgomery, C., Jr.; Jones, P.; Bradley, A.; Caskey, C.T. Hyperuricemia and urate nephropathy in urate oxidase-deficient mice. Proc. Natl. Acad. Sci. USA 1994, 91, 742–746. [Google Scholar] [CrossRef] [Green Version]
- Hosoyamada, M.; Ichida, K.; Enomoto, A.; Hosoya, T.; Endou, H. Function and localization of urate transporter 1 in mouse kidney. J. Am. Soc. Nephrol. 2004, 15, 261–268. [Google Scholar] [CrossRef] [Green Version]
- Hosoyamada, M.; Takiue, Y.; Morisaki, H.; Cheng, J.; Ikawa, M.; Okabe, M.; Morisaki, T.; Ichida, K.; Hosoya, T.; Shibasaki, T. Establishment and analysis of SLC22A12 (URAT1) knockout mouse. Nucleosides Nucleotides Nucleic Acids 2010, 29, 314–320. [Google Scholar] [CrossRef]
- Preitner, F.; Bonny, O.; Laverriere, A.; Rotman, S.; Firsov, D.; Da Costa, A.; Metref, S.; Thorens, B. Glut9 is a major regulator of urate homeostasis and its genetic inactivation induces hyperuricosuria and urate nephropathy. Proc. Natl. Acad. Sci. USA 2009, 106, 15501–15506. [Google Scholar] [CrossRef] [Green Version]
- DeBosch, B.J.; Kluth, O.; Fujiwara, H.; Schurmann, A.; Moley, K. Early-onset metabolic syndrome in mice lacking the intestinal uric acid transporter SLC2A9. Nat. Commun. 2014, 5, 4642. [Google Scholar] [CrossRef]
- Liu, X.; Zhai, T.; Ma, R.; Luo, C.; Wang, H.; Liu, L. Effects of uric acid-lowering therapy on the progression of chronic kidney disease: A systematic review and meta-analysis. Ren. Fail. 2018, 40, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Feig, D.I.; Stack, A.G.; Kang, D.H.; Lanaspa, M.A.; Ejaz, A.A.; Sanchez-Lozada, L.G.; Kuwabara, M.; Borghi, C.; Johnson, R.J. The case for uric acid-lowering treatment in patients with hyperuricaemia and CKD. Nat. Rev. Nephrol. 2019, 15, 767–775. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Co-morbidities associated with hyperuricemia, with an emphasis on conditions that can affect the kidneys. Males tend to have higher serum urate levels, and therefore have an increased risk of associated co-morbidities, while females have lower serum urate levels and are protected from developing associated co-morbidities.
Figure 1.
Co-morbidities associated with hyperuricemia, with an emphasis on conditions that can affect the kidneys. Males tend to have higher serum urate levels, and therefore have an increased risk of associated co-morbidities, while females have lower serum urate levels and are protected from developing associated co-morbidities.

Figure 2.
Effect of sex on urinary urate excretion (UUE) (A), and fractional excretion of urate (FEUA) (B) in participants following inosine load (79 participants; ± SD of mean). Statistical significance evaluated by a two-tailed ANCOVA, adjusted for age, ancestry, and BMI. ACOVA P(time) < 0.0001, P(sex) < 0.0001 for Panels (A,B), ACOVA P(sex*time) = 0.038 (A) and P(sex*time) = 0.45 (B). Adapted from Hoque et al., Nature Communications [130] as allowed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Figure 2.
Effect of sex on urinary urate excretion (UUE) (A), and fractional excretion of urate (FEUA) (B) in participants following inosine load (79 participants; ± SD of mean). Statistical significance evaluated by a two-tailed ANCOVA, adjusted for age, ancestry, and BMI. ACOVA P(time) < 0.0001, P(sex) < 0.0001 for Panels (A,B), ACOVA P(sex*time) = 0.038 (A) and P(sex*time) = 0.45 (B). Adapted from Hoque et al., Nature Communications [130] as allowed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).

Figure 3.
Female metabolic comparisons and ABCG2 abundance in ABCG2-Q140K mice. (A) Serum urate levels for male and female mice: WT and Q140K+/+ animals (WT females n = 9; Q140K+/+ n = 10; p = 0.7113, two-tailed Student’s t-test; males, n = 14 WT, and n = 19 Q140K+/+; * p < 0.0001, ± SEM). (B) Fractional excretion of urate (FEUA) measurements from female WT (n = 7) and female Q140K+/+ (n = 7, p = 0.6263, two-tailed Student’s t-test; males, n = 10 WT, and n = 12 Q140K+/+; * p = 0.0196, ± SEM). (C) Summary data from Western blots of total kidney homogenate from female WT (n = 8) and female Q140K+/+ (n = 8) mice showed no change in abundance of the Q140K+/+ protein (p = 0.0835, two-tailed Student’s t-test. (Males, n = 15 WT, and n = 14 Q140K+/+; * p < 0.0001, ± SEM). NS = non-significant. Adapted from Hoque et al., Nature Communications [130] as allowed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).
Figure 3.
Female metabolic comparisons and ABCG2 abundance in ABCG2-Q140K mice. (A) Serum urate levels for male and female mice: WT and Q140K+/+ animals (WT females n = 9; Q140K+/+ n = 10; p = 0.7113, two-tailed Student’s t-test; males, n = 14 WT, and n = 19 Q140K+/+; * p < 0.0001, ± SEM). (B) Fractional excretion of urate (FEUA) measurements from female WT (n = 7) and female Q140K+/+ (n = 7, p = 0.6263, two-tailed Student’s t-test; males, n = 10 WT, and n = 12 Q140K+/+; * p = 0.0196, ± SEM). (C) Summary data from Western blots of total kidney homogenate from female WT (n = 8) and female Q140K+/+ (n = 8) mice showed no change in abundance of the Q140K+/+ protein (p = 0.0835, two-tailed Student’s t-test. (Males, n = 15 WT, and n = 14 Q140K+/+; * p < 0.0001, ± SEM). NS = non-significant. Adapted from Hoque et al., Nature Communications [130] as allowed under the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/).


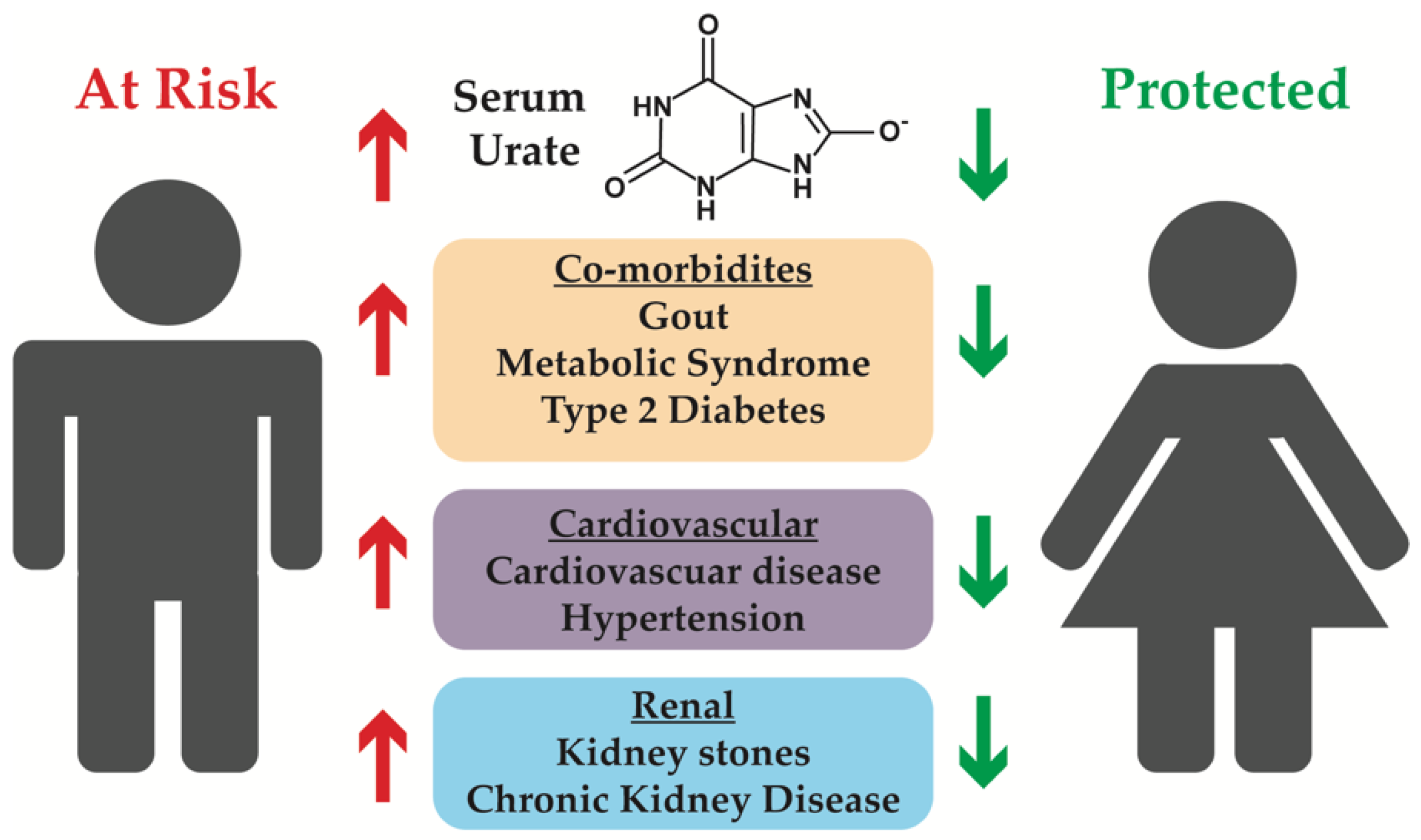{kind=link}
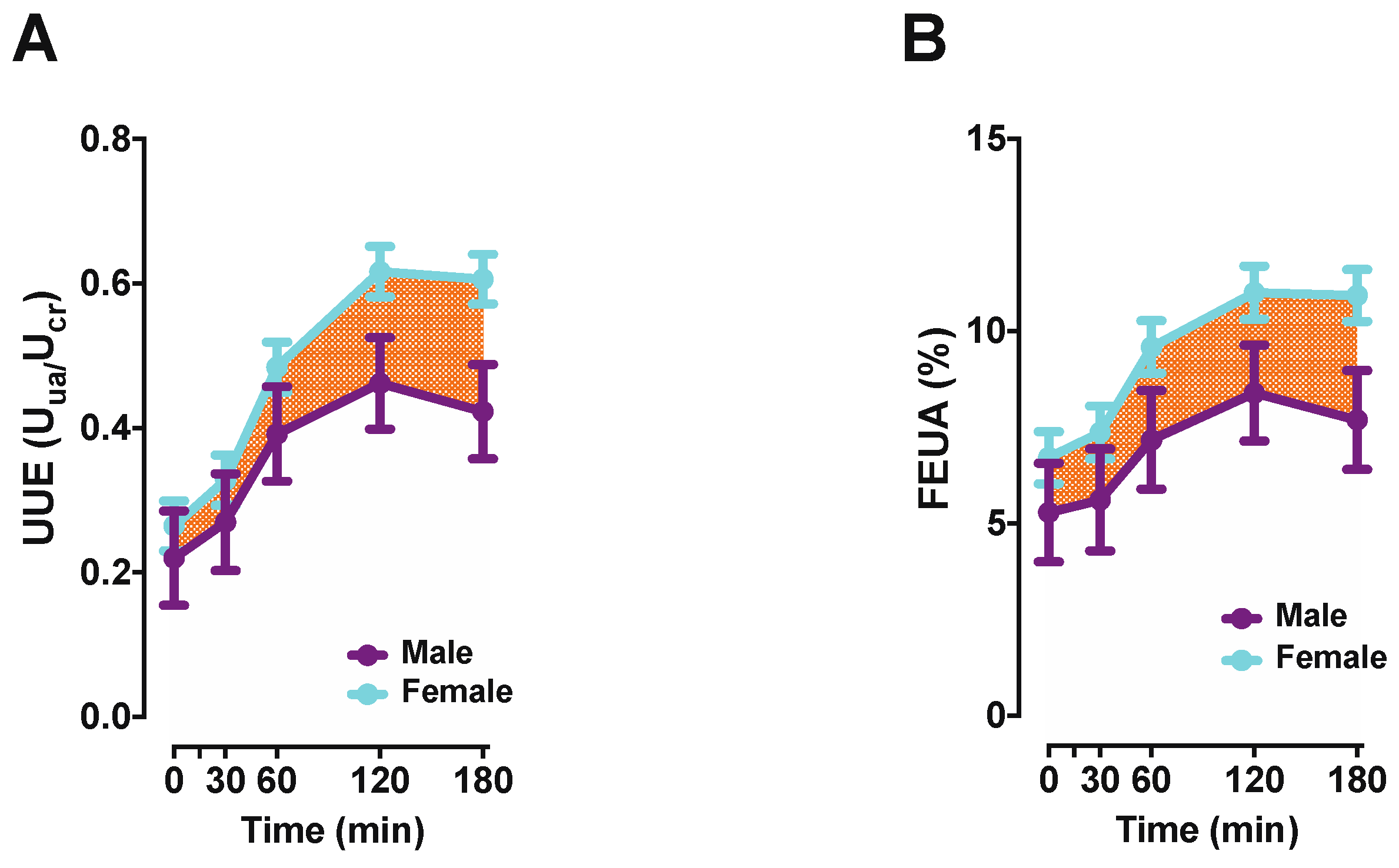{kind=link}
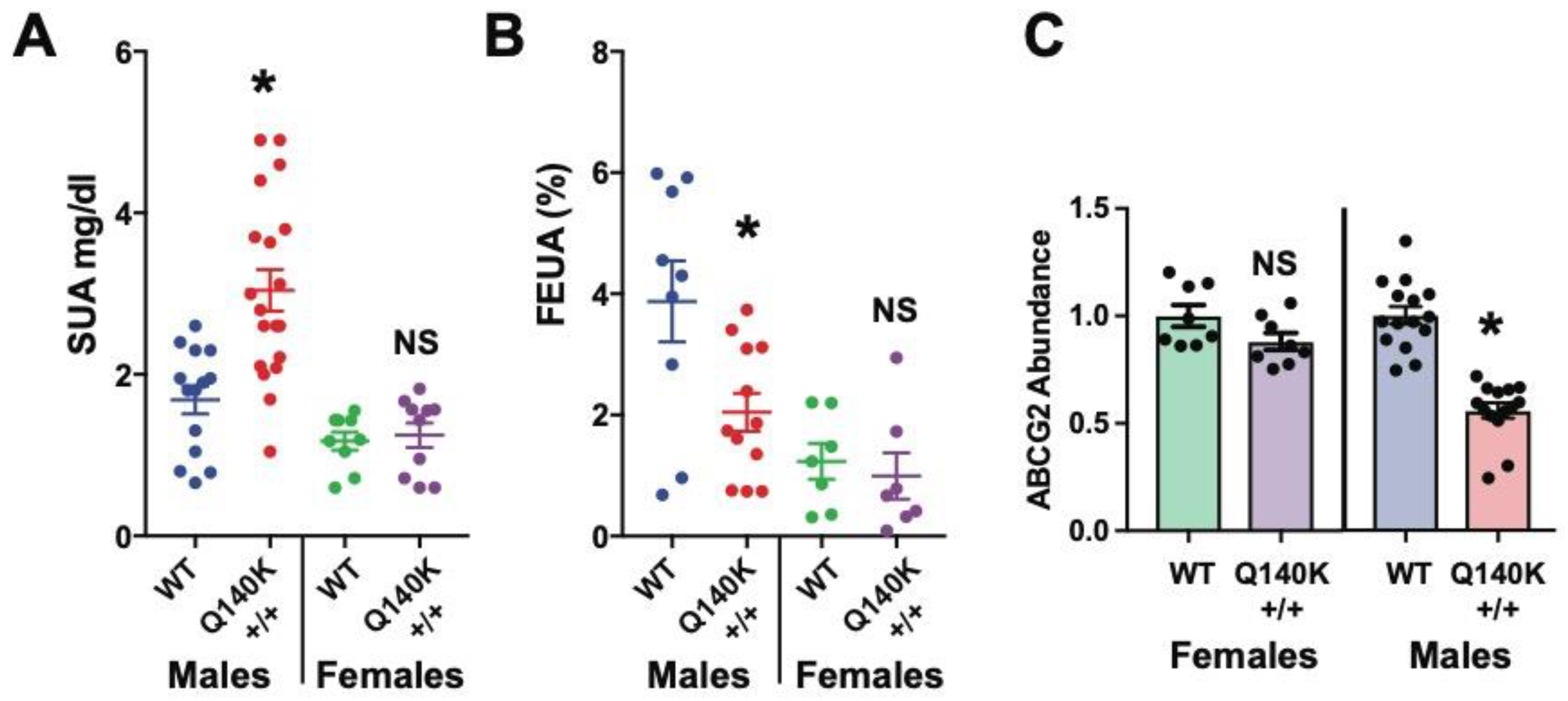{kind=link}
Table 1.
Sex differences in HUA associated phenotypes.
| Study [Ref] | Non-HUA: N | SUA (mg/dL) | Non-HUA: Pathology | HUA: N | SUA (mg/dL) | HUA: Pathology | Increase with HUA |
|---|---|---|---|---|---|---|---|
| Kidney Disease | |||||||
| ESRD: Chang HY, et al. 2010 [50] | Men: 2320 | <5 | 10% | 1825 | ≥7 | 34% | 3.4-fold |
| Women: 4248 | <4 | 9% | 1879 | ≥6 | 47% | 5.2-fold | |
| CKD: Kuwabara M, et al. 2017 [51] | Men: 1582 | ≤7.0 | 12.4% | 282 | >7.0 | 17.4% | 1.4-fold |
| Women: 2902 | <6.0 | 4.8% | 133 | ≥6.0 | 16.5% | 3.4-fold | |
| CKD: Redon P, et al. 2019 [52] | Men: 1238 | <6 | 2% | 381 | >7 | 8% | 4-fold |
| Women: 1142 | <5 | 1% | 445 | >6 | 6% | 6-fold | |
| Hypertension 1 | |||||||
| Yoshitomi R, et al. 2013 [53] (CKD patients) | Men: 29 | <6 | 66% | 29 | >8 | 100% | 1.5-fold |
| Women: 23 | <5 | 30% | 27 | >7 | 93% | 3.1-fold | |
| Kuwabara M, et al. 2017 [51] | Men: 1582 | ≤7.0 | 9.2% | 282 | >7.0 | 14.2% | 1.5-fold |
| Women: 2902 | <6.0 | 10.0% | 133 | ≥6.0 | 22.6% | 2.3-fold | |
| Lin, et al. 2020 [57] (BP > 140/90) | Men: 4150 | <7 | 8.3% | 2588 | >7 | 12.3% | 1.5-fold |
| Women: 676 | <5.7 | 1.3% | 90 | >5.7 | 5.6% | 4.3-fold | |
| Diabetes Mellitus (Type 2) | |||||||
| Kuwabara M, et al. 2017 [51] | Men: 1582 | ≤7.0 | 1.6% | 282 | >7.0 | 1.4% | No change |
| Women: 2902 | <6.0 | 0.5% | 133 | ≥6.0 | 2.3% | 4.6-fold | |
| Yamada T, et al. 2011 [54] | Men: 1561 | <5.2 | 7.2% | 1536 | ≥6.7 | 10.5% | 1.5-fold |
| Women: 1543 | <3.7 | 2.2% | 1238 | ≥4.8 | 7.1% | 3.2-fold | |
| Redon P, et al. 2019 [52] | Men: 1238 | <6 | 25% | 381 | >7 | 34% | 1.4-fold |
| Women: 1142 | <5 | 31% | 445 | >6 | 39% | 1.3-fold | |
1 Hypertension defined as >130/80 mmHg unless otherwise specified; ERSD: end stage renal disease; CKD: chronic kidney disease; SBP: systolic blood pressure; DBP: diastolic blood pressure; BP: blood pressure.
Table 2.
Reported sex differences in UA associated transporters and regulators involved in UA handling.
Table 2.
Reported sex differences in UA associated transporters and regulators involved in UA handling.
| Gene | Protein | Description/Role | SNP Associated Sex Differences | Ref 1 |
|---|---|---|---|---|
| ABCG2 | ABCG2 | UA secretion in the kidney, major UA transporter in the intestine and liver [76,110,111] | • SNP rs2231142 is reported to have gene–sex interactions in European adults, demonstrating a stronger interaction with a greater effect in men than in women [93,100], increasing SUA 0.270 mg/dL in men and only 0.181 mg/dL in women [94] • SNP rs2199936 similarly demonstrates a greater effect size in European men than in women [93] | [82,92,93,94,95,96,97,98,99,100,101,108,112] |
| SLC2A9 | GLUT9 | Glucose, fructose transporter, also involved in UA reabsorption [104,113] | • SNP rs734553 is most significantly associated with SUA in European men, while intron SNP rs12498742 is most significantly associated with SUA in European women [93] • SNP rs7442295 yielded a decrease in SUA of –0.503 mg/dL in women, while only –0.202 mg/dL in men, with a higher heritability component seen in European women [104] • SNP rs11722228 associates with UA levels significantly in Chinese men but not women [114] | [82,92,93,94,95,96,98,99,100,101,102,108,112] |
| SLC16A9 | MCT9 | pH-dependent, sodium-sensitive monocarboxylate transporter, with a reported affinity for creatine [115]. No evidence for direct UA transport. | • Missense variant rs2242206 (K258T) is associated with an increased risk of renal overload gout in Japanese men only (women not included in this study) [116] | [93,94,100,101] |
| SLC17A1 | NPT1 | UA secretion in the renal proximal tubule [77,78] | • Major alleles for rs1165196, rs1179086, and rs3757131 associated with higher UA levels in both sexes, where Japanese men also demonstrate high homocysteine and low folic acid levels, while women do not [117] | [82,93,94,96,97,99,101,108] |
| SLC22A7 | OAT2 | Organic anion/dicarboxylate exchanger located in the renal proximal tubule [118], potential role in UA secretion [119] | • No sex differences reported in humans | [99,101] |
| SLC22A11 | OAT4 | UA excretion [83] | • Common variant rs17300741 associates with renal underexcretion type gout in Japanese men only (women not included in this study) [83] | [93,94,100] |
| SLC22A12 | URAT1 | UA reabsorption [81] | • SNP rs893006 major allele associates with higher SUA levels in Japanese men, but not women [120] • SNP rs506338 shows marginally significant association with SUA in Chinese women but not men [114] | [92,93,94,97,100,101,108,112] |
| PDZK1 | PDZK1 | Scaffold protein, stabilizes UA transporters in the apical compartment [121] | • Variant rs1471633 is associated with increased gout risk in European men but not women [100] | [93,94,96,100,101] |
| HNF4A | HNF4α | Can directly regulate expression of UA transporters ABCG2 [101] and SLC2A9 [122] | • Increased activity in the pancreas [123] and liver [107] of women as compared to men | [99,101] |
| HNF4G | HNF4ᵧ | Associations with pro-inflammatory response [124] | • No sex differences reported | [82,94,96,99,100,101] |
1 Studies that demonstrate gene associations with SUA are referenced.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Halperin Kuhns, V.L.; Woodward, O.M. Sex Differences in Urate Handling. Int. J. Mol. Sci. 2020, 21, 4269. https://doi.org/10.3390/ijms21124269
AMA Style
Halperin Kuhns VL, Woodward OM. Sex Differences in Urate Handling. International Journal of Molecular Sciences. 2020; 21(12):4269. https://doi.org/10.3390/ijms21124269
Chicago/Turabian StyleHalperin Kuhns, Victoria L., and Owen M. Woodward. 2020. "Sex Differences in Urate Handling" International Journal of Molecular Sciences 21, no. 12: 4269. https://doi.org/10.3390/ijms21124269
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.