
Circulating Cell-Free Tumour DNA in the Management of Cancer

Abstract
:1. Introduction
2. Molecular Biomarkers in Tissue
3. Melanoma
4. Non-Small Cell Lung Cancer
5. Colorectal Carcinoma
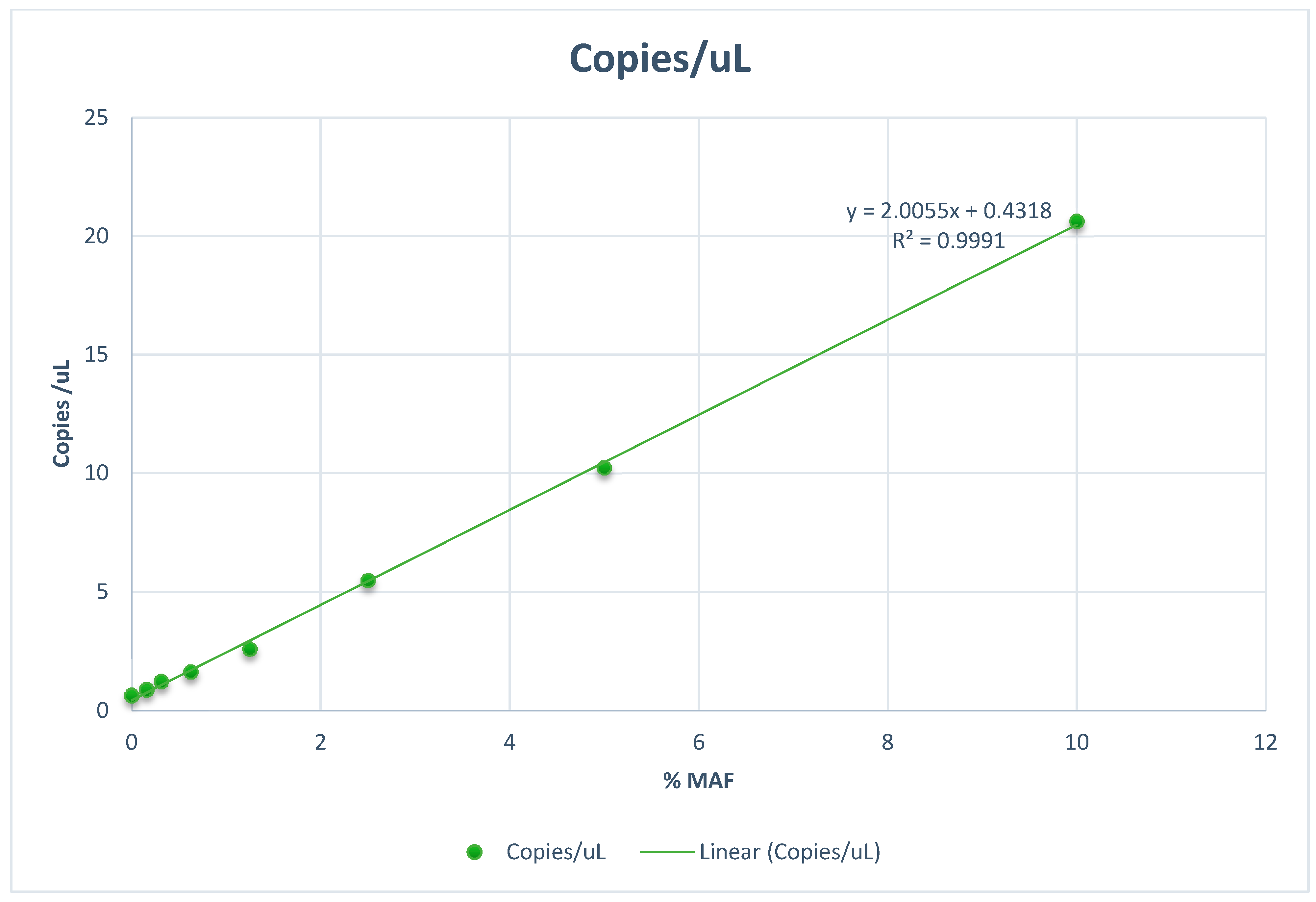
6. Technology


{kind=link}
| Technology | Sensitivity | Tumour Type | Gene/Mutations | Clinical Utility | References |
|---|---|---|---|---|---|
| Droplet Digital PCR (ddPCR) | ~0.001%–0.03% | NSCLC | EGFR T790M | Treatment Decision, Resistance | [40,41] |
| 0.04% | NSCLC | EGFR | Treatment Decision | [42] | |
| 0.01% | Breast | PIK3CA | Prognosis | [43] | |
| 0.1% | CRC | KRAS | Treatment Decision | [40] | |
| 0.005% | Melanoma | BRAF V600E | Tumour Burden | [41] | |
| COLD-ddPCR | 0.2%–1.2% | Multiple | EGFR, TP53 | Monitoring | [44] |
| Nanofluidic Digital PCR | 0.05% | NSCLC | EGFR T790M | Treatment Decision, Resistance | [45] |
| BEAMing Digital PCR | 0.01% | Breast | PIK3CA | Treatment Decision | [46] |
| castPCR | 0.1%–1% | Ovarian | Beta-globin | Prognosis | [47] |
| ARMS-PCR | ~1% | CRC | KRAS, BRAF | Prognosis | [48,49] |
| Breast | PIK3CA | Treatment Decision | [50] | ||
| Clamping PCR | 0.1%–1% | CRC | KRAS | Treatment Decision, Prognosis | [51,52] |
| NSCLC | EGFR | Diagnosis | [53] | ||
| Synchronous TES | 0.01% | NSCLC | EGFR T790M | Treatment Decision, Resistance | [54] |
| NGS | <5% | Ovarian | TP53 | Tumour Burden | [55] |
| 0.5% | Breast | Multiple | Treatment Decision | [56] |
6.1. Digital PCR
6.2. BEAMing
6.3. Synchronous Thermal-Electrophoretic Separation
7. Cell-Free DNA
8. Applications
8.1. Monitoring of Tumour Burden
8.2. Monitoring of Minimal Residual Disease
8.3. Monitoring of Tumour Heterogeneity
8.4. Monitoring of Molecular Resistance
8.5. Early Diagnosis of Tumours
9. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Allred, D.C.; Harvey, J.M.; Berardo, M.; Clark, G.M. Prognostic and predictive factors in breast cancer by immunohistochemical analysis. Mod. Pathol. 1998, 11, 155–168. [Google Scholar] [PubMed]
- Jones, H.B. On a new substance occurring in the urine of a patient with mollities ossium. Philos. Trans. R. Soc. 1848, 138, 55–62. [Google Scholar] [CrossRef]
- Marzese, D.M.; Hirose, H.; Hoon, D.S. Diagnostic and prognostic value of circulating tumor-related DNA in cancer patients. Exp. Rev. Mol. Diagn. 2013, 13, 827–844. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Menzies, A.M.; Nagrial, A.M.; Haydu, L.E.; Hamilton, A.L.; Mann, G.J.; Hughes, T.M.; Thompson, J.F.; Scolyer, R.A.; Kefford, R.F. Prognostic and clinicopathologic associations of oncogenic BRAF in metastatic melanoma. J. Clin. Oncol. 2011, 29, 1239–1246. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.T.; Puzanov, I.; Kim, K.B.; Ribas, A.; McArthur, G.A.; Sosman, J.A.; O’Dwyer, P.J.; Lee, R.J.; Grippo, J.F.; Nolop, K.; et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 2010, 363, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Kwong, L.N.; Davies, M.A. Targeted therapy for melanoma: Rational combinatorial approaches. Oncogene 2014, 33, 1–9. [Google Scholar] [CrossRef] [PubMed]
- AIHW. Cancer in Australia: An. Overview; AIHW, 2012. Available online: http://www.aihw.gov.au/publication-detail/?id=60129542359 (accessed on 19 June 2015).
- AIHW. Lung Cancer in Australia An. Overview; AIHW, 2011. Available online: http://www.aihw.gov.au/publication-detail/?id=10737420419 (accessed on 19 June 2015).
- Luo, S.Y.; Lam, D.C.L. Oncogenic driver mutations in lung cancer. Transl. Respir. Med. 2013, 1, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Kung, H.J.; Mack, P.C.; Gandara, D.R. Genotyping and genomic profiling of non-small-cell lung cancer: Implications for current and future therapies. J. Clin. Oncol. 2013, 31, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- NCCN. Non-Small Cell. Lung Cancer; NCCN, 2014; p. NSCL-16. Available online: http://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf (accessed on 17 June 2015).
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar]
- Roengvoraphoj, M.; Tsongalis, G.J.; Dragnev, K.H.; Rigas, J.R. Epidermal growth factor receptor tyrosine kinase inhibitors as initial therapy for non-small cell lung cancer: Focus on epidermal growth factor receptor mutation testing and mutation-positive patients. Cancer Treat. Rev. 2013, 39, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Dacic, S. Molecular genetic testing for lung adenocarcinomas: A practical approach to clinically relevant mutations and translocations. J. Clin. Pathol. 2013, 66, 870–874. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Popat, S. Toward precision medicine with next-generation EGFR inhibitors in non-small-cell lung cancer. Pharmgenomics. Pers. Med. 2014, 7, 285–295. [Google Scholar] [PubMed]
- Lai, Y.; Zhang, Z.; Li, J.; Sun, D.; Zhou, Y.; Jiang, T.; Han, Y.; Huang, L.; Zhu, Y.; Li, X.; et al. EGFR mutations in surgically resected fresh specimens from 697 consecutive Chinese patients with non-small cell lung cancer and their relationships with clinical features. Int. J. Mol. Sci. 2013, 14, 24549–24559. [Google Scholar] [CrossRef] [PubMed]
- Tibaldi, C.; Giovannetti, E.; Vasile, E.; Boldrini, L.; Gallegos-Ruiz, M.I.; Bernardini, I.; Incensati, R.; Danesi, R.; Cappuzzo, F.; Peters, G.J.; et al. Inherited germline T790M mutation and somatic epidermal growth factor receptor mutations in non-small cell lung cancer patients. J. Thorac. Oncol. 2011, 6, 395–396. [Google Scholar] [CrossRef] [PubMed]
- Prudkin, L.; Tang, X.; Wistuba, I.I. Germ-line and somatic presentations of the EGFR T790M mutation in lung cancer. J. Thorac. Oncol. 2009, 4, 139–141. [Google Scholar] [CrossRef] [PubMed]
- Prudkin, L.; Wistuba, I.I. Epidermal growth factor receptor abnormalities in lung cancer. Pathogenetic and clinical implications. Ann. Diagn. Pathol. 2006, 10, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.Y.; Yu, C.J.; Chang, Y.C.; Yang, C.H.; Shih, J.Y.; Yang, P.C. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non-small cell lung cancer. Clin. Cancer Res. 2011, 17, 3812–3821. [Google Scholar] [CrossRef] [PubMed]
- Cooper, W.A.; Lam, D.C.; O’Toole, S.A.; Minna, J.D. Molecular biology of lung cancer. J. Thorac. Dis. 2013, 5, S479–S490. [Google Scholar] [PubMed]
- Wang, H.L.; Lopategui, J.; Amin, M.B.; Patterson, S.D. KRAS mutation testing in human cancers: The pathologistʼs role in the era of personalized medicine. Adv. Anat. Pathol. 2010, 17, 23–32. [Google Scholar] [PubMed]
- Allegra, C.J.; Jessup, J.M.; Somerfield, M.R.; Hamilton, S.R.; Hammond, E.H.; Hayes, D.F.; McAllister, P.K.; Morton, R.F.; Schilsky, R.L. American society of clinical oncology provisional clinical opinion: Testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J. Clin. Oncol. 2009, 27, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Van Cutsem, E.; Nordlinger, B.; Cervantes, A. ESMO Guidelines Working Group. Advanced colorectal cancer: ESMO Clinical Practice Guidelines for treatment. Ann. Oncol. 2010, 21, v93–v97. [Google Scholar] [CrossRef] [PubMed]
- Aubin, F.; Gill, S.; Burkes, R.; Colwell, B.; Kamel-Reid, S.; Koski, S.; Pollett, A.; Samson, B.; Tehfe, M.; Wong, R.; et al. Canadian Expert Group consensus recommendations: KRAS testing in colorectal cancer. Curr. Oncol. 2011, 18, 180–184. [Google Scholar] [CrossRef]
- Sharma, S.G.; Gulley, M.L. BRAF mutation testing in colorectal cancer. Arch. Pathol. Lab. Med. 2010, 134, 1225–1228. [Google Scholar] [PubMed]
- Kothari, N.; Schell, M.J.; Teer, J.K.; Yeatman, T.; Shibata, D.; Kim, R. Comparison of KRAS mutation analysis of colorectal cancer samples by standard testing and next-generation sequencing. J. Clin. Pathol. 2014, 67, 764–767. [Google Scholar] [CrossRef] [PubMed]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta. Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Wong, N.A.; Gonzalez, D.; Salto-Tellez, M.; Butler, R.; Diaz-Cano, S.J.; Ilyas, M.; Newman, W.; Shaw, E.; Taniere, P.; Walsh, S.V. RAS testing of colorectal carcinoma-a guidance document from the Association of Clinical Pathologists Molecular Pathology and Diagnostics Group. J. Clin. Pathol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, V.; von Weikersthal, L.F.; Decker, T.; Kiani, A.; Vehling-Kaiser, U.; Al-Batran, S.E.; Heintges, T.; Lerchenmuller, C.; Kahl, C.; Seipelt, G.; et al. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): A randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1065–1075. [Google Scholar] [CrossRef]
- Sclafani, F.; Gonzalez, D.; Cunningham, D.; Hulkki Wilson, S.; Peckitt, C.; Tabernero, J.; Glimelius, B.; Cervantes, A.; Dewdney, A.; Wotherspoon, A.; et al. TP53 mutational status and cetuximab benefit in rectal cancer: 5-year results of the EXPERT-C trial. J. Natl. Cancer Inst. 2014, 106, dju121. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, M.; Sedmak, D.; Jewell, S. Effect of fixatives and tissue processing on the content and integrity of nucleic acids. Am. J. Pathol. 2002, 161, 1961–1971. [Google Scholar] [CrossRef]
- Do, H.; Wong, S.Q.; Li, J.; Dobrovic, A. Reducing sequence artifacts in amplicon-based massively parallel sequencing of formalin-fixed paraffin-embedded DNA by enzymatic depletion of uracil-containing templates. Clin. Chem. 2013, 59, 1376–1383. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.S.; Start, R.D.; Smith, J.H. Does delay in fixation affect the number of mitotic figures in processed tissue? J. Clin. Pathol. 1990, 43, 597–599. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.S.; Start, R.D. Estimating mitotic activity in tumours. Histopathology 1996, 29, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Kapp, J.R.; Diss, T.; Spicer, J.; Gandy, M.; Schrijver, I.; Jennings, L.J.; Li, M.M.; Tsongalis, G.J.; Gonzalez de Castro, D.; Bridge, J.A.; et al. Variation in pre-PCR processing of FFPE samples leads to discrepancies in BRAF and EGFR mutation detection: A diagnostic RING trial. J. Clin. Pathol. 2015, 68, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Dressman, D.; Yan, H.; Traverso, G.; Kinzler, K.W.; Vogelstein, B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc. Natl. Acad. Sci. USA 2003, 100, 8817–8822. [Google Scholar] [CrossRef] [PubMed]
- Marziali, A.; Pel, J.; Bizzotto, D.; Whitehead, L.A. Novel electrophoresis mechanism based on synchronous alternating drag perturbation. Electrophoresis 2005, 26, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Taly, V.; Pekin, D.; Benhaim, L.; Kotsopoulos, S.K.; Le Corre, D.; Li, X.; Atochin, I.; Link, D.R.; Griffiths, A.D.; Pallier, K.; et al. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 2013, 59, 1722–1731. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Fernandez-Landazuri, S.; Rodriguez, C.; Zarate, R.; Lozano, M.D.; Zubiri, L.; Perez-Gracia, J.L.; Martin-Algarra, S.; Gonzalez, A. Quantitative cell-free circulating BRAFV600E mutation analysis by use of droplet digital PCR in the follow-up of patients with melanoma being treated with BRAF inhibitors. Clin. Chem. 2015, 61, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Ye, X.; Dong, Z.; Lu, Y.C.; Sun, Y.; Liu, Y.; McCormack, R.; Gu, Y.; Liu, X. Highly sensitive droplet digital PCR method for detection of EGFR activating mutations in plasma cell-free DNA from patients with advanced non-small cell lung cancer. J. Mol. Diagn. 2015, 17, 265–272. [Google Scholar] [CrossRef] [PubMed]
- Oshiro, C.; Kagara, N.; Naoi, Y.; Shimoda, M.; Shimomura, A.; Maruyama, N.; Shimazu, K.; Kim, S.J.; Noguchi, S. PIK3CA mutations in serum DNA are predictive of recurrence in primary breast cancer patients. Breast Cancer Res. Treat. 2015, 150, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rizaldos, E.; Paweletz, C.; Song, C.; Oxnard, G.R.; Mamon, H.; Janne, P.A.; Makrigiorgos, G.M. Enhanced ratio of signals enables digital mutation scanning for rare allele detection. J. Mol. Diagn. 2015, 17, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Isobe, K.; Hata, Y.; Tochigi, N.; Kaburaki, K.; Kobayashi, H.; Makino, T.; Otsuka, H.; Ishida, F.; Hirota, N.; Sano, G.; et al. Usefulness of nanofluidic digital PCR arrays to quantify T790M mutation in EGFR-mutant lung adenocarcinoma. Cancer Genomics Proteomics 2015, 12, 31–37. [Google Scholar] [PubMed]
- Higgins, M.J.; Jelovac, D.; Barnathan, E.; Blair, B.; Slater, S.; Powers, P.; Zorzi, J.; Jeter, S.C.; Oliver, G.R.; Fetting, J.; et al. Detection of tumor PIK3CA status in metastatic breast cancer using peripheral blood. Clin. Cancer Res. 2012, 18, 3462–3469. [Google Scholar] [CrossRef] [PubMed]
- Kamat, A.A.; Baldwin, M.; Urbauer, D.; Dang, D.; Han, L.Y.; Godwin, A.; Karlan, B.Y.; Simpson, J.L.; Gershenson, D.M.; Coleman, R.L.; et al. Plasma cell-free DNA in ovarian cancer: An independent prognostic biomarker. Cancer 2010, 116, 1918–1925. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Hu, T.; Chen, Y.; Zhang, X.; Gu, X.; Guan, M. Development and validation of a tetra-primer amplification refractory mutation system-polymerase chain reaction combined with melting analysis-assay for clinical JAK2 V617F mutation detection. Mol. Diagn. Ther. 2014, 18, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.L.; Pallisgaard, N.; Vogelius, I.; Jakobsen, A. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin. Cancer Res. 2012, 18, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Board, R.E.; Wardley, A.M.; Dixon, J.M.; Armstrong, A.C.; Howell, S.; Renshaw, L.; Donald, E.; Greystoke, A.; Ranson, M.; Hughes, A.; et al. Detection of PIK3CA mutations in circulating free DNA in patients with breast cancer. Breast Cancer Res. Treat. 2010, 120, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.M.; Liu, X.J.; Ge, F.J.; Lin, L.; Wang, Y.; Sharma, M.R.; Liu, Z.Y.; Tommasi, S.; Paradiso, A. KRAS mutations in tumor tissue and plasma by different assays predict survival of patients with metastatic colorectal cancer. J. Exp. Clin. Cancer Res. 2014, 33, 104. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Wu, J.; Xu, S.; Tan, G.; Liu, B.; Feng, J. Modified PNA-PCR method: A convenient and accurate method to screen plasma KRAS mutations of cancer patients. Cancer Biol. Ther. 2012, 13, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.R.; Lee, S.Y.; Hyun, D.S.; Lee, M.K.; Lee, H.K.; Choi, C.M.; Yang, S.H.; Kim, Y.C.; Lee, Y.C.; Kim, S.Y.; et al. Detection of EGFR mutations in circulating free DNA by PNA-mediated PCR clamping. J. Exp. Clin. Cancer Res. 2013, 32, 50. [Google Scholar] [CrossRef] [PubMed]
- Genomics, B. Performance. 2015. Available online: http://www.borealgenomics.com/technology/performance/ (accessed on 9 June 2015).
- Forshew, T.; Murtaza, M.; Parkinson, C.; Gale, D.; Tsui, D.W.; Kaper, F.; Dawson, S.J.; Piskorz, A.M.; Jimenez-Linan, M.; Bentley, D.; et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl. Med. 2012, 4, 136ra68. [Google Scholar] [CrossRef] [PubMed]
- Rothe, F.; Laes, J.F.; Lambrechts, D.; Smeets, D.; Vincent, D.; Maetens, M.; Fumagalli, D.; Michiels, S.; Drisis, S.; Moerman, C.; et al. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann. Oncol. 2014, 25, 1959–1965. [Google Scholar] [CrossRef] [PubMed]
- Huggett, J.F.; Whale, A. Digital PCR as a novel technology and its potential implications for molecular diagnostics. Clin. Chem. 2013, 59, 1691–1693. [Google Scholar] [CrossRef] [PubMed]
- Diehl, F.; Li, M.; He, Y.; Kinzler, K.W.; Vogelstein, B.; Dressman, D. BEAMing: Single-molecule PCR on microparticles in water-in-oil emulsions. Nat. Methods 2006, 3, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, G.D.; Pribish, D.M.; Valone, F.H.; Memoli, V.A.; Bzik, D.J.; Yao, S.L. Soluble normal and mutated DNA sequences from single-copy genes in human blood. Cancer Epidemiol. Biomark. Prev. 1994, 3, 67–71. [Google Scholar]
- Vasioukhin, V.; Anker, P.; Maurice, P.; Lyautey, J.; Lederrey, C.; Stroun, M. Point mutations of the N-ras gene in the blood plasma DNA of patients with myelodysplastic syndrome or acute myelogenous leukaemia. Br. J. Haematol. 1994, 86, 774–779. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Tsui, D.W.; Murtaza, M.; Biggs, H.; Rueda, O.M.; Chin, S.F.; Dunning, M.J.; Gale, D.; Forshew, T.; Mahler-Araujo, B.; et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 368, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Dowler Nygaard, A.; Spindler, K.L.; Pallisgaard, N.; Andersen, R.F.; Jakobsen, A. Levels of cell-free DNA and plasma KRAS during treatment of advanced NSCLC. Oncol. Rep. 2014, 31, 969–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kukita, Y.; Uchida, J.; Oba, S.; Nishino, K.; Kumagai, T.; Taniguchi, K.; Okuyama, T.; Imamura, F.; Kato, K. Quantitative identification of mutant alleles derived from lung cancer in plasma cell-free DNA via anomaly detection using deep sequencing data. PLoS ONE 2013, 8, e81468. [Google Scholar] [CrossRef] [PubMed]
- Mouliere, F.; El Messaoudi, S.; Gongora, C.; Guedj, A.S.; Robert, B.; del Rio, M.; Molina, F.; Lamy, P.J.; Lopez-Crapez, E.; Mathonnet, M.; et al. Circulating cell-free DNA from colorectal cancer patients may reveal high KRAS or BRAF mutation load. Transl. Oncol. 2013, 6, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Yap, T.A.; Pope, L.; Cassidy, A.M.; Dukes, J.P.; Riisnaes, R.; Massard, C.; Cassier, P.A.; Miranda, S.; Clark, J.; et al. Multi-purpose utility of circulating plasma DNA testing in patients with advanced cancers. PLoS ONE 2012, 7, e47020. [Google Scholar] [CrossRef] [PubMed]
- Spindler, K.G.; Appelt, A.L.; Pallisgaard, N.; Andersen, R.F.; Jakobsen, A. KRAS-mutated plasma DNA as predictor of outcome from irinotecan monotherapy in metastatic colorectal cancer. Br. J. Cancer 2013, 109, 3067–3072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spindler, K.L.; Sorensen, M.M.; Pallisgaard, N.; Andersen, R.F.; Havelund, B.M.; Ploen, J.; Lassen, U.; Jakobsen, A.K. Phase II trial of temsirolimus alone and in combination with irinotecan for KRAS mutant metastatic colorectal cancer: Outcome and results of KRAS mutational analysis in plasma. Acta Oncol. 2013, 52, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Fleischhacker, M.; Schmidt, B. Circulating nucleic acids (CNAs) and cancer—A survey. Biochim. Biophys. Acta 2007, 1775, 181–232. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and monitoring of cell-free DNA in blood of patients with colorectal cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Diaz, L.A., Jr.; Williams, R.T.; Wu, J.; Kinde, I.; Hecht, J.R.; Berlin, J.; Allen, B.; Bozic, I.; Reiter, J.G.; Nowak, M.A.; et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012, 486, 537–540. [Google Scholar] [PubMed]
- Kauhanen, S.P.; Komar, G.; Seppanen, M.P.; Dean, K.I.; Minn, H.R.; Kajander, S.A.; Rinta-Kiikka, I.; Alanen, K.; Borra, R.J.; Puolakkainen, P.A.; et al. A prospective diagnostic accuracy study of 18F-fluorodeoxyglucose positron emission tomography/computed tomography, multidetector row computed tomography, and magnetic resonance imaging in primary diagnosis and staging of pancreatic cancer. Ann. Surg. 2009, 250, 957–963. [Google Scholar] [CrossRef] [PubMed]
- Casali, M.; Froio, A.; Carbonelli, C.; Versari, A. PET/CT imaging in oncology: Exceptions that prove the rule. Case Rep. Oncol. Med. 2013, 2013, 865032. [Google Scholar] [CrossRef] [PubMed]
- Dondorp, W.; de Wert, G.; Bombard, Y.; Bianchi, D.W.; Bergmann, C.; Borry, P.; Chitty, L.S.; Fellmann, F.; Forzano, F.; Hall, A.; et al. Non-invasive prenatal testing for aneuploidy and beyond: Challenges of responsible innovation in prenatal screening. Eur. J. Hum. Genet. 2015. [Google Scholar] [CrossRef]
- Zhang, H.; Gao, Y.; Jiang, F.; Fu, M.; Yuan, Y.; Guo, Y.; Zhu, Z.; Lin, M.; Liu, Q.; Tian, Z.; et al. Non-invasive prenatal testing for trisomies 21, 18 and 13: Clinical experience from 146 958 pregnancies. Ultrasound Obstet. Gynecol. 2015, 45, 530–538. [Google Scholar] [CrossRef] [PubMed]
- Osborne, C.M.; Hardisty, E.; Devers, P.; Kaiser-Rogers, K.; Hayden, M.A.; Goodnight, W.; Vora, N.L. Discordant noninvasive prenatal testing results in a patient subsequently diagnosed with metastatic disease. Prenat. Diagn. 2013, 33, 609–611. [Google Scholar] [CrossRef] [PubMed]
- Hohaus, S. Genomic imbalances in circulating DNA in Hodgkinʼs lymphoma. Lancet Haematol. 2015, 2, e48–e49. [Google Scholar] [CrossRef]
- Vandenberghe, P.; Wlodarska, I.; Tousseyn, T.; Dehaspe, L.; Dierickx, D.; Verheecke, M.; Uyttebroeck, A.; Bechter, O.; Delforge, M.; Vandecaveye, V.; et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkinʼs lymphoma by sequencing of circulating cell-free DNA: A technical proof-of-principle study. Lancet Haematol. 2015, 2, e55–e65. [Google Scholar] [CrossRef]
- Knight, M. DNA testing fetus leads moms to their own cancer diagnoses. Genetic Literacy Project 2015. Available online: http://www.geneticliteracyproject.org/2015/03/19/dna-testing-fetus-leads-moms-to-their-own-cancer-diagnoses/ (accessed on 9 June 2015).
- Diaz, L.A., Jr.; Bardelli, A. Liquid biopsies: genotyping circulating tumor DNA. J. Clin. Oncol. 2014, 32, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Lebofsky, R.; Decraene, C.; Bernard, V.; Kamal, M.; Blin, A.; Leroy, Q.; Rio Frio, T.; Pierron, G.; Callens, C.; Bieche, I.; et al. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol. Oncol. 2015, 9, 783–790. [Google Scholar] [CrossRef] [PubMed]
- McLarty, J.L.; Yeh, C. Circulating cell-free DNA: The blood biopsy in cancer management. MOJ Cell. Sci. Rep. 2015, 2, 0021. [Google Scholar]
- Diehl, F.; Schmidt, K.; Choti, M.A.; Romans, K.; Goodman, S.; Li, M.; Thornton, K.; Agrawal, N.; Sokoll, L.; Szabo, S.A.; et al. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 2008, 14, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Dawson, S.J.; Rosenfeld, N.; Caldas, C. Circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 2013, 369, 93–94. [Google Scholar] [CrossRef] [PubMed]
- Reinert, T.; Scholer, L.V.; Thomsen, R.; Tobiasen, H.; Vang, S.; Nordentoft, I.; Lamy, P.; Kannerup, A.S.; Mortensen, F.V.; Stribolt, K.; et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut 2015. [Google Scholar] [CrossRef]
- Lipson, E.J.; Velculescu, V.E.; Pritchard, T.S.; Sausen, M.; Pardoll, D.M.; Topalian, S.L.; Diaz, L.A., Jr. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J. Immunother. Cancer 2014, 2, 42. [Google Scholar] [CrossRef] [PubMed]
- Tseng, J.Y.; Yang, C.Y.; Liang, S.C.; Liu, R.S.; Jiang, J.K.; Lin, C.H. Dynamic changes in numbers and properties of circulating tumor cells and their potential applications. Cancers 2014, 6, 2369–2386. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Garcia-Murillas, I.; Schiavon, G.; Hrebien, S.; Osin, P.; Nerurkar, A.; Kozarewa, I.; Garrido, J.A.; Dowsett, M.; Smith, I.E. Tracking tumor-specific mutations in circulating-free DNA to predict early relapse after treatment of primary breast cancer. J. Clin. Oncol. 2014, 32. abstr 511. [Google Scholar]
- Shinozaki, M.; O’Day, S.J.; Kitago, M.; Amersi, F.; Kuo, C.; Kim, J.; Wang, H.J.; Hoon, D.S. Utility of circulating B-RAF DNA mutation in serum for monitoring melanoma patients receiving biochemotherapy. Clin. Cancer Res. 2007, 13, 2068–2074. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.J.; Tsui, D.W.; Gale, D.; Forshew, T.; Piskorz, A.M.; Parkinson, C.; Chin, S.F.; Kingsbury, Z.; Wong, A.S.; et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature 2013, 497, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Bidard, F.C.; Madic, J.; Mariani, P.; Piperno-Neumann, S.; Rampanou, A.; Servois, V.; Cassoux, N.; Desjardins, L.; Milder, M.; Vaucher, I.; et al. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int. J. Cancer 2014, 134, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Hamakawa, T.; Kukita, Y.; Kurokawa, Y.; Miyazaki, Y.; Takahashi, T.; Yamasaki, M.; Miyata, H.; Nakajima, K.; Taniguchi, K.; Takiguchi, S.; et al. Monitoring gastric cancer progression with circulating tumour DNA. Br. J. Cancer 2015, 112, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Lianos, G.D.; Mangano, A.; Cho, W.C.; Dionigi, G.; Roukos, D.H. Circulating tumor DNA: New horizons for improving cancer treatment. Future Oncol. 2015, 11, 545–548. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Waters, J.; Leung, M.L.; Unruh, A.; Roh, W.; Shi, X.; Chen, K.; Scheet, P.; Vattathil, S.; Liang, H.; et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature 2014, 512, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Treacy, A.D.; Karamchandani, J.R.; Streutker, C.J.; Grin, A. HER2 genetic heterogeneity in gastric cancer: Evaluation according to the college of American pathologists breast cancer criteria. Appl. Immunohistochem. Mol. Morphol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.H.; Sledge, G.W. Heterogeneity and cancer. Oncology 2014, 28, 772–778. [Google Scholar] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Steinert, G.; Scholch, S.; Niemietz, T.; Iwata, N.; Garcia, S.A.; Behrens, B.; Voigt, A.; Kloor, M.; Benner, A.; Bork, U.; et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 2014, 74, 1694–1704. [Google Scholar] [CrossRef] [PubMed]
- De Mattos-Arruda, L.; Weigelt, B.; Cortes, J.; Won, H.H.; Ng, C.K.; Nuciforo, P.; Bidard, F.C.; Aura, C.; Saura, C.; Peg, V.; et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: A proof-of-principle. Ann. Oncol. 2014, 25, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Reiter, J.G.; Allen, B.; Antal, T.; Chatterjee, K.; Shah, P.; Moon, Y.S.; Yaqubie, A.; Kelly, N.; Le, D.T.; et al. Evolutionary dynamics of cancer in response to targeted combination therapy. Elife 2013, 2, e00747. [Google Scholar] [CrossRef] [PubMed]
- Arnedos, M.; Soria, J.C.; Andre, F.; Tursz, T. Personalized treatments of cancer patients: A reality in daily practice, a costly dream or a shared vision of the future from the oncology community? Cancer Treat. Rev. 2014, 40, 1192–1198. [Google Scholar] [CrossRef] [PubMed]
- Arnedos, M.; Vielh, P.; Soria, J.C.; Andre, F. The genetic complexity of common cancers and the promise of personalized medicine: Is there any hope? J. Pathol. 2014, 232, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Blair, B.G.; Bardelli, A.; Park, B.H. Somatic alterations as the basis for resistance to targeted therapies. J. Pathol. 2014, 232, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.A.; Jelovac, D.; Balukrishna, S.; Cochran, R.L.; Croessmann, S.; Zabransky, D.J.; Wong, H.Y.; Valda Toro, P.; Cidado, J.; Blair, B.G.; et al. Detection of cancer DNA in plasma of patients with early-stage breast cancer. Clin. Cancer Res. 2014, 20, 2643–2650. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, H.; Nouso, K.; Miyahara, K.; Morimoto, Y.; Dohi, C.; Tsutsumi, K.; Kato, H.; Matsubara, T.; Okada, H.; Yamamoto, K. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 2015. [Google Scholar] [CrossRef]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Francis, G.; Stein, S. Circulating Cell-Free Tumour DNA in the Management of Cancer. Int. J. Mol. Sci. 2015, 16, 14122-14142. https://doi.org/10.3390/ijms160614122
Francis G, Stein S. Circulating Cell-Free Tumour DNA in the Management of Cancer. International Journal of Molecular Sciences. 2015; 16(6):14122-14142. https://doi.org/10.3390/ijms160614122
Chicago/Turabian StyleFrancis, Glenn, and Sandra Stein. 2015. "Circulating Cell-Free Tumour DNA in the Management of Cancer" International Journal of Molecular Sciences 16, no. 6: 14122-14142. https://doi.org/10.3390/ijms160614122