Immune Escape after Hematopoietic Stem Cell Transplantation (HSCT): From Mechanisms to Novel Therapies


1
Department of Molecular Medicine, University of Pavia, 27100 Pavia, Italy
2
Hematology Department, Fondazione IRCCS Policlinico San Matteo, 27100 Pavia, Italy
*
Author to whom correspondence should be addressed.
Cancers 2020, 12(1), 69; https://doi.org/10.3390/cancers12010069
Submission received: 3 November 2019
/
Revised: 21 December 2019
/
Accepted: 24 December 2019
/
Published: 25 December 2019
(This article belongs to the Special Issue Acute Myeloid Leukemia)
Abstract
:Acute myeloid leukemia (AML) is the most common type of acute leukemia in adults. Recent advances in understanding its molecular basis have opened the way to new therapeutic strategies, including targeted therapies. However, despite an improvement in prognosis it has been documented in recent years (especially in younger patients) that allogenic hematopoietic stem cell transplantation (allo-HSCT) remains the only curative treatment in AML and the first therapeutic option for high-risk patients. After allo-HSCT, relapse is still a major complication, and is observed in about 50% of patients. Current evidence suggests that relapse is not due to clonal evolution, but instead to the ability of the AML cell population to escape immune control by a variety of mechanisms including the altered expression of HLA-molecules, production of anti-inflammatory cytokines, relevant metabolic changes and expression of immune checkpoint (ICP) inhibitors capable of “switching-off” the immune response against leukemic cells. Here, we review the main mechanisms of immune escape and identify potential strategies to overcome these mechanisms.
1. Introduction
In acute myeloid leukemia (AML), the highest priority now is the development of more effective treatments that focus on improving patients’ five-year survival rates, which nowadays are still 30% on average. Poor AML clinical outcomes are predominantly due to disease relapse and are frequently observed in patients treated with chemotherapy alone, despite the possibility of precisely defining the underlying genetic defect and the availability of novel drugs targeting peculiar molecular defects [1]. Thus, for most AML patients allo-HSCT remains the most effective curative option with the best chance of achieving durable complete remission (CR) [2]. However, even with this procedure death due to relapse occurs in about 30–40% of patients depending on the disease type and status at the time of transplant [3,4,5]. Most disease recurrences develop within the first six months after allo-HSCT when patients are still on immune-suppressive treatment. Quick tapering in some clinical conditions may be sufficient to re-induce remission through the so-called graft-versus-leukemia (GvL) effect [6]. This well-known observation is at the basis of the concept that allo-HSCT is a form of immunotherapy in which the intimate cross-talk between host antigen-presenting cells (APC) and donor T cells may truly eradicate the leukemic cell population [7]. Currently, the relevance of this tight interaction has been further substantiated by the demonstration that post-transplant AML relapse is not caused by the development of additional genetic alterations as in patients relapsing after chemotherapy [8], but by the loss or down-regulation of major histocompatibility (MHC) class II genes [9,10]. This seminal finding has paved the way for identifying and revealing other novel potential mechanisms of immune escape that are operative not only in the transplant setting [11,12,13,14], but also in the chemotherapy setting [15,16]. Notably, clarifying studies on these immune escape mechanisms have allowed the development of targeted immunotherapies, which are already providing promising results [11,17].
The present review will focus on immune escape mechanisms and potential strategies for their inhibition.
2. Mechanisms of Post-Transplant Relapse
2.1. Impaired Leukemia Cell Recognition: Loss of Mismatched HLA and Downregulation of HLA Molecules
Haploidentical hematopoietic stem cell transplantation (haplo-HSCT) is considered an effective therapeutic option for patients who need an allo-HSCT, but do not have a fully matched donor. Since its introduction, haplo-HSCT has gained the attention of both hematologists and immunologists because of the complex mechanisms underlying graft-versus-host-disease (GvHD) and the GvL effect. These two post-transplant events are considered “two faces of the same coin” and, to date, there are no well-established methods to switch off the first while retaining the latter. Both of these events rely on T cell alloreactivity, defined as the ability of donor T cells to recognize and target non-self antigens. T cell alloreactivity may be directed against either intact mismatched HLA molecules expressed on recipient cells (i.e., direct T cell alloreactivity) [18] or against peptides derived from proteasomal cleavage of mismatched HLA and presented in the antigen-binding groove of matched HLA molecules (i.e., indirect T cell alloreactivity) [19]. The concept that our immune system discriminates between self and non-self non only by the presence of foreign antigens, but also by the absence of normal self markers has been introduced by Kärre in an elegant work published more than 30 years ago [20]. Recognition of mismatched HLA molecules presented by leukemic stem cells (LSC) is a central function of the GvL immunological mechanism, especially in haplo-HSCT, where considerable genomic differences between the donor and recipient should warrant long-term relapse free survival (RFS) [21]. Human natural killer (NK) cells express multiple receptors that interact with HLA class I molecules. The NK cells’ ability to recognize the absence of self MHC class I is used to discriminate normal cells from cells in distress, like tumor cells. This “missing self” recognition in ensured by inhibitory receptors, which dampen NK cell activation upon interaction with their MHC class I ligands. Consistently, it has been shown that loss of mismatched HLA in the genome of recipient LSC represents an effective mechanism of escape from donor T cell alloreactivity. Genomic loss of mismatched HLA molecules occurs through a mechanism of copy-neutral loss of heterozygosity, which eliminates mismatched HLA alleles without decreasing levels of HLA class I molecule expression. This mechanism of immune escape, also known as immune-selection specific loss, explains why the NK cell-mediated response is not activated. It seems to be especially relevant in the context of allo-HSCT from haploidentical and HLA-mismatched donors where it accounts for more than one-third of relapses [9,22,23,24]. Evidence suggests that this phenomenon may be due to the clonal evolution of LSC as a direct consequence of selective immunological pressure by donor alloreactive T cells [25,26,27]. This assumption is further supported by evidence that the loss of HLA genes is a rare event in HLA-matched allo-HSCT [9,28], where the only targets of donor alloreactive T cells are minor histocompatibility antigens (mHAg) [29]. Therefore, even though there is not clear evidence to support this, it is possible that genomic alterations in mHAg could be the immune escape mechanism operating in HLA-matched HSCT.
The critical role of HLA class-II molecule down-regulation as a mechanism of immune-escape has been further strengthened by other studies that have compared the RNA expression of several genes in paired samples obtained from patients with hematological malignancies at initial presentation and at post-transplant relapse. These studies report the differential expression of more than 200 genes with a known or supposed role in immune function. HLA class-II genes were among the most differentially expressed, with an overall down-regulation at the time of post-transplant relapse [10,30,31]. Consistently, AML cells collected at initial presentation were easily recognized by T cells from HLA-mismatched third-party donors, whereas those collected at post-transplant relapse elicited poor donor T cell activation due to the low expression of HLA class-II molecules. More interestingly, functional in vitro studies revealed that the expression of HLA class-II molecules returned to normal levels when AML cells collected at post-transplant relapse were exposed to interferon-γ (IFN-γ), suggesting that down-regulation of HLA class-II molecules could be dependent on epigenetic mechanisms. This result is supported by the efficacy of hypomethylating agents (HMA), such as 5-azacitidine (AZA) and decitabine, in preventing and treating post-transplant disease relapse [32].
2.2. Freezing the Immune Response: Role of Anti-Inflammatory Cytokines
It is well established that leukemic cells can produce anti-inflammatory cytokines to successfully escape immune surveillance [33]. The neoplastic population of chronic myeloid leukemia (CML) cells can produce transforming growth factor (TGF-β) in order to decrease the expression of HLA-class II molecules and in turn become less immunogenic [33,34,35]. This same goal can be achieved by leukemic cells of chronic lymphocytic leukemia (CLL) and AML through the production of interleukin-10 (IL-10) [36,37]. A hypothetical role of interleukin-4 (IL-4) has also been proposed. Some studies have shown that IL-4 production by leukemic cells could promote their immune escape by decreasing the expression of HLA-class II molecules on their surface [33,34,35,36,37]. However, evidence from other studies have demonstrated that IL-4 is able to selectively induce apoptosis of AML cells by upregulating STAT6 target genes [38]. Currently, the exact role of IL-4 in leukemic cell survival is poorly understood and more studies are needed to elucidate the multiple mechanisms of action of this cytokine.
In addition, leukemic cells and normal hematopoietic progenitors may create an anti-inflammatory microenvironment not only by enhancing the production of anti-inflammatory cytokines, but also by blocking the release of pro-inflammatory cytokines. The low levels of IFN-γ associated with high-risk B-lineage acute lymphoblastic leukemia (B-ALL) have piqued the hypothesis that some leukemic cells might exploit defective IFN-γ production to escape immunological recognition [39]. Even though data derive mainly from studies on B-ALL, it is reasonable that the switching-off of the immune response through the enhanced production of anti-inflammatory cytokines and/or the reduced production of pro-inflammatory cytokines could be a relevant mechanism through which AML cells hamper immune activation after allo-HSCT.
Large amounts of interleukin-15 (IL-15) released from normal myeloid progenitors [40] promote leukemia control after allo-HSCT by boosting T cell alloreactivity against leukemia cells, enhancing NK expansion and instructing the generation of new memory stem T cells from naïve precursors [41,42,43]. Instead, low serum levels of IL-15 and interleukin-7 (IL-7), especially in the early post-transplant period, have been associated with a lower risk of acute GvHD (aGvHD) and a higher risk of relapse; thus supporting the role of these cytokines in improving T cell alloreactivity [44]. Despite the lack of definitive evidence, an anti-tumor role of IL-15 has been suggested by results obtained from a phase I single-arm multicenter study, which reported increased activation and expansion of both CD8+ T cells and NK cells when ALT-803, an IL-15 superagonist complex, was administered to patients who relapsed more than 60 days post-allo-HSCT [43]. Furthermore, studies in animal models have demonstrated that internal tandem duplication (ITD) of the tyrosine kinase FLT3 (FLT3-ITD) negatively regulates transcription of the IL-15 gene through the inhibition of its main transcription regulatory factor: Interferon regulatory factor 7 (IRF7). Thus, AML cells harboring the FLT3-ITD mutation express the IL-15 gene at very low levels. Consistently, the pharmacological inhibition of FLT3 counteracts the IRF7 activation block by reducing the expression of transcription factor ATF4 and restores IL-15 expression. Thus, the inhibition of FLT3, together with T cell transfer, might favor long-term disease control by enhancing the elimination of neoplastic cells [45]. In addition, past studies have demonstrated that IL-15 can also lead to mitochondrial reprogramming, thereby enhancing antineoplastic immunity [46]. As evidenced by human in vitro studies, FLT3 inhibition improves cellular mechanisms responsible for energy production (i.e., glycolysis and mitochondrial respiration), whose functions are essential for CD8+ T cell ATP-dependent cellular activities (e.g., killing of leukemic cells), thereby leading to the enhanced anti-leukemic activity of these immune effectors [45].
To date, it has been well documented that the elimination of apoptotic cells by APC is another important process that is enhanced by anti-inflammatory cytokines (e.g., TGF-β and IL-10). The identification of apoptotic cells by APC involves the recognition of phosphatidylserine (PS) on apoptotic cells by the phosphatidylserine receptor (PSR) expressed on APC. This interaction not only leads to phagocytosis of the apoptotic cell, but also to the production and secretion of anti-inflammatory cytokines. Cancer cells from solid tumors can express both PS and PSR and produce a soluble form of PS (sPS) which interacts and activates the PSR on APC and on neighboring cancer cells, an event that leads to the production and secretion of anti-inflammatory cytokines and facilitates their escape from immune surveillance [47]. Further studies are required to investigate whether this pathway could be exploited by AML cells to hinder their recognition and destruction by immune cells.
Even though studies are lacking, other humoral factors may also be exploited by leukemia cells to negatively regulate anti-tumor activity. For example, the suppression of Interleukin-1β (IL-1β) and granulocyte-colony stimulating factor (G-CSF) production is a hypothetical mechanism that could be exploited by tumor cells to evade the immune system [12].
2.3. Immune-Checkpoint Receptors and Co-Inhibitor Receptors
Under physiologic conditions, T cells receive multiple signals from ICP receptors and these signals are counterbalanced by those provided by co-inhibitory receptors (CTLA-4, PD-1, TIM3, LAG-3, and others). In neoplastic disorders, including AML, co-inhibitory receptors and their ligands may block immune responses by abolishing T cell function and may generate a state of immune tolerance [48]. In AML, this immunological block is due to the fact that despite the presence of a similar number of CD4+ CD8+ T lymphocytes in the leukemic and normal bone marrow (BM), the expression of ICP receptors is higher on T cells contained in the leukemic BM than on T cells contained in the healthy BM [49,50,51].
In addition, various studies have shown that in AML evolved from a myelodysplastic syndrome (MDS) and in relapsed AML, disease aggressiveness directly correlates with increased PD-1 expression on T cells and increased PD-L1 expression on leukemic cells [52,53,54]. A current study has confirmed these findings by revealing a similar percentage of T cells in the BM microenvironment of AML patients and healthy donors, but a higher number of regulatory T (Treg) cells, PD-1-positive/OX40-positive T cells and PD-1-positive/CD8-positive T cells that also express TIM3 or LAG3 in the BM of AML patients [55]. AML patients who experienced multiple relapses or relapsed post-transplant had a higher percentage of PD-1-positive/CD8-positive T cells than that of newly diagnosed or first relapsed patients, and their TP-53-mutated leukemic cells more frequently expressed PD-L1. Gene expression analysis demonstrated that leukemic cells from patients relapsed post-transplant present a deregulated expression of different co-stimulatory molecules and changes in circulating T cells [13]. Interestingly, these patients also have a greater number of early-differentiated memory stem T cells and central memory T cells with multiple inhibitory receptors than those of patients who remain in CR. Moreover, relapse is effectively predicted by the early identification of BM memory stem T cells displaying a severely exhausted phenotype (PD1+, Eomes+, T-bet-) [14].
In this context, therapies aimed at blocking ICP receptors might offer an advantage to prevent and treat AML relapse after allo-HSCT (see Section 3.3 “Immune Checkpoint Inhibitors”).
2.4. Evasion from NK Cell Recognition and Destruction
Recent studies have revealed that the down-regulation of ligands for NK cell activating receptors or some NK activating receptors and the activation of NK inhibitory receptors may promote AML cell evasion from NK recognition. Leukemic cells may reduce the expression of NK ligands through an aberrant hyper-methylation of genes encoding ligands for the activating receptor NKG2D (NKG2DL), namely MICA, ULBP1, ULBP2, and ULBP3 genes [56], and may also release a soluble form of NKG2DLs (sNKG2DL) which induces the down-regulation of NKG2D receptor [57]. Both of these mechanisms of immune disruption can be removed by HMA, which is thought to block MICA, MICB and ULBP2 shedding by increasing the expression of TIMP3 and may also reduce the release of sNKG2DLs [58]. In addition, AML cells which present ligands for NK activating receptors, namely CD112 and CD155, can decrease the expression of some other NK cell activating receptors, namely DNAM-1, NKp30, and NKp46, by physically interacting with NK cells [59,60]. In particular, leukemic blasts from patients aged less than 65 years may express high levels of CD112 and CD155 in order to decrease DNAM-1 expression on NK cells [61], an event which leads to an altered degranulation of NK cells causing impaired killing of blasts in AML and MDS [62,63]. Thus, it is not surprising that a reduced expression of these activating receptors at AML diagnosis has been associated with a significant and poor prognosis. This has been demonstrated in AML patients with high NKp30 and NKp46 expression, which have a better overall survival (OS) and event-free survival (EFS) than those with low expression [64,65]. In patients submitted to allo-HSCT, a high expression of NKp46 has been associated with a significantly better prognosis [66,67].
AML may also escape NK recognition thanks to the high expression of co-inhibitory receptors including T cell immunoglobulin and immune-receptor tyrosine-based inhibitory motif (ITIM) domain (TIGIT) which inhibit NK cell activity by reducing IFN- γ release, as well as PVRIG and TACTILE, which may act as checkpoint regulators. NK cells, similarly to exhausted CD8+ T cells, may express high levels of TIGIT, which have been correlated with primary refractory AML and leukemia relapse post-HSCT [68]. Currently, in AML patients submitted to allo-HSCT, high TIGIT expression at engraftment has been associated with a reduced number of NK cells in the BM and in the post-transplant follow-up high TIGIT expression is associated with a reduced incidence of aGVHD, poor OS, and progression-free survival (PFS) [69].
Other inhibitory NK receptors include PVRIG, whose only ligand is CD112, and TACTILE, which controls tumor growth/metastasis and DNAM1 activity in mice, but whose role in humans is still debated [70].
2.5. Other Mechanisms
Theoretically, the acquisition of new driver mutations that provide a proliferative advantage to leukemia cells could be a mechanism that favors immune escape. Genomic analysis of paired samples collected at initial presentation and at relapse after allo-HSCT have demonstrated that leukemic clones at relapse are genetically different from those present at clinical diagnosis. Most mutations involve genes whose products have a tumor suppressive (e.g., PTEN) or a proliferative (e.g., KRAS) function. Thanks to this proliferative advantage leukemic cells may outnumber the anti-tumor activity of T cells, thereby promoting immune escape by these newly mutated clones [71,72].
Leukemia cells can assume specific metabolic features that are useful for impairing T cell functions. For example, kynurenine, a metabolite produced by tryptophan degradation, impairs normal T cell function. T cell function is also impaired in the absence of adequate amounts of tryptophan. Indoleamine 2,3-dioxygenase-1 (IDO1) catalyzes the first, rate limiting step in tryptophan degradation, thereby inversely regulating the amount of tryptophan and of kynurenine. Thus, leukemia cells can negatively regulate T cell function by up-regulating the expression of IDO1 [73]. This is supported by the clinical observation that higher levels of IDO1 expression by leukemia cells correlates with an adverse prognosis in childhood AML [74].
CD39 and CD73 are two ectonucleotidases that cooperate in the degradation of adenosine triphosphate (ATP) to adenosine diphosphate (ADP), adenosine monophosphate (AMP), and adenosine. It has been largely demonstrated that adenosine has an immunosuppressive property. Thus, CD39 and CD73 promote an anti-inflammatory microenvironment through the enhanced production of adenosine. Using CD73-defective mice it has been demonstrated that, after allo-HSCT, low CD73 activity enhances leukemia cell recognition and destruction by alloreactive T cells [75,76].
Finally, arginine is a basic amino acid whose concentration has a significant impact on T cell function and proliferation. It has been demonstrated that leukemia cells can actively produce and secrete the enzyme arginase, which leads to arginine depletion, thereby impairing proliferation and normal function of T cells and polarizing nearby monocytes and macrophages towards a immunosuppressive M2-like phenotype [77,78,79].
3. Potential Treatments
There are several potential treatments aimed at preventing and treating relapse after allo-HSCT. In a schematic and simplified view, they can be classified as drugs or cellular therapies (Table 1).
3.1. Tyrosine Kinase Inhibitors (TKI)
TKI are drugs with an intrinsic antitumor effect based on their ability to target tyrosine kinases with aberrant and exaggerated functions selectively expressed by neoplastic clones in a few, specific, hematological malignancies. However, the antitumor effects of TKI also rely on their immunomodulatory effects that allow them to induce T-cell cytolytic functions, reduce PD-1 expression by T-cells and reduce myeloid-derived suppressor cells [80].
In patients with CML relapsed after allo-HSCT, anti-BCR-ABL TKI (e.g. imatinib) induce more than 60% of molecular remissions [81], whereas results in Ph+ B-ALL relapsed post-transplant are more controversial [82]. However, their use after allo-HSCT is actually recommended by the European Society for Blood and Marrow Transplantation (EBMT) either as prophylaxis or pre-emptive therapy for MRD-negative Ph+ B-ALL patients [83].
In AML the presence of FLT3-ITD at the time of allo-HSCT is predictive of a higher risk of posttransplant relapse (30% vs. 16%) and therapy with anti-FLT3 TKI is of clinical relevance to reduce this risk. Currently, the use of midostaurin, the main FLT3 inhibitor, is approved for AML with mutated FLT3 whereas its use as post-transplant maintenance therapy has been investigated in a phase II clinical trial that reported a 12-month relapse rate of only 9.2% [84]. Sorafenib is another kinase inhibitor that targets a wide range of tyrosine-kinases (e.g., c-KIT, FLT3, VEGFR-2, VEGFR-3, and PDGFR-ß) and serine/threonine-kinases (e.g., BRAF, V600E BRAF, and CRAF) expressed by cancer cells including tumor endothelial cells. Results from animal studies have revealed that the antitumor activity of sorafenib not only relies on its ability to inhibit kinases, but also on its ability to induce IL-15 production thereby enhancing T-cell activation and the GvL effect [43]. A retrospective study that investigated sorafenib as a prophylactic therapy in FLT3-ITD positive AML reported an improved outcome [85]. Currently, other newer anti-FLT3 TKI such as quizartinib, gilteritinib, and crenolanib are under investigation.
3.2. Acting on Epigenetic Factors: Hypomethylating Agents and Histone Deacetylase Inhibition
Since methylation is a crucial process involved in the epigenetic control of gene expression, it is not surprising that malignant cells use hypermethylation to switch off the expression of a variety of genes involved in apoptotic cell death and growth inhibition. Given their ability to regulate cell differentiation and cell growth by inhibition of DNA methyltransferase, HMA like AZA and decitabine are currently used to treat MDS and AML. However, several studies have reported that HMA may also upregulate the expression of HLA molecules and tumor associated antigens (TAAs), thus enhancing the ability of donor T cells to recognize, target and kill tumor cells after allo-HSCT [85,86]. In AML/MDS patients with early post-transplant relapse, salvage treatment with AZA or decitabine, administered as single agents or combined with donor lymphocyte infusion (DLI) may lead to 28% CR [87], a result confirmed by a large EBMT study that also reported a low incidence of aGvHD.
Interestingly, various studies have shown that combining AZA with DLI may lead to sustained remissions in about 30% AML/MDS relapsed post-transplant with an incidence and severity of GvHD lower than that occurring after DLI alone [88,89,90]. It has been suggested that this GvHD control may be due to an AZA-dependent upregulation of transcriptional factor FoxP3 and subsequent expansion of CD4+ Treg cells [91,92,93]. Other evidence has shown that administration of AZA to this subset of patients may convert a decreasing donor cell chimerism into a full-donor cell chimerism [94].
However, the most relevant point is that due to their immunomodulation properties and ability to upregulate inhibitory ICP co-receptors on T cells [95], various ongoing clinical trials are currently using HMA in combination with ICP inhibitors to treat relapsed/refractory AML [96]. The rational for this therapeutic strategy was provided by the observation that PD-1, PDL-1, and PDL-2 were more often over-expressed in MDS/AML patients resistant to AZA than in sensitive patients so that the blockade of the PD-1/PDL-1 interaction was supposed to be clinically effective in AZA-resistant patients [53]. One of these ongoing phase Ib/II clinical trials that combined AZA with nivolumab in relapsed/refractory AML achieved a 35% overall response rate (ORR) including 21% CR that were durable in 9/11 patients. In this study CR correlated with a higher pre-treatment BM infiltration of CD3+ and CD8+ T cells and a progressive BM increase of CD4+ and CD8+ T cells during treatment. Instead, in non-responsive patients, a progressive BM increase of CTLA4+ CD8+ T cells during treatment was noted and was thought to flag resistance to treatment [96]. Thus, large prospective randomized clinical trials are currently investigating the role of HMA as treatment able to prevent post-transplant relapse.
Histone deacetylation is another epigenetic mechanism of gene expression control performed by a group of enzymes called Histone Deacetylase (HDAC). HDAC inhibitors upregulate gene expression by unwinding histone-bound DNA. In patients submitted to allo-HSCT this leads to reduced expression of genes involved in inflammatory cytokine production and increases in CD4+ Treg cells, thereby preventing development of GvHD [97]. Panobinostat, an inhibitor of HDAC, was investigated in two different clinical trials as post-transplant maintenance therapy in MDS/AML patients: 1-year RFS was 66% when panobinostat was combined with DLI and 2-year RFS was 74% when panobinostat was administered as a single agent. A prospective, randomized phase III trial testing panobinostat as post-transplant maintenance therapy is currently ongoing.
3.3. Immune Checkpoint Inhibitors
Since tumor cells upregulate ICP inhibitors to prevent their killing by immune cells, it has been proposed that Checkpoint Inhibitor inhibition should enhance the GvL effect after allo-HSCT. This pharmacological strategy has already been validated outside the HSCT setting in various cases of solid tumors and hematological malignancies such as Hodgkin lymphoma (HL) with drugs that block CTLA-4 and PD-1 [98]. Moreover, other studies have demonstrated enhanced cytotoxicity in adoptive T-cell therapy by combining PD-1 with cytotoxic T-cell infusions [99]
Two anti-PD-1 antibodies, pidilizumab and nivolumab, have been recently investigated. Pidilizumab, administrated as maintenance therapy after autologous HSCT (auto-HSCT) for diffuse large B cell lymphoma (DLBCL) and nivolumab, administrated as rescue therapy for relapsed HL after auto-HSCT, are well tolerated and ensure a high response rate [100]. However, considering the important T-cell alloreactivity in both the GvHD and GvL effect, there are many concerns regarding the use of anti-PD-1 and anti-CTLA-4 antibodies after allo-HSCT. Another multicenter retrospective study investigated the clinical feasibility and effectiveness of PD-1 blockade with anti-PD-1 antibodies in 31 lymphoma patients with post-transplant relapse: ORR was 77% and aGvHD incidence was 54% with eight patients who died due to GvHD-related complications [101]. In a phase Ib/II clinical trial combining nivolumab with AZA in relapsed/refractory AML patients an ORR of 24% was achieved.
In a phase 1/1b clinical trial, the anti-CTLA-4 antibody ipilimumab was administered at a dose of 3 mg/kg or 10 mg/kg to patients with a variety of hematological cancers (43% of patients had AML) relapsed after allo-HSCT. Among 22 patients who received 10 mg/kg of ipilimumab an ORR of 32% was described, including 22% CR. Grade III-IV aGVHD occurred in 14% patients and was completely resolved with systemic steroid therapy [102].
Extramedullary myeloid leukemia is typically resistant to standard therapies, but provides an antigen-rich environment that can promote T-cell alloreactivity against leukemic cells. Indeed, in one study, all patients with extramedullary leukemia (three with leukemia cutis and one with myeloid sarcoma) achieved complete and durable responses after ipilimumab. The observation that patients who developed GvHD during this treatment were those with a better response to ipilimumab treatment further confirms the intimate relationship between GvHD and GvL. In addition, immunohistochemical and gene-expression analyses revealed that patients who responded to ipilimumab had a higher cytotoxic CD8+ T cell count at the disease site as opposed to those without any response to ipilimumab. The latter presented with an in situ cytotoxic function deficit. This result suggests that ipilimumab resistance may be due to local factors that hamper the acquisition of a cytotoxic phenotype by CD8+ T cells. Moreover, in patients responsive to ipilimumab, peripheral blood analyses revealed the reduction and impaired function of CD4+ Tregs and an increase in CD4+ effector T cells [102]. Further studies combining ipilimumab with decitabine (ClinicalTrials.gov identifier: NCT02890329) in the de novo and post-transplant setting for patients with AML or MDS are currently ongoing.
A phase II nonrandomized study aimed at evaluating the safety and efficacy of AZA with either nivolumab or nivolumab + ipilimumab in relapsed/refractory AML is currently recruiting (ClinicalTrials.gov identifier: NCT02397720). Preliminary results reported a CR rate of 43% in patients receiving AZA plus both nivolumab and ipilimumab (versus a CR rate of 22% in the AZA plus nivolumab arm) [103].
Lastly, a recent phase II single-arm study investigated the safety and efficacy of pembrolizumab, another anti-PD-1 antibody, as induction and maintenance therapy after high-dose cytarabine chemotherapy in patients with relapsed/refractory AML (ClinicalTrials.gov identifier: NCT02768792) and demonstrated a CR rate of 40% [104].
Taken together, these data emphasize the potential role of checkpoint inhibitor blockades to prevent and treat post-transplant relapse, but also underscore the need for further studies aimed at clarifying the potential risks of GvHD.
3.4. Donor Lymphocyte Infusions
Evidence from clinical practice supports a strong link between GvL and GvHD and has revealed that chronic GvHD (cGvHD), but not aGvHD protects patients from disease relapse [105]. Various studies have demonstrated that patients without cGvHD develop disease relapse more often than those with cGvHD of any grade. Thus, many research efforts have tried to separate GvL from GvHD and to identify which immune cell populations are responsible for these post-transplant events. These studies have identified αβ-T cells as the primary immune cells involved in these post-transplant outcomes. αβ-T cells initiate the immune reaction against host tissues and are able to completely eradicate residual, resistant LSC through the GvL effect [106].
Based on this seminal knowledge, various procedures including the photodynamic treatment (PDT) of the graft, have been aimed at depleting αβ-T cells from BM or peripheral stem cell grafts in order to prevent GvHD (Figure 1), while other procedures are aimed at exploiting the activity of αβ-T cells in order to prevent post-transplant relapse.
The demonstration that donor alloreactive T cells are clinically effective in preventing post-transplant relapse was first provided by studies on CML patients in molecular relapse. In 2014, an EBMT study evaluated the sensitivity of different hematological malignancies to the graft-versus-host effects in 48,111 patients submitted to a first allo-HSCT during the period 1998–2007. This study reported that in CML, a reduction in relapse risk was tightly linked with the development and severity of aGvHD and cGvHD and that in both ALL and BCR-ABL negative myeloproliferative disorders (MPD) the relapse risk was similar. In contrast, MDS and lympho-proliferative disorders presented an intermediate correlation with GvHD, while AML and plasma cell disorders had a weak sensitivity to GvHD [107]. These results have been confirmed by a more recent literature review which reported that DLI was successful in 70–80% of patients with hematological/cytogenetic relapse. Patients with multiple myeloma (MM), MDS, and MPD achieved intermediate results, whereas those with acute leukemia, especially ALL, achieved disappointing results [7]. In conclusion, the strength of the GvHD/GvL relationship is different in various hematological disorders. Here we review recent evidence on the efficacy and safety of DLI use in MDS/AML patients relapsing after allo-HSCT.
Studies have reported that only 15–20% of MDS and AML patients respond to DLI and that these responses have a short duration due to the rapid growth kinetics of the AML cells. Other potential mechanisms responsible for decreased DLI efficacy in AML include lack of surface expression of costimulatory molecules by leukemic cells, involvement of immunologically privileged sites, defective tumor antigen presentation, downregulation of HLA molecules and other patient-specific major and minor histocompatibility antigens [9], suppression by peripheral blood Treg cells [108] and inhibition of NK cell responses [109,110]. Thus, a more recent study has questioned whether DLI may be an adequate treatment option in MDS/AML relapsed after HSCT [111] demonstrating that DLI may be effective when combined with chemotherapy or AZA in patients with low leukemic burden, molecular but not hematological relapse, mixed chimerism and favorable cytogenetics and when the time between allo-HSCT and relapse is longer than five months. In addition, it was observed that an initial CD3+ T-cell dose greater than 1x108/kg was associated with a high GvHD incidence without any improvement of relapse risk and OS. While an interval between allo-HSCT and DLI infusion of less than two years was associated with less GvHD. Thus, it has been proposed that the starting dose of CD3+ T-cells should be between 1 × 106 and 1 × 107/kg and should be escalated until GvHD development [112]. However, despite these results, the timing of DLI has not been established yet. Usually, DLI is performed eight weeks post-transplant and is not administered in the first three months post-transplant except in case of relapse.
DLI is associated with a 10% mortality rate and may cause pancytopenia in 20% of patients; while GvHD is observed in 60% of patients independently of the grade of HLA donor matching [113]. One study has evaluated pre-emptive DLI (p-DLI) versus DLI performed for late post-transplant relapse (t-DLI) in patients submitted to T-cell depleted HSCT. This study reported an estimated 5-year OS of 80% for p-DLI versus 40% for t-DLI, a 5-year EFS of 65% versus 69% and a 5-year GvHD incidence of 31% versus 41% [114]. Another study treated selected patients (i.e., those who were in CR for at least 120 days from transplantation, off immunosuppression for at least 30 days and free of GvHD) with adjuvant DLI. A 7-year OS of 67% was reported and GvHD was the main clinical complication [115].
3.5. Antitumor Immunotherapy Based on Dendritic Cells
Dendritic cells (DCs) are professional APC able to uptake, process and present antigens, including TAAs, to naïve CD4+ or CD8+ T-cells in order to activate an immune response against the captured antigen. As such, they represent the principal link between innate and acquired immunity [118].
DCs are normally present within the tumor mass in close association with neoplastic cells, but their anti-tumor response is poor due to the immune-suppressive effect of the tumor milieu, which modulates DC maturation and activity [119,120]. Thus, in order to overcome this problem various research efforts have led to the development of DC-based antitumor vaccines that exploit the ability of DCs to actively infiltrate the tumor mass and cross-present TAAs in order to activate host T-cells and NK-cells [121,122]. DC-based vaccines can be obtained through two distinct strategies (Figure 2). The first strategy (i.e., ex vivo modality), allows DCs to be obtained from patients’ peripheral blood thanks to the expansion of CD34+ progenitor cells cultured in vitro with GM-CSF, IL-4, tumor necrosis factor (TNF), FLT3-ligand and CD40 ligand (CD40L) [123]. These DCs can be loaded with the target TAA, activated and then re-infused back into the patient [124]. However, this strategy is frequently not clinically effective because of the poor immunogenicity of the selected antigen and the ability of the neoplastic cells to down regulate the expression of a specific antigen with consequent immune evasion. In order to overcome these drawbacks, newer approaches based on the in vitro DC activation with entire tumor cells have been developed [125,126]. The rational of this methodology consists in achieving immune activation that is not restricted to a single antigen, but against a broader spectrum of antigens. This strategy might be more effective, allowing an anti-tumor immune response that can also target patient’s specific tumor-associated neo-antigens [127,128].
Deeper knowledge of DC biology has allowed further optimization of DC antigen-presenting functions. Specifically, early clinical trials have reported that activated DCs with a mature phenotype are more capable of activating T-cells. Thus, DCs are now exposed to “maturation cocktails” containing Toll-like-receptor (TLR) ligands, specific pathogen-derived molecules and other “type 1” polarizing agents such as interferons (IFNs). These “maturation cocktails” are aimed at improving DC maturation and activation, thus inducing them to secrete large amounts of IL-12, respond to homing signals from lymph nodes and induce antigen-specific T-cell activation. In a recent study it has been shown that in vitro DC exposure to TLR stimulating agents induces them to differentiate into IFN-producing cells rather than into APCs. In the post-transplant setting, IFN-producing DCs enhance the induction of activated antigen-specific T-cells without stimulating allogeneic T-cells, an event that improves GvL without increasing the risk of GvHD [129]. These results have been confirmed in at least four clinical trials, which demonstrated that immunotherapy with DCs may prevent (or at least delay) AML relapse when CR has been already achieved [130,131,132,133]. DCs vaccines have been evaluated as a form pre-emptive therapy in relapsing AML after allo-HSCT. In a recent phase 1 trial, AML patients with early molecular relapse after allo-HSCT were randomly assigned to receive immunotherapy with either DLI or multi-genetically modified DCs derived from autologous peripheral blood monocytes (moDCs). The vaccine was not only found to be safe, but also induced a three-year OS of 48.9% compared with 27.5% in the DLI group. CR was achieved in 13/23 (57%) patients treated with the moDCs and in 12/25 (48%) treated with DLI. When patients in early molecular relapse (but not in full-blown relapse) were treated with moDCs, a CR rate of 83% was observed, thus indicating that the therapeutic utility of DC vaccines in AML is limited in the case of a high leukemic cell load [133,134].
DC vaccination has been investigated in several other hematological (e.g., MM, non-Hodgkin’s lymphoma) and non-hematological (e.g., malignant melanoma, prostate cancer, renal cell carcinoma, lung, colon, and breast carcinomas) tumor types; however, these studies go beyond the scope of this paper.
3.6. Antitumor Strategies Based on NK Cells
NK cells have been recognized as a critical cellular component of the innate immune system [135,136,137,138,139,140,141]. This is based on their ability, after receiving activating stimuli, to kill target cells and to secrete cytokines involved in shaping and guiding both the innate and the adaptive immune response to infections and malignant transformation.
Among NK receptors, one of the most studied are the killer immunoglobulin-like receptors (KIR), which consist of at least fifteen different receptors that map to chromosome 19q13.4 [142,143,144]. KIRs recognize peculiar domains of class I HLA alleles. Depending on the intracellular tyrosine-based motif of the molecules, KIRs may be either activating or inhibitory [142]. Simultaneous binding of both activating and inhibiting KIRs generally inhibits NK cells from killing [145].
While T cell activity against tumor cells depends on the recognition of specific TAAs, NK cell rejection of tumor cells occurs independently of TAA recognition. This property has been exploited by different NK cell-based immunotherapies. Among these, the first and simplest procedure to be applied was the direct infusion of NK cells into patients. This strategy was based on the observation that patients who received allogeneic peripheral blood stem cell grafts containing a higher amount of NK cells had better clinical outcomes in terms of post-transplant infections and non-relapse mortality [146,147]. In one clinical trial, direct NK cell infusions in the haploidentical allogeneic setting was associated with longer RFS [148]. However, in the absence of data from randomized clinical studies demonstrating a survival advantage after NK infusion in AML patients relapsed after allo-HSCT, there are no current indications concerning the use of this therapy outside of clinical trials.
Another NK cell-based immunotherapy consisted of the in vivo expansion of NK cells obtained from leukemic patients after the administration of various recombinant human cytokines including IL-2 at low, intermediate, and high doses [149,150,151]. This strategy was further improved upon by the administration of a tumor specific antibody whose Fc portion was able to bind CD16 expressed on NK cells thereby enhancing the so called antibody-dependent cellular cytotoxicity (ADCC) [151,152,153].
A third NK cell-based immunotherapy was developed with more in depth knowledge of KIR biology [154]. Several years ago, it was observed that the killing activity of NK cells was inversely correlated with the expression of HLA class I molecules on target cells [20]. This breakthrough led to the so called “missing self” hypothesis, which nowadays represents the main mechanism by which NK cells are thought to operate. Subsequently, three main HLA class I allele specificities were identified: “group 1” comprising HLA-C alleles which express Asn80, “group 2” comprising HLA-C alleles which express Lys80 and “group 3” comprising HLA-Bw4 alleles (e.g., HLA-B27). The NK receptor repertoire is dictated and shaped during NK cell development and maturation by the HLA class I genotype in a such way that every mature NK cell expresses at least one inhibitory KIR specific for self HLA class I molecules [142]. Thus, allogenic targets sensitive to NK cytotoxicity are identified by their lack of self HLA class I inhibitory KIR ligands. Despite some conflicting data, it might be possible that, in the allo-HSCT setting, this donor-versus-recipient NK alloreactivity could improve engraftment and protect against GvHD with enhanced recipient survival and in haplo-HSCT it may protect against relapse [110,155].
Retention of HLA class I molecules by tumor cells represents a mechanism of immune escape and has become the target of many strategies. Among these, the manipulation of the relationship between NK receptor and HLA class I molecules with monoclonal antibodies seems to be the most promising [156,157].
3.7. Chimeric Antigen Receptor T Cell Therapies
The remarkable success of chimeric antigen receptor (CAR)-T cells in lymphoproliferative disorders has fueled enthusiasm over developing a similar therapeutic strategy in AML. However, the identification of unique and disease-specific target antigens for CAR-T cells in AML is much more difficult, as myeloid leukemic cells, including LSC, present antigens that are also expressed on normal hematopoietic progenitors. Thus, it is not surprising that CAR-T are still considered investigational treatments in AML due to their toxicity in normal hematopoiesis.
Despite this drawback, some antigens have already been tested as potential targets in various on-going clinical trials. Among them, CD33, a member of the sialic acid-binding Ig-like lectin family, is the most attractive as it is expressed in 90% of adult and pediatric AMLs [158]. Anti-CD33 monoclonal antibodies have proved their clinical efficacy in a peculiar subset of AML patients [159] and preclinical studies with anti-CD33 CAR-T have achieved promising results despite a severe concomitant myelotoxicity [160]. To date, the only patient with relapsed/refractory AML who received anti-CD33 CAR-T developed cytokine release syndrome (CRS), experienced severe pancytopenia and relapsed nine weeks after the CAR-T cell infusion [161]. Currently, various research groups supported by the observation that in animal models CD33 is not absolutely required for normal hematopoietic function [162], have developed different genetic approaches to inactivate CD33 on hematopoietic stem cells (HSC) in order to avoid the severe myelotoxicity associated with anti-CD33 CAR-T infusions and have reported promising results [163,164,165].
CD123, the alpha chain of the interleukin-3 receptor, is another suitable target but, like CD33, its targeting is associated with severe myelotoxicity [166]. However, a very recent study has revealed that in high-risk MDS, CD123 can effectively distinguish between normal HSCs and MDS stem cells and anti-CD123 CAR-T can selectively kill MDS stem cells in vitro [167].
Since FLT3 mutations occur in about 20% of AML patients, FLT3 has been identified as another potential CAR-T target despite the fact that it is also expressed on HSCs and multipotent hematopoietic progenitors [168]. Interestingly, it has been reported that in AML patients carrying the FLT3-ITD mutation, the therapeutic efficacy of anti-FLT3 CAR-T can be dramatically increased with crenolanib, due its ability to increase FLT3 expression on the leukemic cell surface [169].
However, the most promising CAR-T target might be CD44v6 as it is uniquely and specifically expressed on leukemic cells [170,171]. A preclinical study carried out in immunocompromised mice has demonstrated that second generation anti-CD44v6 CAR-T cleared AML and multiple myeloma cells, spared normal HSC and keratinocytes, but determined severe monocytopenia [172]. Regarding this last construct, the dose-limiting effect as well as hyper-acute xenogeneic GvHD were circumvented by the introduction of thymidine kinase [173] and inducible Caspase 9 suicide genes [174] into these CAR-T cells to effectively eliminate them within a few hours.
4. Conclusions
Currently, the demonstration that about two-thirds of post-transplant relapses are caused by unique patterns of immune escape that might be targeted by specific therapies has diminished the conventional wisdom, which for several years has assumed that alloreactive donor T-cells may cure AML patients.
In the near future, this seminal knowledge will require that in post-transplant relapse all potential mechanisms of immune escape should be accurately investigated in order to decide on the best therapeutic procedure for every single patient. Thus, a new type of personalized medicine is now emerging thanks to studies on post-transplant relapse, but its clinical effectiveness will depend on the assessment of precise biological criteria.
Funding
This research received no external funding.
Conflicts of Interest
The authors have no conflict of interest.
Abbreviations
| ADCC | antibody-dependent cellular cytotoxicity |
| ADP | adenosine diphosphate |
| aGvHD | acute graft-versus-host disease |
| AMP | adenosine monophosphate |
| ALL | acute lymphoblastic leukemia |
| Allo-HSCT | allogeneic hematopoietic stem cell transplantation |
| AML | acute myeloid leukemia |
| APC | antigen presenting cells |
| ATP | adenosine triphosphate |
| auto-HSCT | autologous hematopoietic stem cell transplantation |
| AZA | 5-azacytidine |
| BM | bone marrow |
| CAR | chimeric antigen receptor |
| CD40L | CD40 Ligand |
| cGvHD | chronic graft-versus-host disease |
| CLL | chronic lymphocytic leukemia |
| CML | chronic myeloid leukemia |
| CR | complete remission |
| CRS | cytokine release syndrome |
| DC | dendritic cells |
| DLBCL | diffuse large B cell lymphoma |
| DLI | donor lymphocyte infusion |
| EBMT | European Society for Blood and Marrow Transplantation |
| EFS | event-free survival |
| G-CSF | granulocyte-colony stimulating factor |
| GvHD | graft-versus-host disease |
| GvL | graft-versus-leukemia |
| Haplo-HSCT | haploidentical hematopoietic stem cell transplantation |
| HDAC | histone deacetylase |
| HL | Hodgkin lymphoma |
| HMA | hypomethylating agents |
| HSC | hematopoietic stem cell |
| ICP | immune-checkpoint |
| IDO1 | indoleamine 2,3-dioxygenase-1 |
| IFN | interferon |
| IL | interleukin |
| IRF7 | interferon regulatory Factor 7 |
| ITD | Internal Tandem Duplications |
| ITIM | Immune-receptor Tyrosine-based Inhibitory motif |
| KIR | killer immunoglobulin-like receptors |
| LSC | leukemic stem cells |
| MDS | myelodysplastic syndrome |
| mHAg | minor histocompatibility antigens |
| MHC | major histocompatibility |
| MM | multiple myeloma |
| moDCs | multi-genetically modified DCs derived from autologous peripheral blood monocytes |
| MPD | myeloproliferative disorder |
| NK | natural killer |
| NKG2DL | NKG2D ligand |
| ORR | overall response rate |
| OS | overall survival |
| p-DLI | preemptive donor lymphocyte infusion |
| PFS | progression-free survival |
| PS | phosphatidylserine |
| PSR | phosphatidylserine receptor |
| RFS | relapse-free survival |
| sNKG2DL | soluble form of NKG2DL |
| sPS | soluble form of Phosphatidylserine |
| TAA | tumor associated antigens |
| t-DLI | tardive donor lymphocyte infusion |
| TGF | transforming growth factor |
| TIGIT | T cell immunoglobulin and ITIM domain |
| TKI | tyrosine kinase inhibitors |
| TLR | Toll-like receptor |
| TNF | tumor necrosis factor |
| Treg | regulatory T cell. |
References
- Wei, A.H.; Tiong, I.S. Midostaurin, enasidenib, CPX-351, gemtuzumab ozogamicin, and venetoclax bring new hope to AML. Blood 2017, 130, 2469–2474. [Google Scholar] [CrossRef]
- Horowitz, M.; Schreiber, H.; Elder, A.; Heidenreich, O.; Vormoor, J.; Toffalori, C.; Vago, L.; Kroger, N. Epidemiology and biology of relapse after stem cell transplantation. Bone Marrow Transplant. 2018, 53, 1379–1389. [Google Scholar] [CrossRef]
- Van den Brink, M.R.; Porter, D.L.; Giralt, S.; Lu, S.X.; Jenq, R.R.; Hanash, A.; Bishop, M.R. Relapse after allogeneic hematopoietic cell therapy. Biol. Blood Marrow Transplant. 2010, 16, S138–S145. [Google Scholar] [CrossRef] [Green Version]
- Fathi, A.T.; Chen, Y.B. Treatment of relapse of acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. Curr. Hematol. Malig. Rep. 2014, 9, 186–192. [Google Scholar] [CrossRef]
- De Jong, G.; Janssen, J.; Biemond, B.J.; Zeerleder, S.S.; Ossenkoppele, G.J.; Visser, O.; Nur, E.; Meijer, E.; Hazenberg, M.D. Survival of early posthematopoietic stem cell transplantation relapse of myeloid malignancies. Eur. J. Haematol. 2019, 103, 491–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Cai, Y.; Jiang, J.; Wan, L.; Bai, H.; Zhu, J.; Li, S.; Wang, C.; Song, X. Early tapering of immunosuppressive agents after HLA-matched donor transplantation can improve the survival of patients with advanced acute myeloid leukemia. Ann. Hematol. 2018, 97, 497–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolb, H.J. Hematopoietic stem cell transplantation and cellular therapy. HLA 2017, 89, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Vago, L.; Perna, S.K.; Zanussi, M.; Mazzi, B.; Barlassina, C.; Stanghellini, M.T.; Perrelli, N.F.; Cosentino, C.; Torri, F.; Angius, A.; et al. Loss of mismatched HLA in leukemia after stem-cell transplantation. N. Engl. J. Med. 2009, 361, 478–488. [Google Scholar] [CrossRef] [Green Version]
- Christopher, M.J.; Petti, A.A.; Rettig, M.P.; Miller, C.A.; Chendamarai, E.; Duncavage, E.J.; Klco, J.M.; Helton, N.M.; O’Laughlin, M.; Fronick, C.C.; et al. Immune Escape of Relapsed AML Cells after Allogeneic Transplantation. N. Engl. J. Med. 2018, 379, 2330–2341. [Google Scholar] [CrossRef]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Holland, H.K.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Zeiser, R.; Vago, L. Mechanisms of immune escape after allogeneic hematopoietic cell transplantation. Blood 2019, 133, 1290–1297. [Google Scholar] [CrossRef] [PubMed]
- Toffalori, C.; Zito, L.; Gambacorta, V.; Riba, M.; Oliveira, G.; Bucci, G.; Barcella, M.; Spinelli, O.; Greco, R.; Crucitti, L.; et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019, 25, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Noviello, M.; Manfredi, F.; Ruggiero, E.; Perini, T.; Oliveira, G.; Cortesi, F.; de Simone, P.; Toffalori, C.; Gambacorta, V.; Greco, R.; et al. Bone marrow central memory and memory stem T-cell exhaustion in AML patients relapsing after HSCT. Nat. Commun. 2019, 10, 1065. [Google Scholar] [CrossRef]
- Daver, N.; Garcia-Manero, G.; Basu, S.; Boddu, P.C.; Alfayez, M.; Cortes, J.E.; Konopleva, M.; Ravandi-Kashani, F.; Jabbour, E.; Kadia, T.; et al. Efficacy, Safety, and Biomarkers of Response to Azacitidine and Nivolumab in Relapsed/Refractory Acute Myeloid Leukemia: A Nonrandomized, Open-Label, Phase II Study. Cancer Discov. 2019, 9, 370–383. [Google Scholar] [CrossRef] [Green Version]
- Ravandi, F.; Assi, R.; Daver, N.; Benton, C.B.; Kadia, T.; Thompson, P.A.; Borthakur, G.; Alvarado, Y.; Jabbour, E.J.; Konopleva, M.; et al. Idarubicin, cytarabine, and nivolumab in patients with newly diagnosed acute myeloid leukaemia or high-risk myelodysplastic syndrome: A single-arm, phase 2 study. Lancet Haematol. 2019, 6, e480–e488. [Google Scholar] [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef]
- Archbold, J.K.; Ely, L.K.; Kjer-Nielsen, L.; Burrows, S.R.; Rossjohn, J.; McCluskey, J.; Macdonald, W.A. T cell allorecognition and MHC restriction—A case of Jekyll and Hyde? Mol. Immunol. 2008, 45, 583–598. [Google Scholar] [CrossRef]
- Gökmen, M.R.; Lombardi, G.; Lechler, R.I. The importance of the indirect pathway of allorecognition in clinical transplantation. Curr. Opin. Immunol. 2008, 20, 568–574. [Google Scholar] [CrossRef]
- Kärre, K.; Ljunggren, H.G.; Piontek, G.; Kiessling, R. Selective rejection of H-2-deficient lymphoma variants suggests alternative immune defence strategy. Nature 1986, 319, 675–678. [Google Scholar] [CrossRef]
- Kolb, H.J. Graft-versus-leukemia effects of transplantation and donor lymphocytes. Blood 2008, 112, 4371–4383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vago, L.; Toffalori, C.; Ciceri, F.; Fleischhauer, K. Genomic loss of mismatched human leukocyte antigen and leukemia immune escape from haploidentical graft-versus-leukemia. Semin. Oncol. 2012, 39, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterhouse, M.; Pfeifer, D.; Pantic, M.; Emmerich, F.; Bertz, H.; Finke, J. Genome-wide profiling in AML patients relapsing after allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 2011, 17, 1450–1459.e1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toffalori, C.; Cavattoni, I.; Deola, S.; Mastaglio, S.; Giglio, F.; Mazzi, B.; Assanelli, A.; Peccatori, J.; Bordignon, C.; Bonini, C.; et al. Genomic loss of patient-specific HLA in acute myeloid leukemia relapse after well-matched unrelated donor HSCT. Blood 2012, 119, 4813–4815. [Google Scholar] [CrossRef] [Green Version]
- Zheng, P.; Sarma, S.; Guo, Y.; Liu, Y. Two mechanisms for tumor evasion of preexisting cytotoxic T-cell responses: Lessons from recurrent tumors. Cancer Res. 1999, 59, 3461–3467. [Google Scholar]
- Garcia-Lora, A.; Algarra, I.; Gaforio, J.J.; Ruiz-Cabello, F.; Garrido, F. Immunoselection by T lymphocytes generates repeated MHC class I-deficient metastatic tumor variants. Int. J. Cancer 2001, 91, 109–119. [Google Scholar] [CrossRef]
- Garcia-Lora, A.; Martinez, M.; Algarra, I.; Gaforio, J.J.; Garrido, F. MHC class I-deficient metastatic tumor variants immunoselected by T lymphocytes originate from the coordinated downregulation of APM components. Int. J. Cancer 2003, 106, 521–527. [Google Scholar] [CrossRef]
- Hamdi, A.; Cao, K.; Poon, L.M.; Aung, F.; Kornblau, S.; Fernandez Vina, M.A.; Champlin, R.E.; Ciurea, S.O. Are changes in HLA Ags responsible for leukemia relapse after HLA-matched allogeneic hematopoietic SCT? Bone Marrow Transplant. 2015, 50, 411–413. [Google Scholar] [CrossRef] [Green Version]
- Spierings, E. Minor histocompatibility antigens: Past, present, and future. Tissue Antigens 2014, 84, 360–374. [Google Scholar] [CrossRef]
- Dermime, S.; Mavroudis, D.; Jiang, Y.Z.; Hensel, N.; Molldrem, J.; Barrett, A.J. Immune escape from a graft-versus-leukemia effect may play a role in the relapse of myeloid leukemias following allogeneic bone marrow transplantation. Bone Marrow Transplant. 1997, 19, 989–999. [Google Scholar] [CrossRef] [Green Version]
- Stölzel, F.; Hackmann, K.; Kuithan, F.; Mohr, B.; Füssel, M.; Oelschlägel, U.; Thiede, C.; Röllig, C.; Platzbecker, U.; Schetelig, J.; et al. Clonal evolution including partial loss of human leukocyte antigen genes favoring extramedullary acute myeloid leukemia relapse after matched related allogeneic hematopoietic stem cell transplantation. Transplantation 2012, 93, 744–749. [Google Scholar] [CrossRef]
- Schroeder, T.; Rautenberg, C.; Haas, R.; Germing, U.; Kobbe, G. Hypomethylating agents for treatment and prevention of relapse after allogeneic blood stem cell transplantation. Int. J. Hematol. 2018, 107, 138–150. [Google Scholar] [CrossRef] [Green Version]
- Naka, K.; Hoshii, T.; Muraguchi, T.; Tadokoro, Y.; Ooshio, T.; Kondo, Y.; Nakao, S.; Motoyama, N.; Hirao, A. TGF-beta-FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature 2010, 463, 676–680. [Google Scholar] [CrossRef]
- Han, Y.; Zhou, Z.H.; Ransohoff, R.M. TNF-alpha suppresses IFN-gamma-induced MHC class II expression in HT1080 cells by destabilizing class II trans-activator mRNA. J. Immunol. 1999, 163, 1435–1440. [Google Scholar] [PubMed]
- Tarafdar, A.; Hopcroft, L.E.; Gallipoli, P.; Pellicano, F.; Cassels, J.; Hair, A.; Korfi, K.; Jorgensen, H.G.; Vetrie, D.; Holyoake, T.L.; et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood 2017, 129, 199–208. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.H.; Kim, M.; Lee, B.H.; Lim, J.; Kim, Y.; Lee, E.J.; Min, W.S.; Kang, C.S.; Kim, W.I.; Shim, S.I.; et al. Intracellular IL-4, IL-10, and IFN-gamma levels of leukemic cells and bone marrow T cells in acute leukemia. Ann. Clin. Lab. Sci. 2006, 36, 7–15. [Google Scholar] [PubMed]
- Peña-Martínez, P.; Eriksson, M.; Ramakrishnan, R.; Chapellier, M.; Högberg, C.; Orsmark-Pietras, C.; Richter, J.; Andersson, A.; Fioretos, T.; Järås, M. Interleukin 4 induces apoptosis of acute myeloid leukemia cells in a Stat6-dependent manner. Leukemia 2018, 32, 588–596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cloppenborg, T.; Stanulla, M.; Zimmermann, M.; Schrappe, M.; Welte, K.; Klein, C. Immunosurveillance of childhood ALL: Polymorphic interferon-gamma alleles are associated with age at diagnosis and clinical risk groups. Leukemia 2005, 19, 44–48. [Google Scholar] [CrossRef] [Green Version]
- Colpitts, S.L.; Stonier, S.W.; Stoklasek, T.A.; Root, S.H.; Aguila, H.L.; Schluns, K.S.; Lefrançois, L. Transcriptional regulation of IL-15 expression during hematopoiesis. J. Immunol. 2013, 191, 3017–3024. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Wu, D.; Wang, Y.; Cen, J.; Feng, Y.; Sun, A.; Tang, X.; Chang, H.; Zhu, Z. Expanded donor natural killer cell and IL-2, IL-15 treatment efficacy in allogeneic hematopoietic stem cell transplantation. Eur. J. Haematol. 2008, 81, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Cieri, N.; Camisa, B.; Cocchiarella, F.; Forcato, M.; Oliveira, G.; Provasi, E.; Bondanza, A.; Bordignon, C.; Peccatori, J.; Ciceri, F.; et al. IL-7 and IL-15 instruct the generation of human memory stem T cells from naive precursors. Blood 2013, 121, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Romee, R.; Cooley, S.; Berrien-Elliott, M.M.; Westervelt, P.; Verneris, M.R.; Wagner, J.E.; Weisdorf, D.J.; Blazar, B.R.; Ustun, C.; DeFor, T.E.; et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood 2018, 131, 2515–2527. [Google Scholar] [CrossRef] [PubMed]
- Thiant, S.; Yakoub-Agha, I.; Magro, L.; Trauet, J.; Coiteux, V.; Jouet, J.P.; Dessaint, J.P.; Labalette, M. Plasma levels of IL-7 and IL-15 in the first month after myeloablative BMT are predictive biomarkers of both acute GVHD and relapse. Bone Marrow Transplant. 2010, 45, 1546–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathew, N.R.; Baumgartner, F.; Braun, L.; O’Sullivan, D.; Thomas, S.; Waterhouse, M.; Müller, T.A.; Hanke, K.; Taromi, S.; Apostolova, P.; et al. Sorafenib promotes graft-versus-leukemia activity in mice and humans through IL-15 production in FLT3-ITD-mutant leukemia cells. Nat. Med. 2018, 24, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef] [Green Version]
- Kim, R.; Emi, M.; Tanabe, K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol. Ther. 2005, 4, 924–933. [Google Scholar] [CrossRef] [Green Version]
- Desrichard, A.; Snyder, A.; Chan, T.A. Cancer Neoantigens and Applications for Immunotherapy. Clin. Cancer Res. 2016, 22, 807–812. [Google Scholar] [CrossRef] [Green Version]
- Vidriales, M.B.; Orfao, A.; Lopez-Berges, M.C.; Gonzalez, M.; Hernandez, J.M.; Ciudad, J.; Lopez, A.; Moro, M.J.; Martinez, M.; San Miguel, J.F. Lymphoid subsets in acute myeloid leukemias: Increased number of cells with NK phenotype and normal T-cell distribution. Ann. Hematol. 1993, 67, 217–222. [Google Scholar] [CrossRef]
- Wendelbo, O.; Nesthus, I.; Sjo, M.; Paulsen, K.; Ernst, P.; Bruserud, O. Functional characterization of T lymphocytes derived from patients with acute myelogenous leukemia and chemotherapy-induced leukopenia. Cancer Immunol. Immunother. 2004, 53, 740–747. [Google Scholar] [CrossRef]
- Whiteway, A.; Corbett, T.; Anderson, R.; Macdonald, I.; Prentice, H.G. Expression of co-stimulatory molecules on acute myeloid leukaemia blasts may effect duration of first remission. Br. J. Haematol. 2003, 120, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Graf, M.; Reif, S.; Hecht, K.; Pelka-Fleischer, R.; Kroell, T.; Pfister, K.; Schmetzer, H. High expression of costimulatory molecules correlates with low relapse-free survival probability in acute myeloid leukemia (AML). Ann. Hematol. 2005, 84, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Bueso-Ramos, C.; DiNardo, C.; Estecio, M.R.; Davanlou, M.; Geng, Q.R.; Fang, Z.; Nguyen, M.; Pierce, S.; Wei, Y.; et al. Expression of PD-L1, PD-L2, PD-1 and CTLA4 in myelodysplastic syndromes is enhanced by treatment with hypomethylating agents. Leukemia 2014, 28, 1280–1288. [Google Scholar] [CrossRef] [PubMed]
- Schnorfeil, F.M.; Lichtenegger, F.S.; Emmerig, K.; Schlueter, M.; Neitz, J.S.; Draenert, R.; Hiddemann, W.; Subklewe, M. T cells are functionally not impaired in AML: Increased PD-1 expression is only seen at time of relapse and correlates with a shift towards the memory T cell compartment. J. Hematol. Oncol. 2015, 8, 93. [Google Scholar] [CrossRef] [Green Version]
- Williams, P.; Basu, S.; Garcia-Manero, G.; Hourigan, C.S.; Oetjen, K.A.; Cortes, J.E.; Ravandi, F.; Jabbour, E.J.; Al-Hamal, Z.; Konopleva, M.; et al. The distribution of T-cell subsets and the expression of immune checkpoint receptors and ligands in patients with newly diagnosed and relapsed acute myeloid leukemia. Cancer 2019, 125, 1470–1481. [Google Scholar] [CrossRef] [Green Version]
- Raneros, A.B.; Martín-Palanco, V.; Fernandez, A.F.; Rodriguez, R.M.; Fraga, M.F.; Lopez-Larrea, C.; Suarez-Alvarez, B. Methylation of NKG2D ligands contributes to immune system evasion in acute myeloid leukemia. Genes Immun. 2014, 16, 71–82. [Google Scholar] [CrossRef]
- Baragano Raneros, A.; Suarez-Alvarez, B.; Lopez-Larrea, C. Secretory pathways generating immunosuppressive NKG2D ligands: New targets for therapeutic intervention. Oncoimmunology 2014, 3, e28497. [Google Scholar] [CrossRef] [Green Version]
- Raneros, A.B.; Minguela, A.; Rodriguez, R.M.; Colado, E.; Bernal, T.; Anguita, E.; Mogorron, A.V.; Gil, A.C.; Vidal-Castineira, J.R.; Marquez-Kisinousky, L.; et al. Increasing TIMP3 expression by hypomethylating agents diminishes soluble MICA, MICB and ULBP2 shedding in acute myeloid leukemia, facilitating NK cell-mediated immune recognition. Oncotarget 2017, 8, 31959–31976. [Google Scholar] [CrossRef]
- Costello, R.T.; Sivori, S.; Marcenaro, E.; Lafage-Pochitaloff, M.; Mozziconacci, M.J.; Reviron, D.; Gastaut, J.A.; Pende, D.; Olive, D.; Moretta, A. Defective expression and function of natural killer cell-triggering receptors in patients with acute myeloid leukemia. Blood 2002, 99, 3661–3667. [Google Scholar] [CrossRef]
- Fauriat, C.; Just-Landi, S.; Mallet, F.; Arnoulet, C.; Sainty, D.; Olive, D.; Costello, R.T. Deficient expression of NCR in NK cells from acute myeloid leukemia: Evolution during leukemia treatment and impact of leukemia cells in NCRdull phenotype induction. Blood 2007, 109, 323–330. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Gayoso, I.; Bergua, J.M.; Casado, J.G.; Morgado, S.; Solana, R.; Tarazona, R. Decreased expression of DNAM-1 on NK cells from acute myeloid leukemia patients. Immunol. Cell Biol. 2012, 90, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Pende, D.; Castriconi, R.; Romagnani, P.; Spaggiari, G.M.; Marcenaro, S.; Dondero, A.; Lazzeri, E.; Lasagni, L.; Martini, S.; Rivera, P.; et al. Expression of the DNAM-1 ligands, Nectin-2 (CD112) and poliovirus receptor (CD155), on dendritic cells: Relevance for natural killer-dendritic cell interaction. Blood 2006, 107, 2030–2036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlsten, M.; Baumann, B.C.; Simonsson, M.; Jadersten, M.; Forsblom, A.M.; Hammarstedt, C.; Bryceson, Y.T.; Ljunggren, H.G.; Hellstrom-Lindberg, E.; Malmberg, K.J. Reduced DNAM-1 expression on bone marrow NK cells associated with impaired killing of CD34+ blasts in myelodysplastic syndrome. Leukemia 2010, 24, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Bernson, E.; Hallner, A.; Sander, F.E.; Wilsson, O.; Werlenius, O.; Rydström, A.; Kiffin, R.; Brune, M.; Foà, R.; Aurelius, J.; et al. Impact of killer-immunoglobulin-like receptor and human leukocyte antigen genotypes on the efficacy of immunotherapy in acute myeloid leukemia. Leukemia 2017, 31, 2552–2559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martner, A.; Rydström, A.; Riise, R.E.; Aurelius, J.; Brune, M.; Foà, R.; Hellstrand, K.; Thorén, F.B. NK cell expression of natural cytotoxicity receptors may determine relapse risk in older AML patients undergoing immunotherapy for remission maintenance. Oncotarget 2015, 6, 42569–42574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chretien, A.S.; Fauriat, C.; Orlanducci, F.; Rey, J.; Borg, G.B.; Gautherot, E.; Granjeaud, S.; Demerle, C.; Hamel, J.F.; Cerwenka, A.; et al. NKp30 expression is a prognostic immune biomarker for stratification of patients with intermediate-risk acute myeloid leukemia. Oncotarget 2017, 8, 49548–49563. [Google Scholar] [CrossRef] [Green Version]
- Chretien, A.S.; Devillier, R.; Fauriat, C.; Orlanducci, F.; Harbi, S.; Le Roy, A.; Rey, J.; Bouvier Borg, G.; Gautherot, E.; Hamel, J.F.; et al. NKp46 expression on NK cells as a prognostic and predictive biomarker for response to allo-SCT in patients with AML. Oncoimmunology 2017, 6, e1307491. [Google Scholar] [CrossRef] [Green Version]
- Kong, Y.; Zhu, L.; Schell, T.D.; Zhang, J.; Claxton, D.F.; Ehmann, W.C.; Rybka, W.B.; George, M.R.; Zeng, H.; Zheng, H. T-Cell Immunoglobulin and ITIM Domain (TIGIT) Associates with CD8+ T-Cell Exhaustion and Poor Clinical Outcome in AML Patients. Clin. Cancer Res. 2016, 22, 3057–3066. [Google Scholar] [CrossRef] [Green Version]
- Hattori, N.; Kawaguchi, Y.; Sasaki, Y.; Shimada, S.; Murai, S.; Abe, M.; Baba, Y.; Watanuki, M.; Fujiwara, S.; Arai, N.; et al. Monitoring TIGIT/DNAM-1 and PVR/PVRL2 Immune Checkpoint Expression Levels in Allogeneic Stem Cell Transplantation for Acute Myeloid Leukemia. Biol. Blood Marrow Transplant. 2019, 25, 861–867. [Google Scholar] [CrossRef]
- Sanchez-Correa, B.; Valhondo, I.; Hassouneh, F.; Lopez-Sejas, N.; Pera, A.; Bergua, J.M.; Arcos, M.J.; Banas, H.; Casas-Aviles, I.; Duran, E.; et al. DNAM-1 and the TIGIT/PVRIG/TACTILE Axis: Novel Immune Checkpoints for Natural Killer Cell-Based Cancer Immunotherapy. Cancers 2019, 11, 877. [Google Scholar] [CrossRef] [Green Version]
- Quek, L.; Ferguson, P.; Metzner, M.; Ahmed, I.; Kennedy, A.; Garnett, C.; Jeffries, S.; Walter, C.; Piechocki, K.; Timbs, A.; et al. Mutational analysis of disease relapse in patients allografted for acute myeloid leukemia. Blood Adv. 2016, 1, 193–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindsley, R.C.; Saber, W.; Mar, B.G.; Redd, R.; Wang, T.; Haagenson, M.D.; Grauman, P.V.; Hu, Z.H.; Spellman, S.R.; Lee, S.J.; et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N. Engl. J. Med. 2017, 376, 536–547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munn, D.H.; Sharma, M.D.; Baban, B.; Harding, H.P.; Zhang, Y.; Ron, D.; Mellor, A.L. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity 2005, 22, 633–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Folgiero, V.; Goffredo, B.M.; Filippini, P.; Masetti, R.; Bonanno, G.; Caruso, R.; Bertaina, V.; Mastronuzzi, A.; Gaspari, S.; Zecca, M.; et al. Indoleamine 2,3-dioxygenase 1 (IDO1) activity in leukemia blasts correlates with poor outcome in childhood acute myeloid leukemia. Oncotarget 2014, 5, 2052–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulphy, N.; Henry, G.; Hemon, P.; Khaznadar, Z.; Dombret, H.; Boissel, N.; Bensussan, A.; Toubert, A. Contribution of CD39 to the immunosuppressive microenvironment of acute myeloid leukaemia at diagnosis. Br. J. Haematol. 2014, 165, 722–725. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; Chernogorova, P.; Ayata, K.; Gerlach, U.V.; Rughani, A.; Ritchey, J.W.; Ganesan, J.; Follo, M.; Zeiser, R.; Thompson, L.F.; et al. Deficiency of CD73/ecto-5′-nucleotidase in mice enhances acute graft-versus-host disease. Blood 2012, 119, 4554–4564. [Google Scholar] [CrossRef] [Green Version]
- Mussai, F.; De Santo, C.; Abu-Dayyeh, I.; Booth, S.; Quek, L.; McEwen-Smith, R.M.; Qureshi, A.; Dazzi, F.; Vyas, P.; Cerundolo, V. Acute myeloid leukemia creates an arginase-dependent immunosuppressive microenvironment. Blood 2013, 122, 749–758. [Google Scholar] [CrossRef] [Green Version]
- Orleans-Lindsay, J.K.; Barber, L.D.; Prentice, H.G.; Lowdell, M.W. Acute myeloid leukaemia cells secrete a soluble factor that inhibits T and NK cell proliferation but not cytolytic function--implications for the adoptive immunotherapy of leukaemia. Clin. Exp. Immunol. 2001, 126, 403–411. [Google Scholar] [CrossRef]
- Buggins, A.G.; Milojkovic, D.; Arno, M.J.; Lea, N.C.; Mufti, G.J.; Thomas, N.S.; Hirst, W.J. Microenvironment produced by acute myeloid leukemia cells prevents T cell activation and proliferation by inhibition of NF-kappaB, c-Myc, and pRb pathways. J. Immunol. 2001, 167, 6021–6030. [Google Scholar] [CrossRef] [Green Version]
- Carreras, E. The EBMT Handbook; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Olavarria, E.; Siddique, S.; Griffiths, M.J.; Avery, S.; Byrne, J.L.; Piper, K.P.; Lennard, A.L.; Pallan, L.; Arrazi, J.M.; Perz, J.B.; et al. Posttransplantation imatinib as a strategy to postpone the requirement for immunotherapy in patients undergoing reduced-intensity allografts for chronic myeloid leukemia. Blood 2007, 110, 4614–4617. [Google Scholar] [CrossRef] [Green Version]
- Pfeifer, H.; Wassmann, B.; Bethge, W.; Dengler, J.; Bornhauser, M.; Stadler, M.; Beelen, D.; Vucinic, V.; Burmeister, T.; Stelljes, M.; et al. Randomized comparison of prophylactic and minimal residual disease-triggered imatinib after allogeneic stem cell transplantation for BCR-ABL1-positive acute lymphoblastic leukemia. Leukemia 2013, 27, 1254–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giebel, S.; Czyz, A.; Ottmann, O.; Baron, F.; Brissot, E.; Ciceri, F.; Cornelissen, J.J.; Esteve, J.; Gorin, N.C.; Savani, B.; et al. Use of tyrosine kinase inhibitors to prevent relapse after allogeneic hematopoietic stem cell transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia: A position statement of the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Cancer 2016, 122, 2941–2951. [Google Scholar] [CrossRef] [PubMed]
- Schlenk, R.F.; Weber, D.; Fiedler, W.; Salih, H.R.; Wulf, G.; Salwender, H.; Schroeder, T.; Kindler, T.; Lubbert, M.; Wolf, D.; et al. Midostaurin added to chemotherapy and continued single-agent maintenance therapy in acute myeloid leukemia with FLT3-ITD. Blood 2019, 133, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Hambach, L.; Ling, K.W.; Pool, J.; Aghai, Z.; Blokland, E.; Tanke, H.J.; Bruijn, J.A.; Halfwerk, H.; van Boven, H.; Wieles, B.; et al. Hypomethylating drugs convert HA-1-negative solid tumors into targets for stem cell-based immunotherapy. Blood 2009, 113, 2715–2722. [Google Scholar] [CrossRef]
- Goodyear, O.; Agathanggelou, A.; Novitzky-Basso, I.; Siddique, S.; McSkeane, T.; Ryan, G.; Vyas, P.; Cavenagh, J.; Stankovic, T.; Moss, P.; et al. Induction of a CD8+ T-cell response to the MAGE cancer testis antigen by combined treatment with azacitidine and sodium valproate in patients with acute myeloid leukemia and myelodysplasia. Blood 2010, 116, 1908–1918. [Google Scholar] [CrossRef] [Green Version]
- Schroeder, T.; Frobel, J.; Cadeddu, R.P.; Czibere, A.; Dienst, A.; Platzbecker, U.; Bug, G.; Uharek, L.; Fenk, R.; Germing, U.; et al. Salvage therapy with azacitidine increases regulatory T cells in peripheral blood of patients with AML or MDS and early relapse after allogeneic blood stem cell transplantation. Leukemia 2013, 27, 1910–1913. [Google Scholar] [CrossRef] [Green Version]
- Czibere, A.; Bruns, I.; Kroger, N.; Platzbecker, U.; Lind, J.; Zohren, F.; Fenk, R.; Germing, U.; Schroder, T.; Graf, T.; et al. 5-Azacytidine for the treatment of patients with acute myeloid leukemia or myelodysplastic syndrome who relapse after allo-SCT: A retrospective analysis. Bone Marrow Transplant. 2010, 45, 872–876. [Google Scholar] [CrossRef] [Green Version]
- Graef, T.; Kuendgen, A.; Fenk, R.; Zohren, F.; Haas, R.; Kobbe, G. Successful treatment of relapsed AML after allogeneic stem cell transplantation with azacitidine. Leuk. Res. 2007, 31, 257–259. [Google Scholar] [CrossRef]
- Schroeder, T.; Czibere, A.; Platzbecker, U.; Bug, G.; Uharek, L.; Luft, T.; Giagounidis, A.; Zohren, F.; Bruns, I.; Wolschke, C.; et al. Azacitidine and donor lymphocyte infusions as first salvage therapy for relapse of AML or MDS after allogeneic stem cell transplantation. Leukemia 2013, 27, 1229–1235. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Abarca, L.I.; Gutierrez-Cosio, S.; Santamaria, C.; Caballero-Velazquez, T.; Blanco, B.; Herrero-Sanchez, C.; Garcia, J.L.; Carrancio, S.; Hernandez-Campo, P.; Gonzalez, F.J.; et al. Immunomodulatory effect of 5-azacytidine (5-azaC): Potential role in the transplantation setting. Blood 2010, 115, 107–121. [Google Scholar] [CrossRef] [Green Version]
- Edinger, M.; Hoffmann, P.; Ermann, J.; Drago, K.; Fathman, C.G.; Strober, S.; Negrin, R.S. CD4+CD25+ regulatory T cells preserve graft-versus-tumor activity while inhibiting graft-versus-host disease after bone marrow transplantation. Nat. Med. 2003, 9, 1144–1150. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ritchey, J.; Prior, J.L.; Holt, M.; Shannon, W.D.; Deych, E.; Piwnica-Worms, D.R.; DiPersio, J.F. In vivo administration of hypomethylating agents mitigate graft-versus-host disease without sacrificing graft-versus-leukemia. Blood 2010, 116, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Platzbecker, U.; Wermke, M.; Radke, J.; Oelschlaegel, U.; Seltmann, F.; Kiani, A.; Klut, I.M.; Knoth, H.; Rollig, C.; Schetelig, J.; et al. Azacitidine for treatment of imminent relapse in MDS or AML patients after allogeneic HSCT: Results of the RELAZA trial. Leukemia 2012, 26, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Stahl, M.; Kohrman, N.; Gore, S.D.; Kim, T.K.; Zeidan, A.M.; Prebet, T. Epigenetics in Cancer: A Hematological Perspective. PLoS Genet. 2016, 12, e1006193. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Basu, S.; Garcia-Manero, G.; Cortes, J.E.; Ravandi, F.; Jabbour, E.; Hendrickson, S.; Brandt, M.; Pierce, S.; Gordon, T.; et al. Phase IB/II study of nivolumab with azacytidine (AZA) in patients (pts) with relapsed AML. J. Clin. Oncol. 2017, 35, 7026. [Google Scholar] [CrossRef]
- Choi, S.W.; Braun, T.; Chang, L.; Ferrara, J.L.; Pawarode, A.; Magenau, J.M.; Hou, G.; Beumer, J.H.; Levine, J.E.; Goldstein, S.; et al. Vorinostat plus tacrolimus and mycophenolate to prevent graft-versus-host disease after related-donor reduced-intensity conditioning allogeneic haemopoietic stem-cell transplantation: A phase 1/2 trial. Lancet Oncol. 2014, 15, 87–95. [Google Scholar] [CrossRef] [Green Version]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Deng, R.; Fan, F.Y.; Yi, H.; Liu, F.; He, G.C.; Sun, H.P.; Su, Y. PD-1 blockade potentially enhances adoptive cytotoxic T cell potency in a human acute myeloid leukaemia animal model. Hematology 2018, 23, 740–746. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin’s lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: A multicentre, multicohort, single-arm phase 2 trial. Lancet Oncol. 2016, 17, 1283–1294. [Google Scholar] [CrossRef] [Green Version]
- Haverkos, B.M.; Abbott, D.; Hamadani, M.; Armand, P.; Flowers, M.E.; Merryman, R.; Kamdar, M.; Kanate, A.S.; Saad, A.; Mehta, A.; et al. PD-1 blockade for relapsed lymphoma post-allogeneic hematopoietic cell transplant: High response rate but frequent GVHD. Blood 2017, 130, 221–228. [Google Scholar] [CrossRef]
- Davids, M.S.; Kim, H.T.; Bachireddy, P.; Costello, C.; Liguori, R.; Savell, A.; Lukez, A.P.; Avigan, D.; Chen, Y.B.; McSweeney, P.; et al. Ipilimumab for Patients with Relapse after Allogeneic Transplantation. N. Engl. J. Med. 2016, 375, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.G.; Garcia-Manero, G.; Basu, S.; Cortes, J.E.; Ravandi, F.; Kadia, T.M.; Konopleva, M.Y.; Jabbour, E.J.; DiNardo, C.D.; Assi, R.; et al. Safety, efficacy, and biomarkers of response to azacitidine (AZA) with nivolumab (Nivo) and AZA with nivo and ipilimumab (Ipi) in relapsed/refractory acute myeloid leukemia: A non-randomized, phase 2 study. Medicine 2018. [Google Scholar] [CrossRef]
- Zeidner, J.F.; Vincent, B.G.; Ivanova, A.; Foster, M.; Coombs, C.C.; Jamieson, K.; Van Deventer, H.; Scibilia, R.; Blanchard, L.; Frank, C.; et al. Phase II study of high dose cytarabine followed by pembrolizumab in relapsed/refractory acute myeloid leukemia (AML). Blood 2017, 130, 1349. [Google Scholar]
- Weiden, P.L.; Sullivan, K.M.; Flournoy, N.; Storb, R.; Thomas, E.D. Antileukemic effect of chronic graft-versus-host disease: Contribution to improved survival after allogeneic marrow transplantation. N. Engl. J. Med. 1981, 304, 1529–1533. [Google Scholar] [CrossRef] [Green Version]
- Peggs, K.S.; Mackinnon, S. Cellular therapy: Donor lymphocyte infusion. Curr. Opin. Hematol. 2001, 8, 349–354. [Google Scholar] [CrossRef]
- Stern, M.; de Wreede, L.C.; Brand, R.; van Biezen, A.; Dreger, P.; Mohty, M.; de Witte, T.M.; Kroger, N.; Ruutu, T. Sensitivity of hematological malignancies to graft-versus-host effects: An EBMT megafile analysis. Leukemia 2014, 28, 2235–2240. [Google Scholar] [CrossRef]
- Szczepanski, M.J.; Szajnik, M.; Czystowska, M.; Mandapathil, M.; Strauss, L.; Welsh, A.; Foon, K.A.; Whiteside, T.L.; Boyiadzis, M. Increased frequency and suppression by regulatory T cells in patients with acute myelogenous leukemia. Clin. Cancer Res. 2009, 15, 3325–3332. [Google Scholar] [CrossRef] [Green Version]
- Khaznadar, Z.; Boissel, N.; Agaugue, S.; Henry, G.; Cheok, M.; Vignon, M.; Geromin, D.; Cayuela, J.M.; Castaigne, S.; Pautas, C.; et al. Defective NK Cells in Acute Myeloid Leukemia Patients at Diagnosis Are Associated with Blast Transcriptional Signatures of Immune Evasion. J. Immunol. 2015, 195, 2580–2590. [Google Scholar] [CrossRef] [Green Version]
- Ruggeri, L.; Capanni, M.; Urbani, E.; Perruccio, K.; Shlomchik, W.D.; Tosti, A.; Posati, S.; Rogaia, D.; Frassoni, F.; Aversa, F.; et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science 2002, 295, 2097–2100. [Google Scholar] [CrossRef] [Green Version]
- Toprak, S.K. Donor lymphocyte infusion in myeloid disorders. Transfus. Apher. Sci. 2018, 57, 178–186. [Google Scholar] [CrossRef]
- Marks, D.I.; Lush, R.; Cavenagh, J.; Milligan, D.W.; Schey, S.; Parker, A.; Clark, F.J.; Hunt, L.; Yin, J.; Fuller, S.; et al. The toxicity and efficacy of donor lymphocyte infusions given after reduced-intensity conditioning allogeneic stem cell transplantation. Blood 2002, 100, 3108–3114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeidan, A.M.; Forde, P.M.; Symons, H.; Chen, A.; Smith, B.D.; Pratz, K.; Carraway, H.; Gladstone, D.E.; Fuchs, E.J.; Luznik, L.; et al. HLA-haploidentical donor lymphocyte infusions for patients with relapsed hematologic malignancies after related HLA-haploidentical bone marrow transplantation. Biol. Blood Marrow Transplant. 2014, 20, 314–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, P.; Potter, V.T.; Barber, L.D.; Kulasekararaj, A.G.; Lim, Z.Y.; Pearce, R.M.; de Lavallade, H.; Kenyon, M.; Ireland, R.M.; Marsh, J.C.; et al. Outcome of donor lymphocyte infusion after T cell-depleted allogeneic hematopoietic stem cell transplantation for acute myelogenous leukemia and myelodysplastic syndromes. Biol. Blood Marrow Transplant. 2013, 19, 562–568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jedlickova, Z.; Schmid, C.; Koenecke, C.; Hertenstein, B.; Baurmann, H.; Schwerdtfeger, R.; Tischer, J.; Kolb, H.J.; Schleuning, M. Long-term results of adjuvant donor lymphocyte transfusion in AML after allogeneic stem cell transplantation. Bone Marrow Transplant. 2016, 51, 663–667. [Google Scholar] [CrossRef] [PubMed]
- Hicheri, Y.; Bouchekioua, A.; Hamel, Y.; Henry, A.; Rouard, H.; Pautas, C.; Beaumont, J.L.; Kuentz, M.; Cordonnier, C.; Cohen, J.L.; et al. Donor regulatory T cells identified by FoxP3 expression but also by the membranous CD4+CD127low/neg phenotype influence graft-versus-tumor effect after donor lymphocyte infusion. J. Immunother. 2008, 31, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Maury, S.; Aljurf, M. Management of adult patients older than 40 years refractory to at least one immunosuppressive course: HLA-identical sibling HSCT using fludarabine-based conditioning. Bone Marrow Transplant. 2013, 48, 196–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantoff, P.W.; Higano, C.S.; Shore, N.D.; Berger, E.R.; Small, E.J.; Penson, D.F.; Redfern, C.H.; Ferrari, A.C.; Dreicer, R.; Sims, R.B.; et al. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 2010, 363, 411–422. [Google Scholar] [CrossRef] [Green Version]
- Kalinski, P.; Okada, H. Polarized dendritic cells as cancer vaccines: Directing effector-type T cells to tumors. Semin. Immunol. 2010, 22, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.; Schildknecht, A.; Nguyen, L.T.; Ohashi, P.S. Dendritic cells integrate signals from the tumor microenvironment to modulate immunity and tumor growth. Immunol. Lett. 2010, 127, 77–84. [Google Scholar] [CrossRef]
- Palucka, K.; Banchereau, J. Cancer immunotherapy via dendritic cells. Nat. Rev. Cancer 2012, 12, 265–277. [Google Scholar] [CrossRef]
- Anguille, S.; Smits, E.L.; Lion, E.; van Tendeloo, V.F.; Berneman, Z.N. Clinical use of dendritic cells for cancer therapy. Lancet Oncol. 2014, 15, e257–e267. [Google Scholar] [CrossRef]
- Zeng, J.; Wu, C.; Wang, S. Antigenically Modified Human Pluripotent Stem Cells Generate Antigen-Presenting Dendritic Cells. Sci. Rep. 2015, 5, 15262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palucka, K.; Banchereau, J. Dendritic-cell-based therapeutic cancer vaccines. Immunity 2013, 39, 38–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.; Li, H.; Qian, J.; Yang, J.; Lu, Y.; Yi, Q. Optimizing dendritic cell vaccine for immunotherapy in multiple myeloma: Tumour lysates are more potent tumour antigens than idiotype protein to promote anti-tumour immunity. Clin. Exp. Immunol. 2012, 170, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Koido, S.; Homma, S.; Okamoto, M.; Namiki, Y.; Takakura, K.; Uchiyama, K.; Kajihara, M.; Arihiro, S.; Imazu, H.; Arakawa, H.; et al. Fusions between dendritic cells and whole tumor cells as anticancer vaccines. Oncoimmunology 2013, 2, e24437. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.L.; Coukos, G.; Kandalaft, L.E. Whole Tumor Antigen Vaccines: Where Are We? Vaccines 2015, 3, 344–372. [Google Scholar] [CrossRef] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Herblot, S.; Paquin, V.; Cordeiro, P.; Duval, M. Adoptive Transfers of Plasmacytoid Dendritic Cells after Hematopoietic Stem Cell Transplantation Decrease the Risk Acute Lymphoblastic Leukemia Relapse without Increasing the Risk of Graft-Versus-Host-Disease. Blood 2018, 132, 2053. [Google Scholar] [CrossRef]
- Van de Loosdrecht, A.A.; van Wetering, S.; Santegoets, S.; Singh, S.K.; Eeltink, C.M.; den Hartog, Y.; Koppes, M.; Kaspers, J.; Ossenkoppele, G.J.; Kruisbeek, A.M.; et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol. Immunother. 2018, 67, 1505–1518. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.; Stone, R.M.; Uhl, L.; Neuberg, D.; Joyce, R.; Levine, J.D.; Arnason, J.; McMasters, M.; Luptakova, K.; Jain, S.; et al. Individualized vaccination of AML patients in remission is associated with induction of antileukemia immunity and prolonged remissions. Sci. Transl. Med. 2016, 8, 368ra171. [Google Scholar] [CrossRef] [Green Version]
- Anguille, S.; Van de Velde, A.L.; Smits, E.L.; Van Tendeloo, V.F.; Juliusson, G.; Cools, N.; Nijs, G.; Stein, B.; Lion, E.; Van Driessche, A.; et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood 2017, 130, 1713–1721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Huang, X.F.; Hong, B.; Song, X.T.; Hu, L.; Jiang, M.; Zhang, B.; Ning, H.; Li, Y.; Xu, C.; et al. Efficacy of intracellular immune checkpoint-silenced DC vaccine. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawlowska, A.B.; Hashino, S.; McKenna, H.; Weigel, B.J.; Taylor, P.A.; Blazar, B.R. In vitro tumor-pulsed or in vivo Flt3 ligand-generated dendritic cells provide protection against acute myelogenous leukemia in nontransplanted or syngeneic bone marrow-transplanted mice. Blood 2001, 97, 1474–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, M.A.; Fehniger, T.A.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Cooper, M.A.; Fehniger, T.A.; Turner, S.C.; Chen, K.S.; Ghaheri, B.A.; Ghayur, T.; Carson, W.E.; Caligiuri, M.A. Human natural killer cells: A unique innate immunoregulatory role for the CD56(bright) subset. Blood 2001, 97, 3146–3151. [Google Scholar] [CrossRef] [Green Version]
- Campbell, J.J.; Hedrick, J.; Zlotnik, A.; Siani, M.A.; Thompson, D.A.; Butcher, E.C. Chemokines and the arrest of lymphocytes rolling under flow conditions. Science 1998, 279, 381–384. [Google Scholar] [CrossRef]
- Chen, J.J.; Yao, P.L.; Yuan, A.; Hong, T.M.; Shun, C.T.; Kuo, M.L.; Lee, Y.C.; Yang, P.C. Up-regulation of tumor interleukin-8 expression by infiltrating macrophages: Its correlation with tumor angiogenesis and patient survival in non-small cell lung cancer. Clin. Cancer Res. 2003, 9, 729–737. [Google Scholar]
- Sutton, A.; Friand, V.; Brule-Donneger, S.; Chaigneau, T.; Ziol, M.; Sainte-Catherine, O.; Poire, A.; Saffar, L.; Kraemer, M.; Vassy, J.; et al. Stromal cell-derived factor-1/chemokine (C-X-C motif) ligand 12 stimulates human hepatoma cell growth, migration, and invasion. Mol. Cancer Res. 2007, 5, 21–33. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, R.; Ghobrial, I.M.; Roodman, G.D. Chemokines in multiple myeloma. Exp. Hematol. 2006, 34, 1289–1295. [Google Scholar] [CrossRef] [Green Version]
- Kalinkovich, A.; Tavor, S.; Avigdor, A.; Kahn, J.; Brill, A.; Petit, I.; Goichberg, P.; Tesio, M.; Netzer, N.; Naparstek, E.; et al. Functional CXCR4-expressing microparticles and SDF-1 correlate with circulating acute myelogenous leukemia cells. Cancer Res. 2006, 66, 11013–11020. [Google Scholar] [CrossRef] [Green Version]
- Farag, S.S.; Fehniger, T.A.; Ruggeri, L.; Velardi, A.; Caligiuri, M.A. Natural killer cell receptors: New biology and insights into the graft-versus-leukemia effect. Blood 2002, 100, 1935–1947. [Google Scholar] [CrossRef] [PubMed]
- Litwin, V.; Gumperz, J.; Parham, P.; Phillips, J.H.; Lanier, L.L. NKB1: A natural killer cell receptor involved in the recognition of polymorphic HLA-B molecules. J. Exp. Med. 1994, 180, 537–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagtmann, N.; Rajagopalan, S.; Winter, C.C.; Peruzzi, M.; Long, E.O. Killer cell inhibitory receptors specific for HLA-C and HLA-B identified by direct binding and by functional transfer. Immunity 1995, 3, 801–809. [Google Scholar] [CrossRef] [Green Version]
- Uhrberg, M.; Valiante, N.M.; Shum, B.P.; Shilling, H.G.; Lienert-Weidenbach, K.; Corliss, B.; Tyan, D.; Lanier, L.L.; Parham, P. Human diversity in killer cell inhibitory receptor genes. Immunity 1997, 7, 753–763. [Google Scholar] [CrossRef] [Green Version]
- Klingemann, H.G. Natural killer cell-based immunotherapeutic strategies. Cytotherapy 2005, 7, 16–22. [Google Scholar] [CrossRef]
- Kim, D.H.; Sohn, S.K.; Lee, N.Y.; Baek, J.H.; Kim, J.G.; Won, D.I.; Suh, J.S.; Lee, K.B.; Shin, I.H. Transplantation with higher dose of natural killer cells associated with better outcomes in terms of non-relapse mortality and infectious events after allogeneic peripheral blood stem cell transplantation from HLA-matched sibling donors. Eur. J. Haematol. 2005, 75, 299–308. [Google Scholar] [CrossRef]
- Miller, J.S.; Soignier, Y.; Panoskaltsis-Mortari, A.; McNearney, S.A.; Yun, G.H.; Fautsch, S.K.; McKenna, D.; Le, C.; Defor, T.E.; Burns, L.J.; et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood 2005, 105, 3051–3057. [Google Scholar] [CrossRef] [Green Version]
- Farag, S.S.; George, S.L.; Lee, E.J.; Baer, M.; Dodge, R.K.; Becknell, B.; Fehniger, T.A.; Silverman, L.R.; Crawford, J.; Bloomfield, C.D.; et al. Postremission therapy with low-dose interleukin 2 with or without intermediate pulse dose interleukin 2 therapy is well tolerated in elderly patients with acute myeloid leukemia: Cancer and Leukemia Group B study 9420. Clin. Cancer Res. 2002, 8, 2812–2819. [Google Scholar]
- Bernstein, Z.P.; Porter, M.M.; Gould, M.; Lipman, B.; Bluman, E.M.; Stewart, C.C.; Hewitt, R.G.; Fyfe, G.; Poiesz, B.; Caligiuri, M.A. Prolonged administration of low-dose interleukin-2 in human immunodeficiency virus-associated malignancy results in selective expansion of innate immune effectors without significant clinical toxicity. Blood 1995, 86, 3287–3294. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.D.; Emmanouilides, C.; Benson, D.M., Jr.; Hurst, D.; Garcia, P.; Michelson, G.; Milan, S.; Ferketich, A.K.; Piro, L.; Leonard, J.P.; et al. A phase 2 study of rituximab in combination with recombinant interleukin-2 for rituximab-refractory indolent non-Hodgkin’s lymphoma. Clin. Cancer Res. 2006, 12, 7046–7053. [Google Scholar] [CrossRef] [Green Version]
- Carson, W.E.; Parihar, R.; Lindemann, M.J.; Personeni, N.; Dierksheide, J.; Meropol, N.J.; Baselga, J.; Caligiuri, M.A. Interleukin-2 enhances the natural killer cell response to Herceptin-coated Her2/neu-positive breast cancer cells. Eur. J. Immunol. 2001, 31, 3016–3025. [Google Scholar] [CrossRef]
- Ansell, S.M.; Witzig, T.E.; Kurtin, P.J.; Sloan, J.A.; Jelinek, D.F.; Howell, K.G.; Markovic, S.N.; Habermann, T.M.; Klee, G.G.; Atherton, P.J.; et al. Phase 1 study of interleukin-12 in combination with rituximab in patients with B-cell non-Hodgkin lymphoma. Blood 2002, 99, 67–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sentman, C.L.; Barber, M.A.; Barber, A.; Zhang, T. NK cell receptors as tools in cancer immunotherapy. Adv. Cancer Res. 2006, 95, 249–292. [Google Scholar] [CrossRef] [PubMed]
- Bellucci, R.; Alyea, E.P.; Chiaretti, S.; Wu, C.J.; Zorn, E.; Weller, E.; Wu, B.; Canning, C.; Schlossman, R.; Munshi, N.C.; et al. Graft-versus-tumor response in patients with multiple myeloma is associated with antibody response to BCMA, a plasma-cell membrane receptor. Blood 2005, 105, 3945–3950. [Google Scholar] [CrossRef] [PubMed]
- Gleason, M.K.; Ross, J.A.; Warlick, E.D.; Lund, T.C.; Verneris, M.R.; Wiernik, A.; Spellman, S.; Haagenson, M.D.; Lenvik, A.J.; Litzow, M.R.; et al. CD16 × CD33 bispecific killer cell engager (BiKE) activates NK cells against primary MDS and MDSC CD33+ targets. Blood 2014, 123, 3016–3026. [Google Scholar] [CrossRef] [PubMed]
- Wiernik, A.; Foley, B.; Zhang, B.; Verneris, M.R.; Warlick, E.; Gleason, M.K.; Ross, J.A.; Luo, X.; Weisdorf, D.J.; Walcheck, B.; et al. Targeting natural killer cells to acute myeloid leukemia in vitro with a CD16 x 33 bispecific killer cell engager and ADAM17 inhibition. Clin. Cancer Res. 2013, 19, 3844–3855. [Google Scholar] [CrossRef] [Green Version]
- Laing, A.A.; Harrison, C.J.; Gibson, B.E.S.; Keeshan, K. Unlocking the potential of anti-CD33 therapy in adult and childhood acute myeloid leukemia. Exp. Hematol. 2017, 54, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef]
- Kenderian, S.S.; Ruella, M.; Shestova, O.; Klichinsky, M.; Aikawa, V.; Morrissette, J.J.; Scholler, J.; Song, D.; Porter, D.L.; Carroll, M.; et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia 2015, 29, 1637–1647. [Google Scholar] [CrossRef]
- Wang, Q.S.; Wang, Y.; Lv, H.Y.; Han, Q.W.; Fan, H.; Guo, B.; Wang, L.L.; Han, W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015, 23, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Brinkman-Van der Linden, E.C.; Angata, T.; Reynolds, S.A.; Powell, L.D.; Hedrick, S.M.; Varki, A. CD33/Siglec-3 binding specificity, expression pattern, and consequences of gene deletion in mice. Mol. Cell Biol. 2003, 23, 4199–4206. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.Y.; Yu, K.R.; Kenderian, S.S.; Ruella, M.; Chen, S.; Shin, T.H.; Aljanahi, A.A.; Schreeder, D.; Klichinsky, M.; Shestova, O.; et al. Genetic Inactivation of CD33 in Hematopoietic Stem Cells to Enable CAR T Cell Immunotherapy for Acute Myeloid Leukemia. Cell 2018, 173, 1439–1453.e1419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borot, F.; Wang, H.; Ma, Y.; Jafarov, T.; Raza, A.; Ali, A.M.; Mukherjee, S. Gene-edited stem cells enable CD33-directed immune therapy for myeloid malignancies. Proc. Natl. Acad. Sci. USA 2019, 116, 11978–11987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humbert, O.; Laszlo, G.S.; Sichel, S.; Ironside, C.; Haworth, K.G.; Bates, O.M.; Beddoe, M.E.; Carrillo, R.R.; Kiem, H.P.; Walter, R.B. Engineering resistance to CD33-targeted immunotherapy in normal hematopoiesis by CRISPR/Cas9-deletion of CD33 exon 2. Leukemia 2019, 33, 762–808. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Lee, E.M.; Ramshaw, H.S.; Busfield, S.J.; Peoppl, A.G.; Wilkinson, L.; Guthridge, M.A.; Thomas, D.; Barry, E.F.; Boyd, A.; et al. Monoclonal antibody-mediated targeting of CD123, IL-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell 2009, 5, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Stevens, B.M.; Zhang, W.; Pollyea, D.A.; Winters, A.; Gutman, J.; Smith, C.; Budde, E.; Forman, S.J.; Jordan, C.T.; Purev, E. CD123 CAR T cells for the treatment of myelodysplastic syndrome. Exp. Hematol. 2019, 74, 52–63.e53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Xu, Y.; Li, S.; Liu, J.; Xing, Y.; Xing, H.; Tian, Z.; Tang, K.; Rao, Q.; Wang, M.; et al. Targeting FLT3 in acute myeloid leukemia using ligand-based chimeric antigen receptor-engineered T cells. J. Hematol. Oncol. 2018, 11, 60. [Google Scholar] [CrossRef] [Green Version]
- Jetani, H.; Garcia-Cadenas, I.; Nerreter, T.; Thomas, S.; Rydzek, J.; Meijide, J.B.; Bonig, H.; Herr, W.; Sierra, J.; Einsele, H.; et al. CAR T-cells targeting FLT3 have potent activity against FLT3(-)ITD(+) AML and act synergistically with the FLT3-inhibitor crenolanib. Leukemia 2018, 32, 1168–1179. [Google Scholar] [CrossRef]
- Neu, S.; Geiselhart, A.; Sproll, M.; Hahn, D.; Kuci, S.; Niethammer, D.; Handgretinger, R. Expression of CD44 isoforms by highly enriched CD34-positive cells in cord blood, bone marrow and leukaphereses. Bone Marrow Transplant. 1997, 20, 593–598. [Google Scholar] [CrossRef] [Green Version]
- Legras, S.; Gunthert, U.; Stauder, R.; Curt, F.; Oliferenko, S.; Kluin-Nelemans, H.C.; Marie, J.P.; Proctor, S.; Jasmin, C.; Smadja-Joffe, F. A strong expression of CD44-6v correlates with shorter survival of patients with acute myeloid leukemia. Blood 1998, 91, 3401–3413. [Google Scholar] [CrossRef]
- Casucci, M.; Nicolis di Robilant, B.; Falcone, L.; Camisa, B.; Norelli, M.; Genovese, P.; Gentner, B.; Gullotta, F.; Ponzoni, M.; Bernardi, M.; et al. CD44v6-targeted T cells mediate potent antitumor effects against acute myeloid leukemia and multiple myeloma. Blood 2013, 122, 3461–3472. [Google Scholar] [CrossRef] [PubMed]
- Ciceri, F.; Bonini, C.; Stanghellini, M.T.; Bondanza, A.; Traversari, C.; Salomoni, M.; Turchetto, L.; Colombi, S.; Bernardi, M.; Peccatori, J.; et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): A non-randomised phase I-II study. Lancet Oncol. 2009, 10, 489–500. [Google Scholar] [CrossRef]
- Di Stasi, A.; Tey, S.K.; Dotti, G.; Fujita, Y.; Kennedy-Nasser, A.; Martinez, C.; Straathof, K.; Liu, E.; Durett, A.G.; Grilley, B.; et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 2011, 365, 1673–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tashiro, H.; Sauer, T.; Shum, T.; Parikh, K.; Mamonkin, M.; Omer, B.; Rouce, R.H.; Lulla, P.; Rooney, C.M.; Gottschalk, S.; et al. Treatment of Acute Myeloid Leukemia with T Cells Expressing Chimeric Antigen Receptors Directed to C-type Lectin-like Molecule 1. Mol. Ther. 2017, 25, 2202–2213. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Chen, S.; Xiao, W.; Li, W.; Wang, L.; Yang, S.; Wang, W.; Xu, L.; Liao, S.; Liu, W.; et al. CAR-T cells targeting CLL-1 as an approach to treat acute myeloid leukemia. J. Hematol. Oncol. 2018, 11, 7. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Photodynamic treatment (PDT) of the graft consists of a first phase in which donor and patient peripheral blood mononucleated cells are collected by apheresis. Then, patient cells are gamma-irradiated and donor and patient cells are co-cultured to stimulate activation of host alloreactive T-cells. GvHD causing T-cells from the donor are activated by the presence of “foreign” cells from the patient. TH9402, a photosensitizing reagent, is added to the culture and is retained only in activated donor T-cells. This mixture is then exposed to light, which activates TH9402 leading to production of oxygen radicals, resulting in self-destruction of GvHD causing donor T-cells. The remaining mixture (named ATIR101) is then infused into the patient. Color of the nucleus indicates different cell types: grey, irradiated patient cells; green, virus-specific T cells; blue, inactivated GvHD causing donor T cells; red, activated GvHD causing donor T cells; yellow, other immune cells (e.g., NK cells). Cells that retain the TH9402 reagent have orange colored cytoplasm. P: patient, D: donor.
Figure 1.
Photodynamic treatment (PDT) of the graft consists of a first phase in which donor and patient peripheral blood mononucleated cells are collected by apheresis. Then, patient cells are gamma-irradiated and donor and patient cells are co-cultured to stimulate activation of host alloreactive T-cells. GvHD causing T-cells from the donor are activated by the presence of “foreign” cells from the patient. TH9402, a photosensitizing reagent, is added to the culture and is retained only in activated donor T-cells. This mixture is then exposed to light, which activates TH9402 leading to production of oxygen radicals, resulting in self-destruction of GvHD causing donor T-cells. The remaining mixture (named ATIR101) is then infused into the patient. Color of the nucleus indicates different cell types: grey, irradiated patient cells; green, virus-specific T cells; blue, inactivated GvHD causing donor T cells; red, activated GvHD causing donor T cells; yellow, other immune cells (e.g., NK cells). Cells that retain the TH9402 reagent have orange colored cytoplasm. P: patient, D: donor.

Figure 2.
DC-based vaccines exploit the ability of DCs to cross-present TAAs in order to activate a T cell immune response against neoplastic cells. (a) CD34+ progenitors obtained from peripheral blood of the patient are exposed in vitro to a mixture of cytokines and differentiate into DCs. These are loaded with a selected target TAA, activated and re-infused back into the patient. However, the poor immunogenicity of the selected TAA and the ability of tumor cells to actively down regulate specific antigen expression under the selective pressure of monoclonal T cells can hinder an effective antitumor response. (b) DCs are activated using entire tumor cells: these DCs, when activated and re-infused, are able to stimulate a polyclonal and more effective T cell response against tumor cells.
Figure 2.
DC-based vaccines exploit the ability of DCs to cross-present TAAs in order to activate a T cell immune response against neoplastic cells. (a) CD34+ progenitors obtained from peripheral blood of the patient are exposed in vitro to a mixture of cytokines and differentiate into DCs. These are loaded with a selected target TAA, activated and re-infused back into the patient. However, the poor immunogenicity of the selected TAA and the ability of tumor cells to actively down regulate specific antigen expression under the selective pressure of monoclonal T cells can hinder an effective antitumor response. (b) DCs are activated using entire tumor cells: these DCs, when activated and re-infused, are able to stimulate a polyclonal and more effective T cell response against tumor cells.


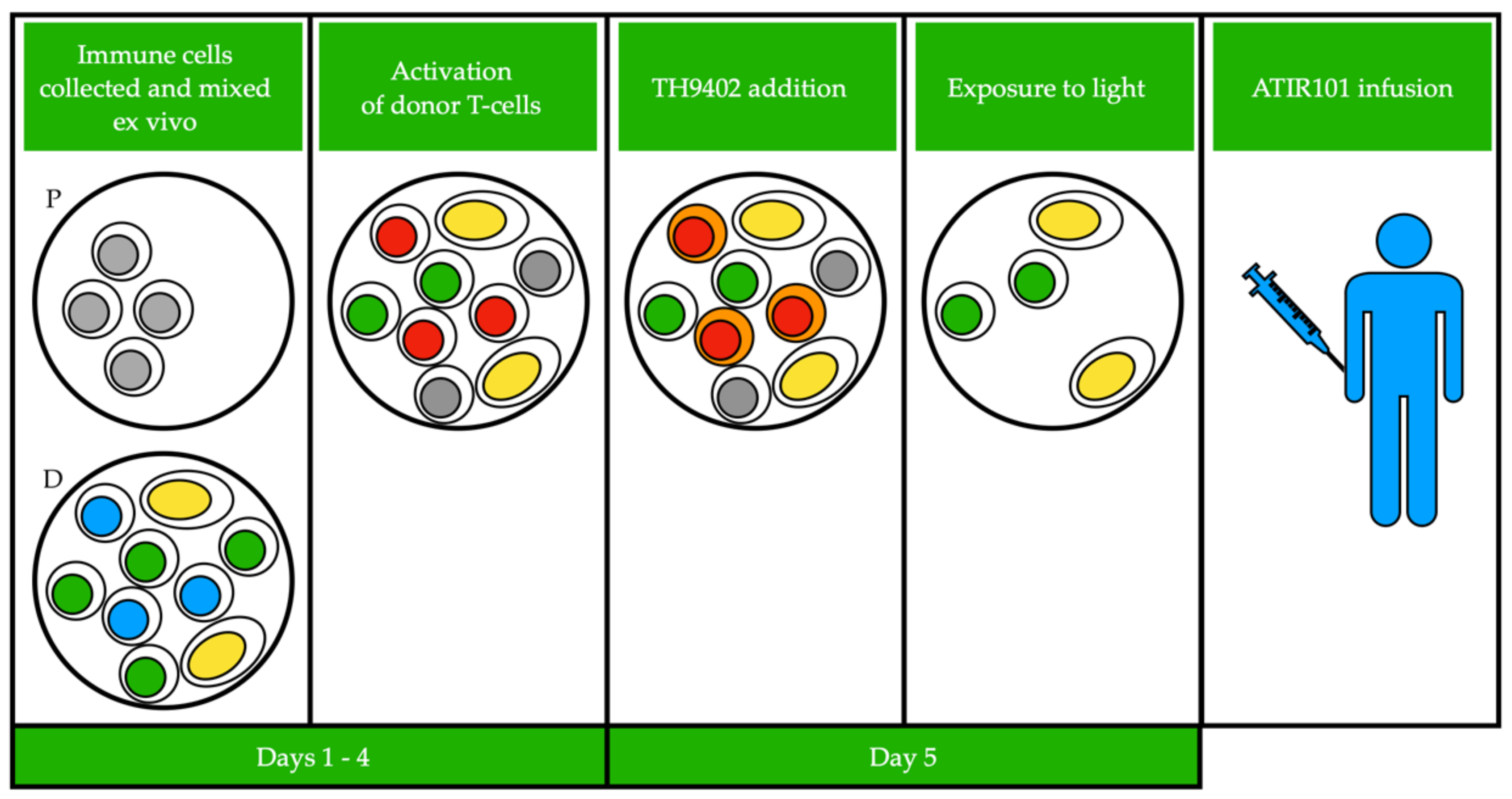{kind=link}
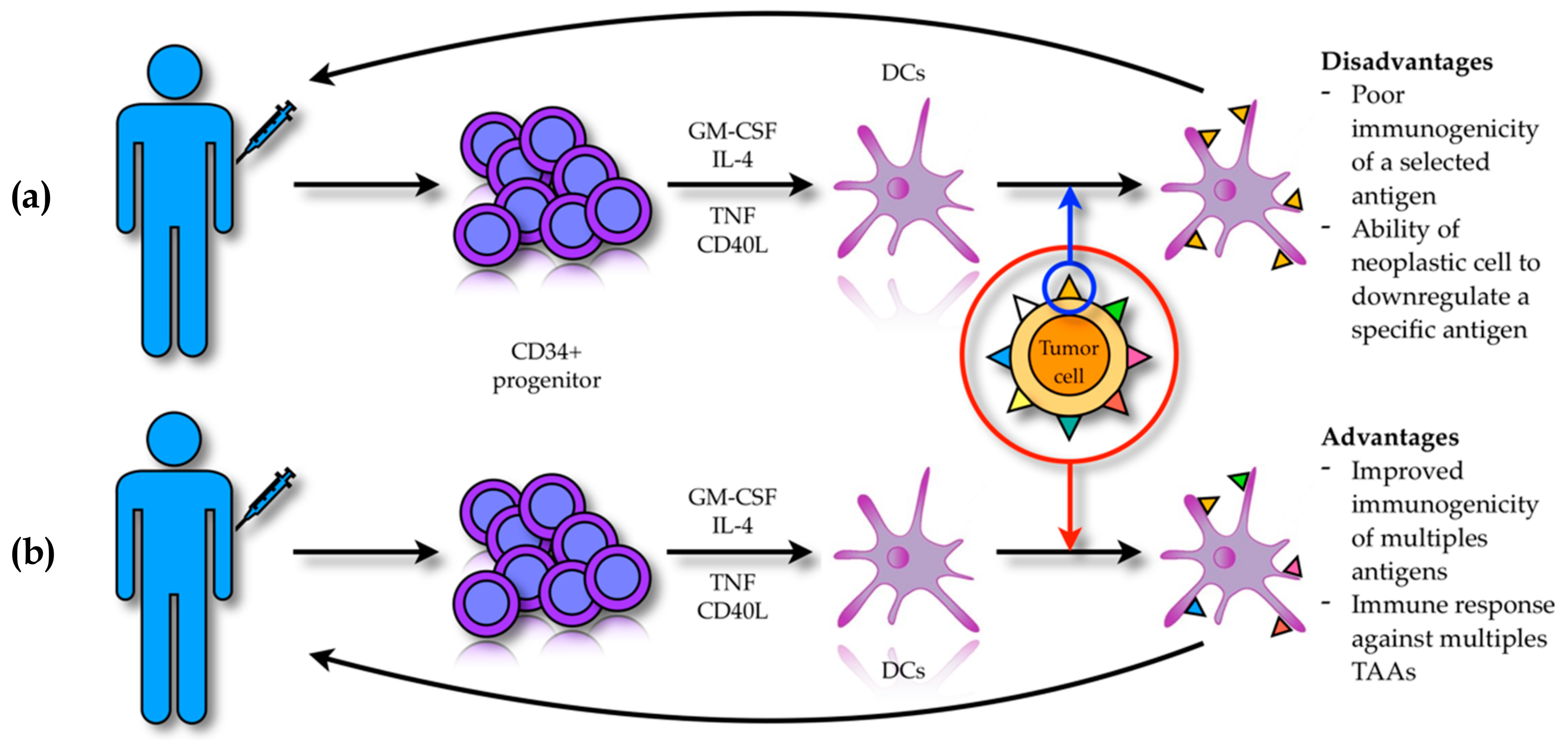{kind=link}
Table 1.
Schematic view of the main drugs and cellular therapies used to prevent and treat relapse after allo-HSCT.
Table 1.
Schematic view of the main drugs and cellular therapies used to prevent and treat relapse after allo-HSCT.
| Therapeutic Strategy | Mechanisms of Action | Examples | |
|---|---|---|---|
| Drugs | TKI |
| imatinib midostaurin quizartinib gilteritinib crenolanib sorafenib |
| HMA |
| azacytidine decitabine | |
| HDACi |
| panobinostat | |
| ICP inhibitors |
| nivolumab ipilimumab pidilizumab | |
| Cellular Therapies | DLI |
| |
| DC infusion |
| Sipuleucel-T | |
| NK cell based therapies |
| ULBP2-BB4 | |
| CAR-T cell based therapies |
| ||
CAR: chimeric antigen receptor, DC: dendritic cells, DLI: donor lymphocyte infusion, HMA: hypomethylating agents, HDACi: inhibitors of histone deacetylase, ICP: immune-checkpoint, NK: natural killer, TAA: tumor associated antigens, TKI: tyrosine kinase inhibitors.
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Bernasconi, P.; Borsani, O. Immune Escape after Hematopoietic Stem Cell Transplantation (HSCT): From Mechanisms to Novel Therapies. Cancers 2020, 12, 69. https://doi.org/10.3390/cancers12010069
AMA Style
Bernasconi P, Borsani O. Immune Escape after Hematopoietic Stem Cell Transplantation (HSCT): From Mechanisms to Novel Therapies. Cancers. 2020; 12(1):69. https://doi.org/10.3390/cancers12010069
Chicago/Turabian StyleBernasconi, Paolo, and Oscar Borsani. 2020. "Immune Escape after Hematopoietic Stem Cell Transplantation (HSCT): From Mechanisms to Novel Therapies" Cancers 12, no. 1: 69. https://doi.org/10.3390/cancers12010069
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.