Systematic Review on the Efficacy and Safety of Oral Janus Kinase Inhibitors for the Treatment of Atopic Dermatitis
 Michelle Le1
Michelle Le1  Melissa Berman-Rosa1
Melissa Berman-Rosa1  Feras M. Ghazawi2 Marc Bourcier3 Loretta Fiorillo4
Feras M. Ghazawi2 Marc Bourcier3 Loretta Fiorillo4  Melinda Gooderham5
Melinda Gooderham5  Lyn Guenther6 Sameh Hanna7 H. Chih-Ho Hong8
Lyn Guenther6 Sameh Hanna7 H. Chih-Ho Hong8  Ian Landells9 Perla Lansang10 Danielle Marcoux11 Marni C. Wiseman12 Jensen Yeung10,13 Charles Lynde7*
Ian Landells9 Perla Lansang10 Danielle Marcoux11 Marni C. Wiseman12 Jensen Yeung10,13 Charles Lynde7*  Ivan V. Litvinov1*
Ivan V. Litvinov1*- 1Division of Dermatology, McGill University Health Centre, Montreal, QC, Canada
- 2Division of Dermatology, University of Ottawa, Ottawa, ON, Canada
- 3Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC, Canada
- 4Division of Pediatric Dermatology, University of Alberta, Edmonton, AB, Canada
- 5SKiN Centre for Dermatology, Probity Medical Research, and Queens University, Peterborough, ON, Canada
- 6Division of Dermatology, University of Western Ontario, London, ON, Canada
- 7Dermatology on Bloor, Toronto, ON, Canada
- 8Department of Dermatology and Skin Sciences, University of British Columbia, Vancouver, BC, Canada
- 9Division of Dermatology, Memorial University of Newfoundland, St. John's, NL, Canada
- 10Division of Dermatology, University of Toronto, Toronto, ON, Canada
- 11Division of Pediatric Dermatology, University of Montreal, Montreal, QC, Canada
- 12Section of Dermatology, Department of Medicine, University of Manitoba, Winnipeg, MB, Canada
- 13Probity Medical Research, Waterloo, ON, Canada
Background: Atopic dermatitis is a chronic, relapsing and remitting disease that can be difficult to treat despite a recently approved biologic therapy targeting IL-4/IL-13 receptor. Oral janus kinase inhibitors (JAKi) represent a novel therapeutic class of targeted therapy to treat moderate-to-severe atopic dermatitis (AD).
Objective: To review the efficacy, safety, and pharmacokinetic characteristics of oral JAKi in the treatment of AD.
Methods: A PRISMA systematic review was conducted using MEDLINE, EMBASE (Ovid), and PubMed databases for studies assessing the efficacy, safety, and/or pharmacokinetic properties of oral forms of JAKi in the treatment of AD in pediatric or adult populations from inception to June 2021.
Results: 496 papers were reviewed. Of 28 articles that underwent full text screening, 11 met our inclusion criteria for final qualitative review. Four studies examined abrocitinib; three studies examined baricitinib; three examined upadacitinib and one examined gusacitinib (ASN002). Significant clinical efficacy and a reassuring safety profile was reported for all JAKi agents reviewed. Rapid symptom control was reported for abrocitinib, baricitinib and upadacitinib.
Limitations: Given the relatively limited evidence for each JAKi and the differences in patient eligibility criteria between studies, the data was not deemed suitable for a meta-analysis at this time.
Conclusion: Given their ability to achieve rapid symptom control with a reassuring safety profile, we recommend considering the use of JAKi as a reliable systemic treatment option for adult patients with moderate-to-severe AD, who are unresponsive to topical or skin directed treatments.
Introduction
Atopic dermatitis (AD), is a chronic and relapsing inflammatory skin condition that affects up to 20% of children and 10% of adults (1). Pruritus is the hallmark of the disease (2); other signs include erythema, scaling, papules, lichenification, excoriations, crusting and vesicles. At times the affected skin can become impetiginized and/or infected with Herpes simplex virus (eczema herpeticum) or molluscum contangiosum (3) leading to increased disease morbidity. Other complications of AD are well-recognized and were reviewed elsewhere (4). In addition to the physical burden, patients with AD have higher rates of psychosocial distress and a reduced quality of life (5–7).
AD is thought to be a multifactorial disease that arises due to both genetic and environmental factors, although the complete pathophysiology has yet to be elucidated (6). Indeed, a meta-analysis of genome-wide association studies, demonstrated that filaggrin, Inteleukin-13 (IL-13), and Ovo Like Transcriptional Repressor 1 (OVOL1) were found to be the most commonly identified genes associated with an elevated risk of acquiring AD (8). Specifically, IL-13 and OVOL1 regulate filaggrin expression, which is essential for skin barrier protection and plays a crucial role in the pathogenesis of AD. Considering the importance of filaggrin, disruption of the epidermal barrier due to genetic or environmental causes is thought to lead to increased trans-epidermal water loss, making the skin more vulnerable to allergens and pathogen penetration. This, in turn, causes inflammation via the release of chemokines by keratinocytes and subsequent inflammatory cell infiltration (9).
Although historically thought to be a type 2 T helper (Th2) cell driven disease, multiple inflammatory pathways with their respective cytokines have been implicated in AD to varying degrees. These pathways include Th2 (IL-4, IL-5, IL-13, IL-31), Th22 (IL-22), with variable Th1 [interferon (IFN)-γ] and Th17/IL-23 related cytokine involvement (10). These inflammatory pathways have provided potential therapeutic targets for the treatment of AD.
The mainstay for AD management involves the treatment of acute skin flares, management of secondary infections, and prevention of recurrences. General daily skin care for AD includes gentle cleansing of skin, restoration of the skin barrier through the regular use of emollients and avoidance of aggravating factors (11). For acute flares, topical corticosteroids (TCS), topical calcineurin inhibitors (TCI), and/or a topical phosphodiesterase-4 (PDE-4) inhibitor are recommended (12). In moderate-to-severe cases of AD, phototherapy and systemic immunosuppressants (corticosteroids, cyclosporine, azathioprine, mycophenolate mofetil, or methotrexate) can be used (12) although none are approved by Health Canada for the treatment of AD, and side effect profiles for certain systemic immunosuppressants can decrease overall adherence to a treatment plan (11, 13). Access to phototherapy is limited for many patients (14). Although multiple therapeutic modalities are available for the treatment of AD, it remains a challenging disease to manage; in a survey conducted by the National Eczema Association, it was reported that 86% of patients were not satisfied with the treatment of AD (15).
Dupilumab, a monoclonal antibody against IL-4 receptor subunit alpha (Rα) has shown efficacy in many patients and was the first approved biologic therapy for AD that revolutionized the treatment landscape (16). However, approximately 4%-14% experience treatment failure with dupilumab due to either AD worsening (in 5%) or side effects such as conjunctivitis (in 3%) and paradoxical facial erythema (17–19). Patients may also discontinue treatment due to a lack of desired response or for other reasons. Therefore, there remains a need for other targeted therapies. Currently, several other monoclonal antibodies are in phase II/III development and/or pending approval for the treatment of AD, including tralokinumab (20) and lebrikizumab (IL-13 receptor signaling inhibitors), nemolizumab (IL-31 receptor signaling inhibitor) (21–23), and etokimab (IL-33 signaling inhibitor) (24). Given the diversity of cytokines implicated in the inflammatory processes of AD, there is a growing interest toward janus kinases inhibitors (JAKi), which could interfere with the signaling of multiple cytokines simultaneously (25).
Janus kinases (JAKs) are signal transduction proteins that are comprised of a family of four proteins: JAK1, JAK2, JAK3, and TYK2 (26). JAKs are recruited to the inflammatory pathways by the binding of cytokines (such as IL-2, IL-4, IL-6, IL-12, IL-21, IL-22, IL-23, or IFN such as IFN- γ) to their cognate receptors that initiate an inflammatory cascade (Figure 1). Recruitment and activation of JAKs results in the phosphorylation of tyrosine residues including residues within the cytokine receptor chains (Figure 1). Consecutively, transcription factors, signal transducer and activator of transcription (STAT) proteins, are recruited and become activated by JAKs phosphorylation. Activated STAT proteins undergo dimerization, which then enables the translocation of these proteins into the nucleus and allows for the transactivation of a broad range of different genes (26, 27). Given that specific JAKs are selectivity activated by different cytokine receptors, this selectivity enables JAKi to demonstrate a defined specificity and different capacities to block cytokine receptor signaling. For instance, while pan-JAKi have a broad inhibitory effect against multiple cytokines, JAKi that selectively target JAK1, JAK2, or TYK2 proteins exclusively, have a more targeted mode of action (28).
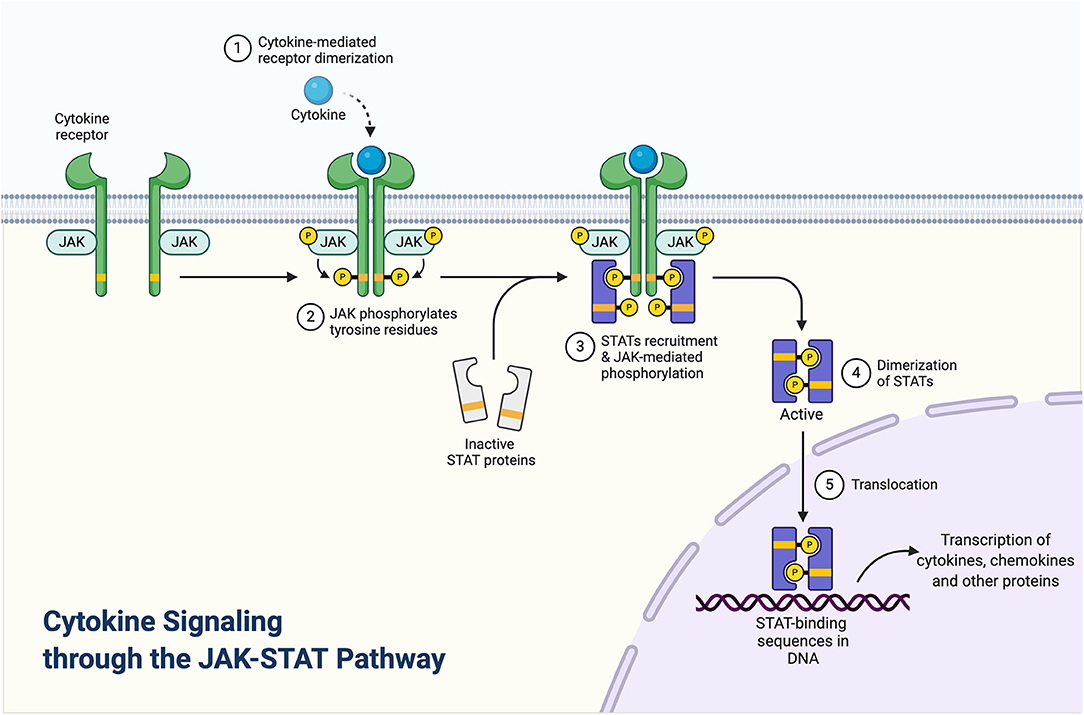
Figure 1. Cytokine signaling through the JAK-STAT pathway. Adapted from “Cytokine signaling through the JAK-STAT pathway,” by BioRender.com (2020). Retrieved from https://app.Biorender.com/biorender-templates.
In this systematic review, we aim to present the current literature on the pharmacokinetics, clinical efficacy and safety of topical and oral JAKi that were recently approved or are currently under investigation for AD.
Methods
Search Strategy
We systematically searched Ovid MEDLINE, EMBASE (Ovid), and PubMed for studies assessing the efficacy, safety, and/or pharmacokinetic properties of oral forms of JAKi in the treatment of AD in pediatric or adult populations from inception to June 2021. We combined free-text search terms for the concept of JAKi (“Janus kinases” OR “JAK inhibitor” or “janus tyrosine kinase inhibitor”) and AD (“atopic dermatitis” or “atopic eczema” or “eczema atopica” or “eczema endogenous” or “eczema infantum” or “eczema, infantile” or “endogenous eczema” or “infantile eczema” or “neurodermatitis constitutionalis” or “neurodermatitis disseminata” or “neurodermatitis, atopic constitutional”). A sample of the search strategy is shown in detail (Supplementary Text 1).
Study Selection
Two researchers (ML and MB-R) independently assessed study eligibility by title and abstract. When a study was deemed potentially eligible for inclusion, the full text article was obtained and assessed by the reviewers independently. Additional reviewer (FG) was consulted when consensus could not be reached.
We restricted our inclusion criteria to randomized-controlled trials (RCTs) that examined the efficacy or safety of JAKi in the treatment AD as measured by changes from baseline in the Eczema Area and Severity Index (EASI), Investigator's Global Assessment (IGA) score, and the peak pruritus numeric rating scale (PP-NRS). Specifically, proportion of patients achieving EASI-75 and EASI-50, defined as a 75 and 50% reduction from baseline in the EASI score, respectively; achieving an IGA score of 0 or 1 (i.e., clear or almost clear) with an improvement of ≥2 grades from baseline (later referred to as an IGA response); and achieving a PP-NRS score improvement of ≥4-point from baseline (later referred to as PP-NRS response), were required as measures of clinical efficacy. Percentage or absolute value changes in these outcomes were accepted. For safety, qualitative reports of adverse events (AEs) and/or side effects were accepted. We set no restrictions on the concentrations or duration of administrations of experimental compounds. As comparators, we accepted any other type of management for AD, including active surveillance.
We accepted studies published in English or French without date restrictions. Non-randomized trials were excluded. Studies were not included if they were only available as abstracts from conference proceedings or if published in a language other than English or French.
Quality Assessment and Data Extraction
Two reviewers (ML and MB-R) independently conducted data extraction and methodology quality assessment for all included studies. We extracted the study type, study time frame, type of population (pediatric vs. adult vs. elderly); method of randomization; whether trial was blinded; sample size; follow-up time; the specific JAKi and comparator treatment employed as well as the dosing and regimens; outcome definitions and method for ascertaining treatment effectiveness; and efficacy. For safety, we extracted the most common and most serious treatment emergent adverse events (TEAEs). As a secondary outcome, we extracted the drug's pharmacokinetic characteristics, when reported.
For all studies, the main measures of interest were the efficacy of JAKi on reducing the severity and extent of involvement of AD compared with any other treatments. If reported, efficacy from intention-to-treat analyses (ITT) was preferred and extracted over per-protocol estimates.
We used the modified Cochrane Collaboration tool to assess risk of bias of RCTs (RoB 2) (29), considered the gold-standard for quality assessment of RCTs (30). The tool is structured into five domains through which bias may be introduced into the results: (1) bias arising from the randomization process; (2) bias due to deviations from intended interventions; (3) bias due to missing outcome data; (4) bias in measurement of outcome(s); and (5) bias in the selection of reported results (31). The overall bias assessment within each domain is characterized as “low risk,” “some concerns,” or “high risk of bias,” according to responses provided to the signaling questions within each domain (Supplementary Table 1).
Data Analysis
We summarized the included reports through descriptive analyses to provide an overview of studies' characteristics, quality, effectiveness, and safety profile of JAKi. Because of the heterogeneity in dosing, length of treatment and length of follow-up, conducting a meta-analysis was not considered. We followed the Preferred Reporting Items for Systematic Reviews and Meta-Analysis (PRISMA) in this systematic review reporting (32).
Results
Study Characteristics
Our initial search yielded a total of 614 studies (Figure 2). After removing duplicates, 496 articles remained and were screened by title and abstract. Of the 29 articles that underwent full text screening, 11 met our inclusion criteria. Of these, all examined the efficacy of various JAKi in the treatment of moderate-to-severe AD in adults and adolescents: four examined abrocitinib; three baricitinib; three upadacitinib, and one ASN002. In 8/11 studies inclusion criteria were restricted to patients with moderate-to-severe AD, defined as: an EASI score ≥ 16, IGA score ≥ 3, more than 10% Body Surface Area (BSA) involvement ± a score of ≥ 4 in the PP-NRS (33–38). In the remaining 2 studies, subjects needed to demonstrate an EASI ≥ 12, IGA-score ≥ 3 and BSA involvement > than 10% for inclusion (Table 1). All studies included safety assessment in the form of reported TEAEs (Table 2), and two included evaluation of pharmacokinetics. Physicochemical properties of oral JAK inhibitors are listed in Tables 3, 4.
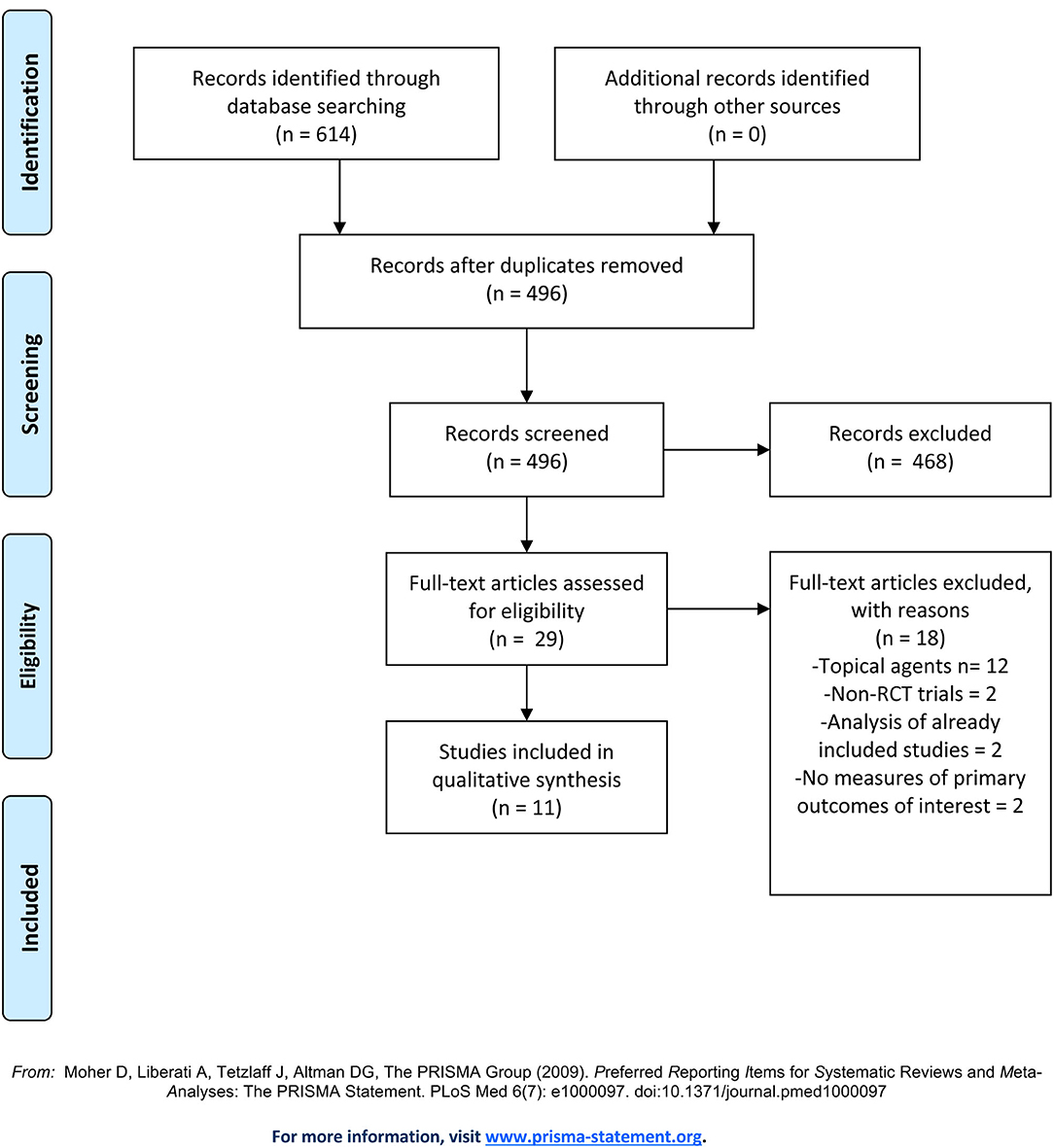
Figure 2. PRISMA Flow Diagram.
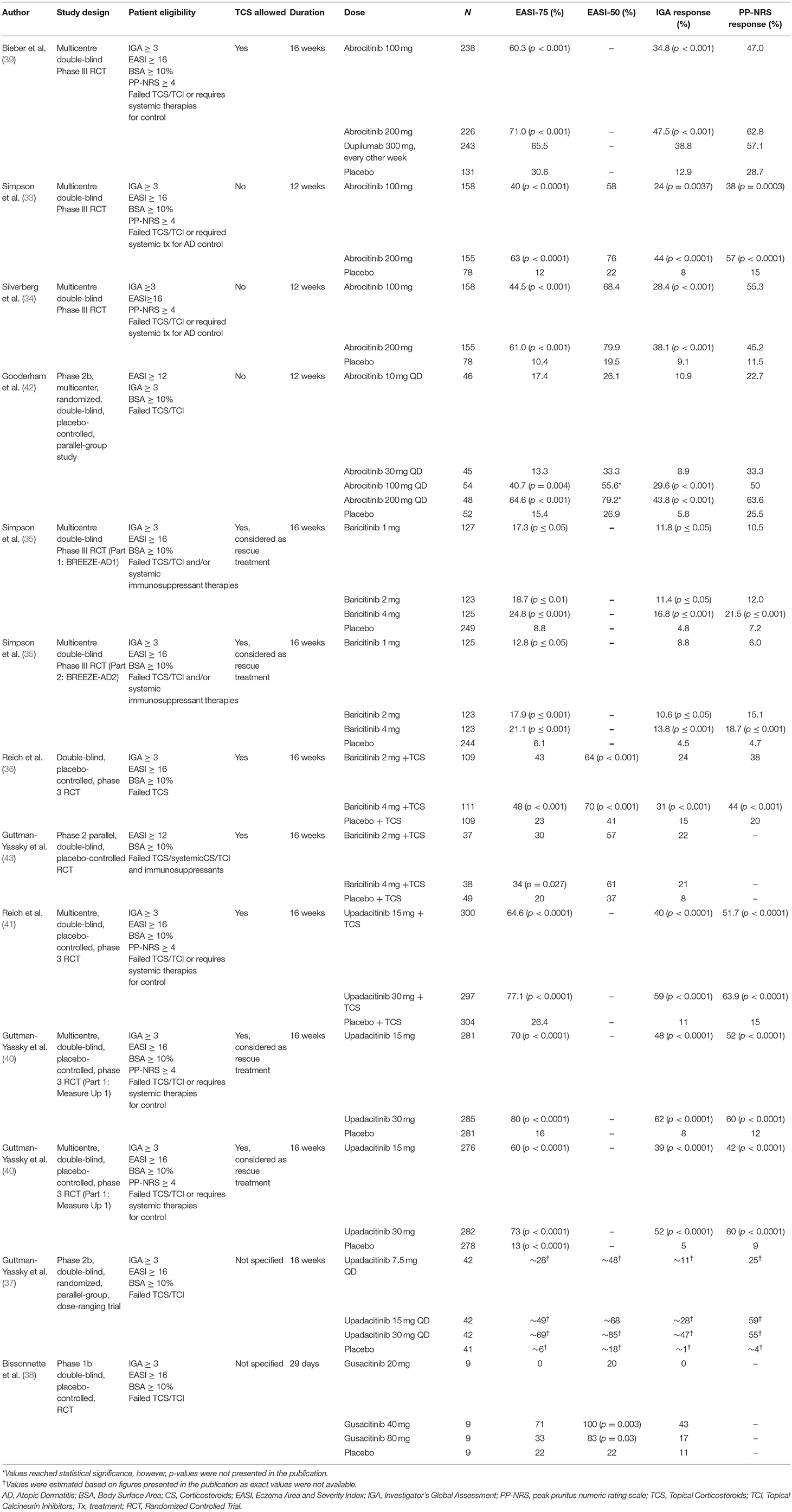
Table 1. Summary of clinical efficacy of published oral JAK inhibitor trials.
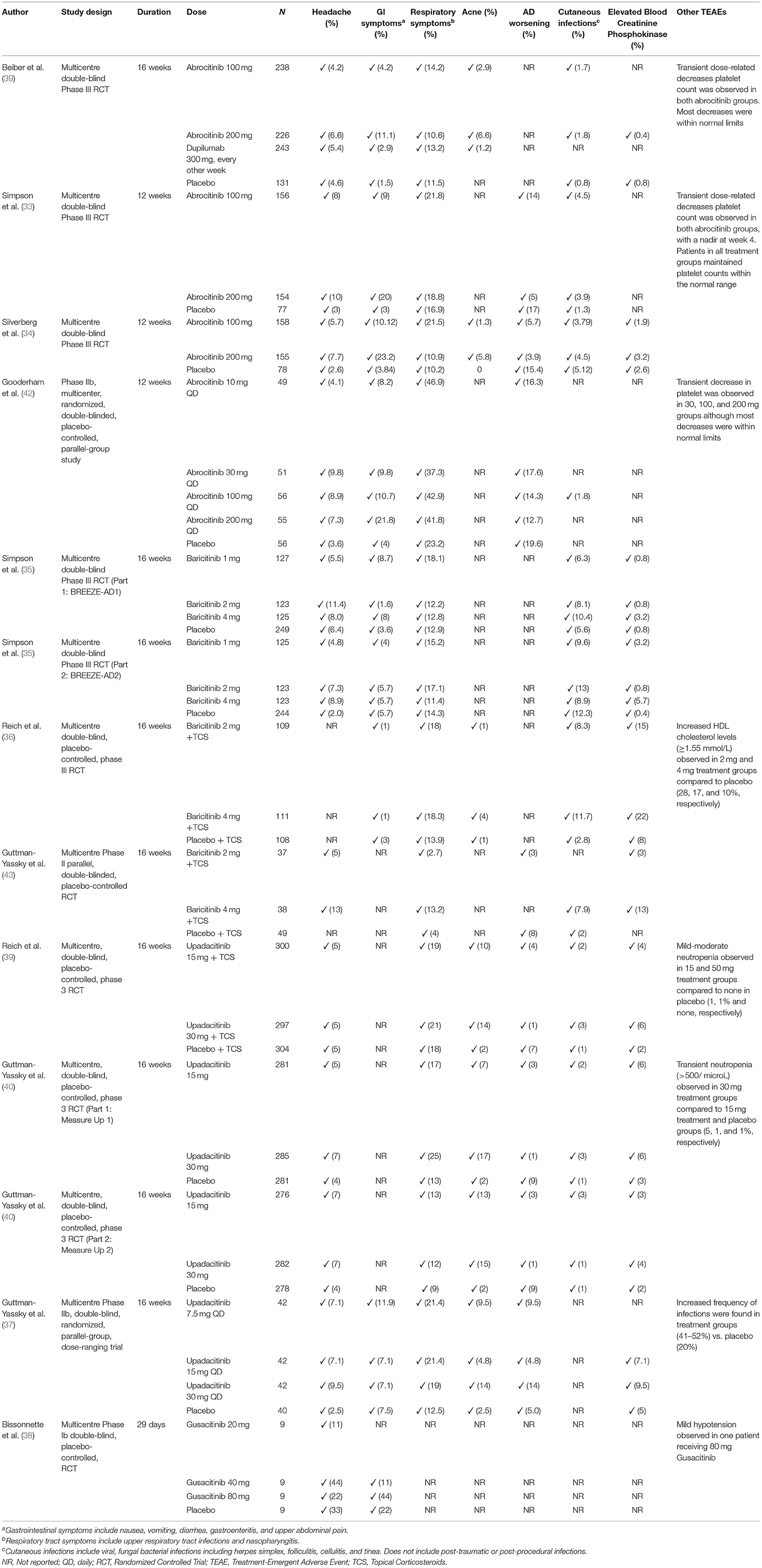
Table 2. Summary of common TEAEs in published oral JAK inhibitor trials.
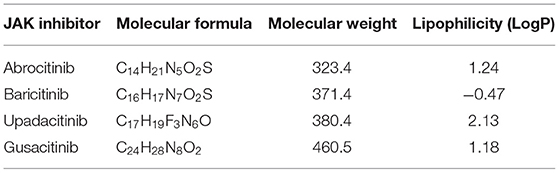
Table 3. Summary of physicochemical properties of oral JAK inhibitors.
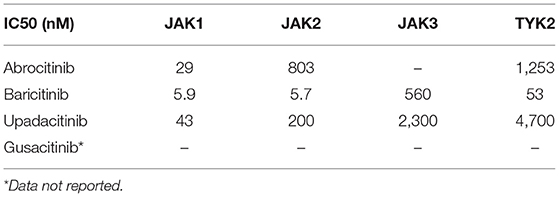
Table 4. Summary of half maximal inhibitory concentration (IC50) of oral JAK inhibitors.
Quality Assessment
Overall, we rated six out of eleven studies as low risk of bias (33, 35, 36, 39–41), and the remainder were considered as higher risk (34, 38, 42–44). All included studies employed a central randomization scheme and stipulated blinding of treatment assignment for investigators, patients and study personnel. Post-randomization, two studies (38, 44) had remaining imbalances between groups in IGA-measured-baseline severity of AD and were thus rated as “higher risk of bias” on the “randomization process” domain of the quality assessment tool. Most studies assessed efficacy of treatment by either an ITT analysis, or a modified version of ITT analysis that included all randomized participants who have received at least one-dose of the study's intervention. A single study (38) reported the per-protocol treatment efficacy estimates and was therefore considered at higher risk of bias for the “deviation from intended intervention” element (Supplementary Table 1).
Across all studies, attrition rates were high (9 to 50%, depending on study group) an effect compounded by relatively small sample sizes (range: 36–847). In evaluating the potential effects of missing outcome values on the assessment of treatment efficacy, we considered the stated reasons for attrition within each group. If these reasons differed between groups within a given study, we considered the potential for bias in outcome assessment as “high.” If attrition rates were high but reasons for discontinuation were relatively similar between groups, we deemed the risk of bias in measurement as low. With this reasoning, five studies (34, 38, 42–44) were evaluated as having a high risk for bias in the assessment of treatment efficacy. Since most studies abided by an ITT-analysis for the measurement of efficacy, these missing values likely biased the results toward the null hypothesis of no effect. Therefore, efficacy estimates reported in these studies are considered likely to represent an under-estimation, rather than an over-estimation, of the treatment's true efficacy.
Abrocitinib
Clinical Efficacy
Based on in-vitro studies, the JAK1 half maximal inhibitory concentration (IC50) of abrocitinib is 29 nM (45) (Table 4). The clinical efficacy of abrocitinib, an oral selective JAK1 inhibitor, was studied in four independent clinical trials, of which three were Phase III trials, while one was a Phase II trial (33, 34, 42) (Table 1). In the recent Phase III trial by Bieber et al. (39), a total of 838 adult patients (aged ≥18 years) with moderate-to-severe AD, who failed treatment with TCS or TCI or required systemic therapy to control their disease, were enrolled. Moderate-to-severe AD was defined as IGA ≥ 3, EASI Score ≥ 16, BSA ≥ 10%, and PP-NRS ≥ 4. Patients were randomized in a 2:2:2:1 ratio to 100 mg abrocitinib, 200 mg abrocitinib, 300 mg dupilumab (every other week), or placebo groups and assessed over 16 weeks. While a significantly higher percentage of patients in the 100 and 200 mg abrocitinib groups achieved an EASI-75 score and IGA response at 16 weeks in comparison to a placebo (p < 0.001), this significant increase was not noted when comparing outcome measures with the dupilumab treatment group (Table 1). Specifically, an EASI-75 was achieved in 71% of patients in the 200 mg abrocitinib group, 60.3% of the 100 mg abrocitinib group, 65.5% of the dupilumab group and 30.6% of the placebo group at 16 weeks. An IGA response was observed in 47.5% of the 200 mg abrocitinib group, 34.8% of the 100 mg abrocitinib group, 38.8% of the dupilumab group and 12.9% of the placebo group. Interestingly, a significantly higher (p < 0.001) proportion of patients achieving a PP-NRS response as early as week 2 was observed in the 200-mg abrocitinib compared to the dupilumab group. However, this response was not observed in the 100 mg abrocitinib group.
In another Phase III JADE MONO-1 trial by Simpson et al. (33) a total of 387 patients (aged ≥12 years) with moderate-to-severe AD, defined as IGA ≥ 3, EASI Score ≥ 16, BSA ≥ 10%, and PP-NRS ≥ 4, were enrolled. Patients were randomized to receive either placebo, 100 or 200 mg of abrocitinib daily for a total treatment duration of 12 weeks. At the end of treatment, the authors found that the proportion of patients who had achieved an IGA response, was significantly higher in the abrocitinib 100 mg group than in the placebo group [37 (24%) of 156 patients vs. six (8%) of 76 patients; p = 0.0037], and in the abrocitinib 200 mg group compared with the placebo group [67 [(44%) of 153 patients vs. six (8%) of 76 patients; p < 0.0001]. Additionally, the proportion of patients who achieved an EASI-75 response was significantly higher in the abrocitinib 100 mg group [62 (40%) of 156 patients vs. nine (12%) of 76 patients; p < 0.0001] and abrocitinib 200 mg group [96 (63%) of 153 patients vs. nine (12%) of 76 patients; p < 0.0001]. Interestingly, a significant difference in the proportion of patients achieving a PP-NRS response for 100 mg and 200 mg abrocitinib groups vs. placebo was achieved by the second week of treatment [20, 46, and 3%, respectively, with p = 0.0004 (100 mg abrocitinib vs. placebo) and p < 0.0001 (200 mg abrocitinib vs. placebo)]. This significant difference in PP-NRS response was maintained at week 12 [38, 57, and 15% of patients in the 100, 200 mg abrocitinib and placebo groups, respectively, with p = 0.0003 (100 mg abrocitinib vs. placebo) and p < 0.0001 (200 mg abrocitinib vs. placebo)] (Table 1).
Using the same patient inclusion criteria as the JADE MONO-1 trial, Silverberg et al. (34) also examined abrocitinib at 100 and 200 mg concentrations in adults and adolescent patients (12 to 18 years inclusively) with moderate-to-severe AD. A total of 391 patients were randomized to receive either abrocitinib 100 or 200 mg vs. a placebo intervention for 12-weeks duration. Compared to placebo, the proportion of participants achieving an IGA response were 28.7% higher (p < 0.001) for the 200 mg group and 19.3% higher (p < 0.001) in the 100 mg group. At the end of the 12-weeks, the 200 and 100 mg groups had achieved an EASI-75 response that was 50.5% (p < 0.001) and 33.9% (p < 0.001) higher than placebo, respectively. Percentage decreases in EASI scores from baseline were greater for both abrocitinib doses than for placebo at all time points. Significant differences in PP-NRS scores between both doses of abrocitinib and placebo were observed by day 2 of treatment, with decreases of 0.7 [95% Confidence Interval (CI), −0.9–0.5] and 0.6 (95% CI, −0.8–0.4) for the 200 mg and 100 mg doses respectively, vs. 0.1 decrease (95% CI, −0.4–0.2) for placebo.
Similar findings of clinical efficacy were demonstrated in the Phase IIb RCT investigating various dosages of abrocitinib vs. placebo in adult patients (≥18 years of age) with moderate-to-severe AD by Gooderham et al. (42) At week 12, 21 of 48 patients receiving 200 mg of abrocitinib (43.8%; p < 0.001), 16 of 54 patients receiving 100 mg of abrocitinib (29.6%; p < 0.001), and 3 of 52 patients receiving placebo (5.8%) achieved an IGA response. Additionally, through logistic regression modeling, authors estimated that a greater proportion of patients achieved an EASI-75 response in the 200 mg [estimated 31 of 48 (63.7%), p < 0.001] and 100 mg [estimated 22 of 54 (41.6%), p = 0.004] groups when compared to a placebo group [estimated 8 of 52 (15.6%)]. Significant differences from placebo in percentage reduction in EASI score from baseline were observed as early as week 1 (first postbaseline assessment) in the 200 mg group [least squares mean (LSM) difference from placebo, −28.3%; p < 0.001], and at week 2 in the 100 mg group (−14.9%; p = 0.03). Decreases from baseline in EASI score for the 200 mg and 100 mg groups were found to plateau by weeks 4 to 6 and were maintained through week 12.
Safety
In the four trials (33, 34, 39, 42), gastrointestinal and respiratory symptoms were found to be the most frequently reported TEAEs in the abrocitinib 100 and 200 mg groups, followed by a headache. AD worsening was found to be more common in placebo compared to abrocitinib groups. Moreover, in all four trials, transient dose-related numeric decreases in median platelet count were observed in patients receiving abrocitinib, with a nadir observed at week 4 and a return toward baseline values thereafter. Nevertheless, the majority of patients in treatment groups maintained platelet counts within the normal range (Table 2).
Specifically, in the Bieber et al. (39) trial, nausea was the most frequently reported TEAE in each of the 100 mg, 200 mg abrocinitib and dupilumab groups. Mild to moderate acne was also more frequently reported in abrocitinib groups (6.6 and 2.9% for 200 and 100 mg abrocitinib, respectively), in comparison to dupilumab or placebo groups (1.2 and 0%, respectively) (Table 2). Two malignancies were reported in this study: one cutaneous squamous-cell carcinoma in the in the 200-mg abrocitinib group, and one invasive intraductal breast neoplasia in the dupilumab group. The authors did not comment whether or not these malignancies were considered to be treatment-related. No deaths, or venous thromboembolisms (VTEs) were observed during this trial.
In the Phase III JADE MONO-1 trial (33), the most frequently reported TEAE in the abrocitinib 100 mg and 200 mg groups were nausea (9% in 100 mg and 20% in 200 mg groups) and nasopharyngitis (15% in 100 mg and 12% in 200 mg groups). Other common TEAEs included headache, and upper respiratory tract infection (URTI) symptoms (≥5% in any treatment group). Herpes virus infections were reported in all treatment groups, albeit uncommon [one (<1%) of 156 patients in the abrocitinib 100 mg group, and three (~2%) of 154 patients in the abrocitinib 200 mg group].
Serious adverse events (SAEs) were reported in five (3%) of 156 patients in the abrocitinib 100 mg group, five (3%) of 154 patients in the abrocitinib 200 mg group, and three (4%) of 77 patients in the placebo group. Among these patients, only two SAEs were considered treatment-related: in one patient receiving the abrocitinib 200 mg, who developed chronic inflammatory bowel disease, abrocitinib was permanently discontinued leading to full recovery; the other patient was in the abrocitinib 100 mg group and developed acute pancreatitis during the treatment period. Thus, abrocitinib was permanently discontinued, and the patient recovered. In this study, no cases of VTE, malignancies, major adverse cardiovascular events, changes in blood creatinine phosphokinase (CPK) levels or deaths were observed.
In Silverberg's Phase III trial of abrocitinib in adults and adolescents (34), the most frequent treatment TEAEs of any causality included nausea in the 200 mg group (14.2%), nasopharyngitis in the 100 mg group (12.7%) and worsening AD in the placebo group (15.4%). Other TEAEs of interest were acne (5.8% in the 200 mg group, 1.3% in 100 mg group and none in placebo group), folliculitis (3.2% in in 200 mg group and 2.6% in placebo group), vomiting (5.2% in 200 mg group, 1.3% in both 100 mg and placebo groups), and upper abdominal pain (3.9% in 200 mg vs. 1.3% and 0 in 100 mg and placebo respectively). SAEs that were considered related to treatment were reported for two patients in the 100 mg group (herpangina and pneumonia) and two patients in the placebo group (eczema herpeticum and a case of staphylococcal infection). None were observed in the 200 mg group. An elderly participant with pre-existing aortic valve sclerosis and untreated hypertension experienced a sudden cardiac death 3-weeks after discontinuation of abrocitinib. The event was not considered related to treatment. Furthermore, no cases of thromboembolisms or malignant neoplasms were reported in any treatment groups. The authors also reported a dose-related increase of ~10% in high-and-low-density lipoprotein levels, as well as an increase in CPK levels, for both the 200 mg and 100 mg groups compared to placebo (34).
The most frequently reported of TEAEs (≥3 patients in any treatment group) in Gooderham et al. Phase IIb study of abrocitinib included diarrhea, nausea, viral URTI, headache, and worsening atopic dermatitis (42). Two of 267 patients experienced SAEs that were considered related to treatment; one patient in the 200 mg group developed pneumonia during follow-up after initiation of cyclosporine, which was continued, and treated with antibiotics; and one patient in the 100 mg group developed eczema herpeticum during the treatment period, abrocitinib was permanently discontinued. One patient in the 200 mg group reported a pulmonary embolism (PE) after traveling a long distance by car with baseline laboratory values within normal limits. One patient receiving 10mg dose developed a melanoma, which was deemed not related to treatment. No treatment-related trends in serum lipids and transaminase levels were observed in the trial. CPK levels were, unfortunately, not reported.
Baricitinib
Clinical Efficacy
Baricitinib is an oral selective JAK1 and JAK2 inhibitor that blocks the downstream action of several cytokines in AD pathogenesis, including thymic stromal lymphopoietin, IL-4, IL-5, IL-13, IL-22, and IL-31 (36). According to in-vitro analyses, the baricitinib IC50 values were reported as 5.9 and 5.7 nM for JAK1 and JAK2 inhibition, respectively (46) (Table 4). In the 16-week Phase III independent BREEZE-AD1, BREEZE-AD2 trials by Simpson et al. (35) the efficacy of baricitinib vs. placebo was assessed in a total of 1,239 adult patients with moderate-to-severe AD at varying doses. In both BREEZE-AD1 and BREEZE-AD2 studies, moderate-to-severe AD was defined as IGA ≥ 3, EASI ≥ 16, BSA ≥ 10%. Eligible patients also had to demonstrate an inadequate response to TCS/TCIs and/or systemic immunosuppressant therapies. In total, 624 patients were enrolled in BREEZE-AD1, where they were randomized to daily placebo (n = 249), 1 mg (n = 127), 2 mg (n = 123), or 4 mg (n = 125) baricitinib groups. Similarly, a total of 615 patients were enrolled in BREEZE-AD2, where patients were randomized to similar groups [placebo (n = 244), 1 mg (n = 125), 2 mg (n = 123), or 4 mg (n = 123)].
In both trials, 2 mg and 4 mg of baricitinib achieved the study's primary efficacy outcome: a significant improvement vs. placebo for the proportion of patients achieving an IGA response at week 16. The percentage of patients achieving IGA response was 4.8% for placebo, 11.4% for baricitinib 2 mg, and 16.8% for baricitinib 4 mg (baricitinib 2 mg, p ≤ 0.05; baricitinib 4 mg, p ≤ 0.001 vs. placebo) in BREEZE-AD1, and 4.5% for placebo, 10.6% for baricitinib 2 mg, and 13.8% for baricitinib 4 mg (baricitinib 2 mg, p ≤ 0.05; baricitinib 4 mg, p ≤ 0.001 vs. placebo) in BREEZE-AD2 (Table 1).
In both studies, 4 mg baricitinib treatment was found to lead to significant improvement for all secondary study endpoints, including a significantly higher proportion of patients achieving a PP-NRS response compared to placebo (p ≤ 0.001) at weeks 1, 2, 4, and 16; proportion of patients achieving EASI-75; and percentage change from baseline EASI score. Similarly, 2 mg baricitinib treatment also demonstrated a significant improvement for the aforementioned secondary endpoints in both trials, except for the proportion of patients achieving a PP-NRS response compared to placebo at week 1. However, 1 mg of baricitinib treatment led to inconsistent clinical outcomes in primary and secondary endpoints in both trials.
In another Phase III clinical trial by Reich et al. (36) (BREEZE-AD7), a total of 329 patients with moderate-to-severe AD were randomly assigned (1:1:1) to receive 2 mg of baricitinib once daily (n = 109), 4 mg of baricitinib once daily (n = 111), or placebo (n = 109) for 16 weeks. The use of low-to-moderate potency TCSs as well as TCIs and crisaborole for active lesions was allowed throughout the trial. Rescue therapy with high- or ultrahigh-potency TCSs or systemic therapies were available for patients who experienced worsening and unacceptable AD symptoms after 2 weeks of treatment.
The proportion of patients who achieved the primary endpoint of IGA response at week 16 was significantly higher for patients treated with 4 mg of baricitinib vs. placebo [34 of 111 (31%); p = 0.004]. Unlike the BREEZE-AD1 and BREEZE-AD2 trials, the primary end point for 2 mg of baricitinib was not met [26 of 109 (24%); P = 0.08]. As such, secondary endpoints were only evaluated for the 4 mg of baricitinib group. Specifically, the 4 mg of baricitinib group experienced a significant improvement compared with the placebo group (p < 0.001) for a proportion of patients, who achieved an EASI-75 response at week 16 [53 of 111 (48%) in the 4 mg group, vs. 25 of 109 (23%) in the placebo group], proportion of patients who achieved a PP-NRS response at week 4 [52 of 100 (52%) for the 4 mg group vs. 11 of 104 (11%) for the placebo group] and week 16 [44 of 100 (44%) for the 4 mg group, 37 of 97 (38%) vs. 21 of 104 (20%) for the placebo group] (Table 1).
Findings of a Phase II RCT investigating the clinical efficacy of 2 mg and 4 mg baricitinib vs. placebo in adult patients with moderate-to-severe AD by Guttman-Yassky et al. (43) demonstrated similar results. In this study, however, moderate-to-severe AD was defined by EASI ≥ 12, BSA ≥ 10% and eligible patients had to fail treatment with either TCS, TCI, systemic corticosteroids or other conventional immunosuppressants. Additionally, triamcinolone 0.1% cream was used throughout the study according to label instructions or as recommended by the investigator. Significantly more patients who received 4 mg baricitinib, achieved EASI-50 than did patients who were assigned to a placebo arm [61 vs. 37% (p = 0.027)] at 16 weeks. However, the proportion of patients achieving EASI-50 in the 2 mg baricitinib group did not reach statistical significance at 16 weeks (p = 0.065).
Safety
Overall, the most common TEAEs reported in all baricitinib studies reviewed were respiratory symptoms, headache, cutaneous infections, gastrointestinal symptoms and elevation of blood CPK. Specifically, in the study conducted by Simpson et al. (35), the most frequently reported TEAEs (>2% in any treatment group) were nasopharyngitis, URTIs, CPK elevations and headaches. However, there was no increase in the frequency of nasopharyngitis and URTIs, when comparing baricitinib with placebo. Headaches were reported at similar rates in patients treated with 4 mg baricitinib and placebo in BREEZE-AD1 (8.0 and 8.9%, respectively), while a greater percentage of patients reported headaches in the 4 mg baricitinib group compared to placebo in the BREEZE-AD2 trial (6.4 and 2.0%, respectively). Nevertheless, reported headaches were mild (76% of reported cases) and short-lived (median duration of ≤ 5 days), with none requiring study-drug interruption or discontinuation. Herpes simplex was also observed more frequently with baricitinib in BREEZE-AD1 trail. Most cases were of mild or moderate severity in both studies and did not cause SAEs or required drug discontinuation. Although CPK elevations were common, most cases (16 of 20) were asymptomatic and either resolved to below the upper limit of normal or were resolving during the study without treatment interruptions. Three patients treated with baricitinib had temporary treatment interruption with resolution of CPK elevations and one patient discontinued the study. No changes in serum lipids were reported. No deaths or VTEs (including PE and Deep Venous Thrombosis [DVT]) were reported in any group. There were no malignancies reported in baricitinib treatment groups.
In the BREEZE-AD7 trial, the most frequently reported (≥2% in any treatment group) TEAEs for 4 mg and 2 mg baricitinib doses compared with placebo were nasopharyngitis, folliculitis, oral herpes, URTI, acne, diarrhea, and back pain (36). One 51-year-old female patient in the 4 mg baricitinib group experienced a PE in the context of receiving oral contraceptives and having a previous history of smoking (7 pack-years). The patient subsequently discontinued treatment and recovered from the event. No major adverse cardiovascular events, malignant tumors, or deaths were reported.
CPK levels were elevated with baricitinib compared with placebo, with most increases being classified as Common Terminology Criteria for Adverse Events (CTCAE) grades 1 and 2 (increase of CPK of <2.5 times and 2.5–5 times the upper limit of normal, respectively) (36). Additionally, CPK elevations were not associated with evidence of muscle injury (e.g., rhabdomyolysis). Although changes were seen in lipid levels, including increases in high-density lipoprotein (HDL) level (≥60 mg/dL; 4 mg group, 28%; 2 mg group, 17%; and placebo group, 10%), changes in low-density lipoprotein (LDL) levels were similar in all groups (≥160 mg/dL; 4 mg group, 3%; 2 mg group, 3%; and placebo group, 4%).
Similarly, in the phase II trial by Guttman-Yassky et al. headaches and nasopharyngitis were reported as common TEAEs (43). Additionally, it was noted that infections were not increased in the groups treated with 2 mg or 4 mg baricitinib compared with those who received a placebo. In both 2 mg and 4 mg baricitinib-plus-TCS groups, the authors observed asymptomatic increases in CPK levels of ≥ 30 U/L at week 16 (43). No deaths, VTEs or malignancies were reported.
Upadacitinib
Clinical Efficacy
The clinical efficacy of upadacitinib, a selective JAK1 inhibitor, has recently been determined by two Phase III studies by Reich et al. (41) and Guttman-Yassky et al. (40). In the study by Reich et al. (41) the efficacy of upadacitinib at 15 and 30 mg daily with TCS vs. placebo with TCS was assessed in 901 adolescent (aged 12 to 17 years old) and adult (aged 18 to 75 years) patients with moderate to severe atopic dermatitis, as defined by the Hanefin and Rajka criteria. At 16 weeks, authors found that the proportion of patients who had achieved an EASI-75, was significantly higher in the 15 mg and 30 mg upadacitinib with TCS treatment groups than in the placebo with TCS group (64.6, 77.1, and 26.4%, respectively; p < 0.0001; Table 1). Additionally, a significantly higher proportion of patients achieved an IGA response at week 16 in both 15 and 30 mg upadacinitib with TCS treatment groups in comparison to placebo alone (Table 1).
The proportion of patients achieving a PP-NRS response as early as 1 week was significantly higher in patients receiving 15 and 30 mg upadacitinib with TCS treatments than in the placebo with TCS treated group (12.2, 19.2, 3.1%, respectively; p < 0.0001). A similar trend was noted in the proportion of patients achieving an EASI-75 score at 2 weeks. Reich et al. (41) documented 31.0% of patients achieving an EASI-75 score in the 15 mg upadacitinib with TCS group, 44.1% in the 30 mg upadacitinib with TCS group, and 6.9 % in the placebo with TCS group treatment (p < 0.0001 when comparing 15 and 30 mg upadacitinib with TCS treatment groups vs. placebo).
In the study by Guttman-Yassky et al. (40), the efficacy of upadacitinib at 15 and 30 mg daily vs. placebo were assessed in Measure Up 1 and Measure Up 2 replicate Phase III RCTs. A total of 1,683 adolescent (aged 12 to 17 years old) and adult (aged 18 to 75 years) patients with moderate-to-severe AD, defined as IGA ≥ 3, EASI ≥ 16, BSA ≥ 10%, and PP-NRS ≥ 4 were enrolled in both studies.
Eight hundred forty-seven patients participated in Measure Up 1, where they were Randomized to daily placebo (n = 281), 15 mg (n = 281, or 30 mg (n = 285) upadacitinib treatment groups. Eight hundred thirty-six patients participated in Measure Up 2, where patients were randomized to placebo (n = 278), 15 mg (n = 276), or 30 mg (n = 282) upadacitinib treatment groups.
In both Measure Up 1 and 2 trials, patients in the 15 and 30 mg upadacitinib groups demonstrated important efficacy outcomes. Namely, the study showed a significantly higher (p < 0.0001 in all cases) proportion of patients in the 15 or 30 mg of upadacitinib groups achieving an EASI-75 score (coprimary endpoint), IGA response (coprimary endpoint) and PP-NRS at week 16 vs. placebo (Table 1).
Interestingly, a significantly higher proportion (p < 0.0001) of patients in both 15 mg and 30 mg upadacitinib treatment groups achieved a PP-NRS response as early as 1 week in comparison to placebo, in the Measure Up 1 and Measure Up 2 Trials (Measure Up 1: 15.0% in the 15 mg upadacitinib group, 19.6% in the 30 mg group, and 0.4% in the placebo group; Measure Up 2: 7.4% in the 15 mg upadacitinib group, 15.7% in the 30 mg group, and 3.6% in the placebo group). Similarly, a significantly higher proportion (p < 0.0001) of patients in both 15 mg and 30 mg upadacitinib treatment groups achieved an EASI-75 score as early as 2 weeks in comparison to placebo, in the Measure Up 1 and Measure Up 2 Trials (Measure Up 1: 38.1% in the 15 mg upadacitinib group, 47.4% in the 30 mg group, and 3.6% in the placebo group; Measure Up 2: 33.0% in the 15 mg upadacitinib group, 44.0% in the 30 mg group, and 3.6% in the placebo group).
In another recent study by Guttman-Yassky et al. (37), the clinical efficacy of the selective JAK 1 inhibitor, upadacitinib, was investigated over a period of 16 weeks in this Phase IIb, double-blinded, randomized, parallel-group, dose-ranging trial. Patients with moderate-to-severe AD, defined by IGA ≥ 3, EASI ≥ 16, BSA ≥ 10%, and who failed treatment with TCSs/TCIs were randomized 1:1:1:1 to once-daily upadacitinib oral monotherapy 7.5, 15, or 30 mg or placebo groups. Results at 16 weeks demonstrated that EASI-50, EASI-75, and EASI-90 responses were also achieved at week 16. EASI-100 was achieved by 2.4% (1 of 42; p = 0.43), 9.5% (4 of 42; P = 0.05), and 24% (10 of 42; p = 0.001) of patients in the upadacitinib 7.5-, 15-, and 30 mg groups, respectively, vs. none (0 of 41) in the placebo group. Each upadacitinib dose level was significantly superior to placebo for achieving an IGA response and patient assessment of pruritus (achievement of PP-NRS response) at week 16. Interestingly, efficacy at the studied doses was generally demonstrated by weeks 1 to 4, with peak values reached and maintained after weeks 4 or 8.
Pharmacokinetics
In the study by Guttman-Yassky et al. (37), pharmacokinetic measures were investigated, where it was found that upadacitinib exposures were approximately dose proportional over the 7.5- to 30 mg dose range. Upadacitinib median (interquartile range) plasma concentrations around peak and trough periods were consistent with exposures previously observed for the evaluated doses in healthy volunteers [7.5 mg dose: 10.6 (0.8–21.0) and 2.8 (1.4–4.5) ng/mL, respectively; 15 mg dose: 32.5 (22.7–39.3) and 3.6 (1.8–7.0) ng/mL; 30 mg dose: 57.0 (28.1–94.8) and 8.1 (6.6–16.6) ng/mL] (37).
Safety
In both the Reich et al. (41) and Guttman-Yassky et al. (40) Phase III studies, TEAEs were reported more frequently in the upadacitinib treatment groups than the placebo group (Table 2). The most common reported TEAEs (>5% in any treatment group) were acne, respiratory symptoms, headache, elevation in CPK and worsening of AD. The majority of patients who reported treatment-emergent acne had mild to moderate symptoms consisting of inflammatory papules, pustules, comedones, with few cysts and nodules. While in the Reich et al. (41) study, none of the acne events were considered as severe and did not lead to treatment discontinuation, in the Guttman-Yassky et al. (40) study, one acne event was severe, involving >30% of body surface area. Additionally, among patients in the Measure up 1 and 2 trials, one patient in the upadacitinib 15 mg group and one patient in the upadacitinib 30 mg group discontinued study drug because of moderate acne.
With respect to potentially clinically important laboratory findings, most reports of elevations in CPK levels were asymptomatic and associated with exercise. In the Reich et al. (41) study, the elevated CPK levels were reported to be dose related. In the Guttman-Yassky et al. (40) study, only one case of elevated creatinine phosphokinase levels was reported in the upadacitinib 15 mg group, which led to treatment discontinuation. Transient, mild to moderate, neutropenia was also observed more frequently in upadacitinib groups in comparison to placebo in both Phase III studies. Only one event, occurring in the Reich et al., study lead to discontinuation of 30 mg upadacitinib treatment.
No treatment-related deaths or VTEs were reported in the upadacinitib groups in both Phase III RCTs. In the Reich et al. (41) study, two malignancies were reported in the upadacitinib 30 mg with TCS treatment group: one non-melanoma skin cancer (a keratoacanthoma) identified on treatment day 45 and one adenocarcinoma of the colon identified on treatment day 7. The case of colon adenocarcinoma was considered as a non-treatment-related, serious adverse event that lead to the discontinuation of upacitinib. In the Guttman Yassky et al. (40) study, six cases of malignancy were reported in the upadacitinib groups, all of which were determined not to be treatment-related [squamous cell skin carcinoma (n = 2), basal cell skin carcinoma (n = 1), breast cancer (n = 1), gastric cancer (n = 1), and anal cancer (n = 1)].
In the Guttman-Yassky et al. (37) Phase IIb study, TEAEs were reported in 71% (30 of 42), 74% (31 of 42), and 79% (33 of 42) of patients receiving upadacitinib 7.5, 15, and 30 mg, respectively, vs. 63% (25 of 40) of patients receiving a placebo (Table 2). The most frequently reported TEAEs were gastrointestinal and URTI symptoms, followed by acne and AD worsening, all of which were reported as mild or moderate in severity. Additionally, in the 15 mg and 30 mg groups, increased blood CPK was observed in 7.1 and 9.5%, respectively. Nevertheless, the CPK elevations were asymptomatic in patients receiving upadacitinib and reported to be mild to moderate in severity. There was no relationship noted between the dose of upadacitinib and the occurrence of particular TEAEs.
Two patients in the upadacitinib 7.5 mg group had SAEs, namely worsening AD (skin infection and exacerbation of AD) in the context of contact dermatitis and a lower jaw pericoronitis due to recurring tooth infections, not thought to be associated with the treatment. One patient in the upadacitinib 15 mg group had appendicitis. All SAEs in patients who received upadacitinib resolved with treatment.
There were no deaths, opportunistic infections, malignancies, gastrointestinal perforations, herpes zoster, renal dysfunction, active or latent tuberculosis reactivation, adjudicated cardiovascular events, or VTEs. While infections were more common with upadacitinib than with placebo, there were fewer serious infections with upadacitinib.
Gusacitinib
Clinical Efficacy
Bissonnette et al. (38) were the first to demonstrate gusacitinib (ASN002) as an effective AD treatment in a double-blinded, placebo-controlled Phase Ib RCT. Gusacitinib is an oral dual inhibitor of JAK and tyrosine-protein kinase SYK (also known as spleen tyrosine kinase). Patients included had a diagnosis of moderate-to-severe AD defined by an IGA ≥ 3, EASI ≥ 16, BSA ≥ 10%. In this study, 36 patients were randomized at a 3:1 ratio, gusacitinib or placebo, whereby 9 patients were included in the 20, 40, and 80 mg gusacitinib once daily and placebo groups. Each patient received either gusacitinib or placebo once daily for 28 days. In the context of our systematic review, clinical efficacy was determined via EASI-50 and EASI-75 tools over 29 days. Gusacitinib was found to be significantly superior to placebo for the proportion of patients achieving EASI-50 in the 40 mg and 80 mg dose groups at end of treatment (p = 0.003 and p = 0.03, respectively) (Table 1). However, the same efficacy could not be demonstrated in the 20 mg gusacitinib group. The proportion of patients achieving EASI-75 was also greater in the 40 and 80 mg gusacitinib groups compared to placebo, although the difference did not reach statistical significance (p = 0.06 and p = 0.65, respectively).
Pharmacokinetics
The trial by Bissonnette et al. (38) also reported the pharmacokinetic parameters of gusacitinib in AD patients (38). For 20 mg gusacitinib, the mean maximum concentration (Cmax) was found to be 67.8 ng/ml, and half-life of 6.62 h. For 40 mg gusacitinib the mean Cmax was found to be 136 ng/ml, and half-life of 9.10 h. For 80 mg gusacitinib the mean Cmax was found to be 186 ng/ml, and half-life of 11.2 h.
Safety
The most common TEAEs were headache and nausea in 7 and 5% of patients who received gusacitinib, respectively (Table 2) (38). There were 2 TEAEs that led to discontinuation including a subject with mild hypertension and another with low lymphocyte counts. The event of mild hypertension was reported in a patient receiving 80 mg gusacitinib and was classified as being possibly related to treatment. The patient with lymphopenia had had low pre-treatment lymphocyte levels and the AE was not considered to be related to treatment. No clinically significant changes in lipid profile were observed in the study. CPK levels were not reported in this trial. Additionally, no VTEs, malignancies or deaths were noted (38).
Discussion
AD is a common and debilitating inflammatory skin disease driven by barrier dysfunction and abnormal Th cell activation. Multiple inflammatory pathways and their respective cytokines are believed to be involved in the chronicity and relapsing nature of the disease. The JAK-STAT and SYK pathways have been shown to assert a downstream modulating effect on AD-associated cytokines. Therefore, JAKi have introduced a promising novel area of therapeutics in the treatment of AD, as well as in other cytokine-mediated autoimmune and inflammatory diseases. With the growing number of clinical trials evaluating the efficacy of various JAKi for the treatment of AD, we sought to systematically review the literature to synthesize and evaluate the available evidence on efficacy and safety of these new compounds. We identified 11 RCTs evaluating the efficacy and safety of four compounds: abrocitinib, baricitinib, upadacitinib, and gusacitinib. A summary of the physicochemical properties of JAKi discussed are provided in Table 3. Given the relative paucity of evidence for each individual compound and the differences in patient eligibility criteria among studies, the data was not deemed suitable for a meta-analysis at this time. Nevertheless, the presented review provides a comprehensive summary of the evidence, most of which lends support to the use of JAKi in the treatment of AD.
Abrocitinib, baricitinib, and upadacitinib were the most extensively studied JAKi for the treatment of moderate-to-severe AD to date with Phase III data available. While abrocitinib and upadactinib are oral selective JAK1 inhibitors, baricitinib is an oral selective JAK1 and JAK2 inhibitor. This selectivity enables JAKi to demonstrate specificity and different capacities to block cytokine receptor signaling (Figure 3). Given that baricitinib is a JAK1/2 inhibitor, it has the capacity to inhibit the signaling of multiple cytokine receptors including the IL-10 family receptor, cytokine sharing IL-12Rß1, IFN-γ receptor, homodimeric cytokine receptor, among others. By contrast, JAK1 inhibitors can inhibit most cytokine receptors inhibited by JAK1/2 inhibitors except for the cytokine sharing IL-12Rß1 and the homodimeric cytokine receptor (Figure 3). Among the other small molecules reviewed, gusacitinib is a dual JAK/SYK inhibitor.
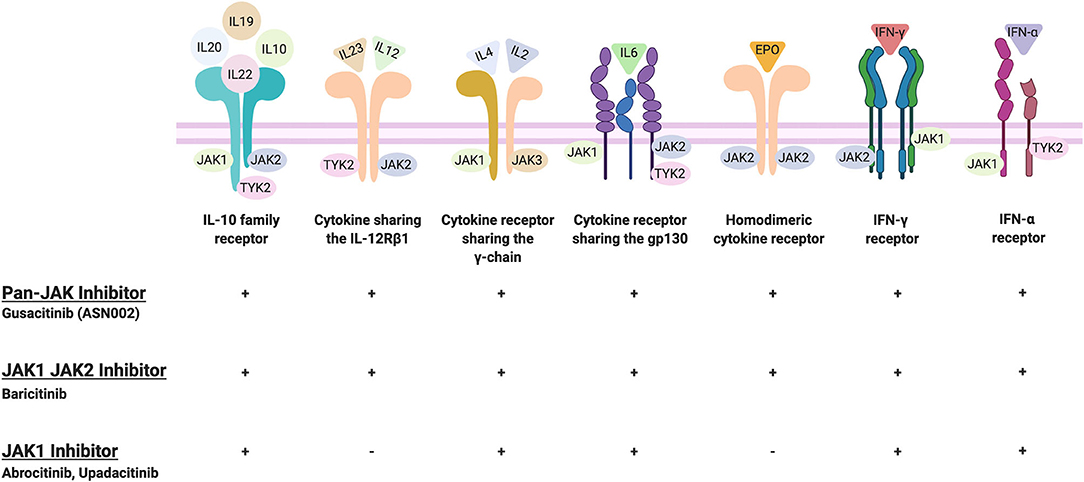
Figure 3. JAKi selectivity toward cytokine receptors. Created and retrieved from https://app.Biorender.com/biorender-templates.
From the studies reviewed, the clinical efficacy of treatment, as defined by achieving a 4-point reduction in PP-NRS score, was achieved with 100 and 200 mg of abrocitinib, 15 and 30 mg of upadacitinib, and 4 mg of baricitinib as early as week 1 (abrocitinib and upadacitinib) and week 2 (baricitinib), respectively (33–36, 39–41). Notably, 200 mg abrocitinib dose was found to be superior to dupilimab in improving itch response at 2 weeks (39). Two mg baricitinib was also found to be effective in quickly controlling pruritus (35). This rapid efficacy is especially welcome for patients with moderate-to-severe AD, who require prompt symptom control. Similar to cyclosporine, JAKi are able to produce a fast response while demonstrating a side effect profile superior to cyclosporine. Summary of JAKi clinical efficacy by multiple outcome measures are presented in Figure 4.
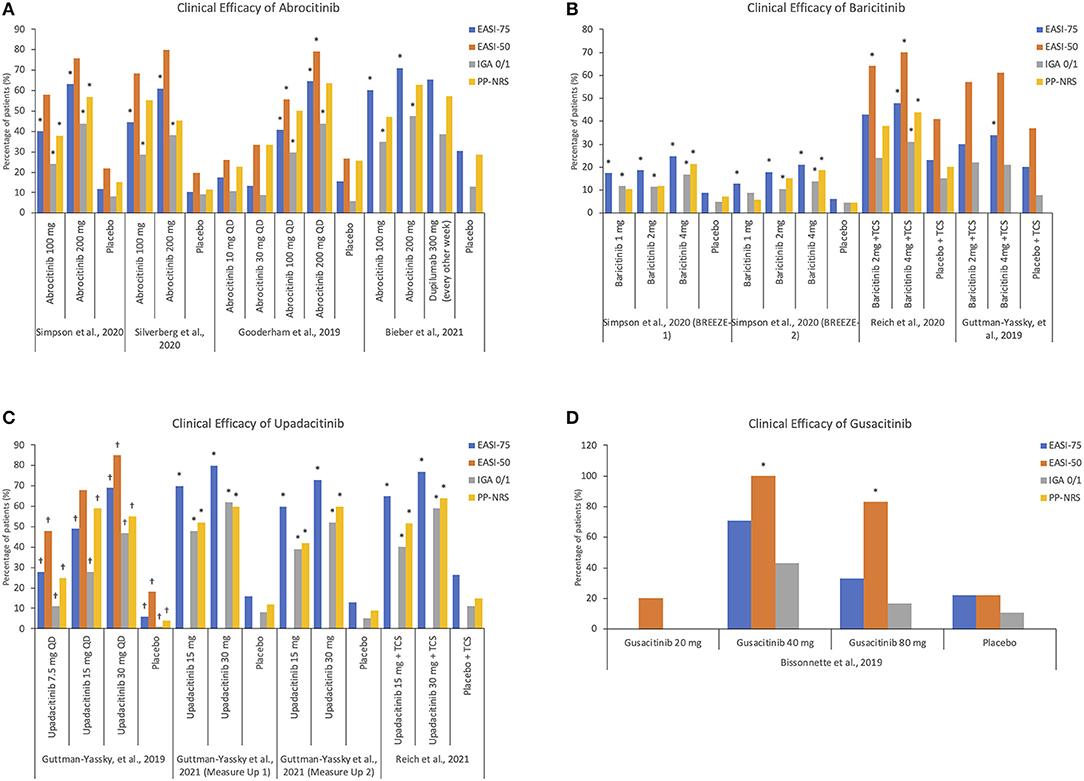
Figure 4. Summary of JAKi clinical efficacy by multiple outcome measures. (A) Clinical efficacy of abrocitinib (B) Clinical efficacy of baricitinib (C) Clinical efficacy of upadacitinib (D) Clinical efficacy of gusacitinib. *Values reached statistical significance; †Values were estimated based on figures presented in the publication as exact values were not available. EASI, Eczema Area and Severity Index; IGA, Investigator's Global Assessment; PP-NRS, Peak Pruritus Numerical Rating Scale.
The safety profile for reviewed JAKi small molecules is reassuring with most TEAE being mild and transient in nature, and amenable to symptomatic treatment. Nevertheless, it is important to note that asymptomatic increases in serum CPK levels were observed in the trials with all JAKi (34, 37, 43). These increases mirror the findings from previous studies of JAKi, including tofacitinib (47, 48), baricitinib (49), and upadacitinib (50–52), used in the treatment for a range of other inflammatory diseases. In all previous trials, CPK increases have not been associated with clinically overt myopathy or rhabdomyolysis. However, a recent case report from Australia described two patients with rheumatoid arthritis (RA), who were treated with baricitinib and developed muscle pain and joint swelling coupled to moderate CPK elevation (53). In both cases, clinical and biochemical resolution occurred rapidly after baricitinib discontinuation (53). Increases in serum lipids were also reported in response to abrocitinib and baricitinib in studies included in this review (34, 36). These findings are congruent with previous reports, particularly in response to tofacitinib (54) and baricitinib (55). Yet, while both compounds increase both LDL and HDL cholesterols, they do not appear to alter the LDL:HDL ratio (54, 56). Thus, further evaluation of cardiovascular event rates during long-term treatment is warranted to elucidate the clinical implications of these findings. Overall, it appears that the increases of CPK and lipid levels likely represent class effects, although minor differences deserving special attention in future trials might exists between JAKi compounds.
Two studies within our review reported cases of thromboembolic TEAEs after treatment with 200 mg abrocitinib or 4 mg baricitinib. In both cases, the patients had pre-existing risk factors, and one case also had a history of immobilization. Thromboembolism was first identified as a potential clinically important TEAE of JAKi during the baricitinib approval process for RA (57). In 2017, the European Medicines Agency moved to include DVT and PE as possible side effects of baricitinib, cautioning against its use in patients with risk factors for DVT or PE (58). In 2019, the Food and Drug Administration (FDA) issued a safety warning regarding the preliminary results of safety trial of tofacitinib in the treatment of RA. In patients with RA and at least one cardiovascular risk factor (CVRF), the 10 mg dose of tofacitinib given twice daily was associated with higher rates of thrombosis and all cause-mortality compared to 5 mg given twice daily or TNF-α inhibitors (59). In the absence of a mechanistic explanation for the observed increase in thromboembolic risk in patients with pre-existing CVRFs, regulatory health bodies have moved toward broadening the black box warning to include other JAKi, such as upadacitinib (which is currently approved for RA). The warning is also expected to be added to all future JAKi entering the market, including abrocitinib and gusacitinib. While there is sufficient evidence to conclude that JAKi increase the risk of thromboembolic events, it has been challenging to quantify the magnitude of this association. In one large systematic review and meta-analysis, Oliviera et al. examined the safety of JAKi in patients with inflammatory bowel disease or other immune-mediated diseases (60). Risk of DVT was assessed in 17 studies, including a total of 24,128 patients exposed to a JAKi. The overall incidence rate of VTEs was 0.31 per 100 patient-years, but the results differed between compounds. In healthy individuals, the frequency of thromboembolic events is cited at around 0.1 to 0.2 cases per 100 patient-years, increasing to about 0.5 per 100-patient-years in those aged ≥75. With such low-level frequency rates in the general population, only very large field studies could offer enough evidence for robust conclusions as to the strength of this association. Until then, JAKi should be used judiciously in patients with pre-existing cardiovascular comorbidities.
While no clinical trials within our review reported definitive treatment-related malignancies in JAKi treatment groups, a squamous cell carcinoma and keratoacanthoma were diagnosed in two patients receiving abrocitinib and updacitinib, respectively (39, 41). Nevertheless, we were unable to accurately evaluate the risk of malignancy given that these clinical trials were limited in duration. Previous studies have suggested a link between the use of tofacitinib for RA and the development of malignancies. Of 5,677 adult patients who participated in phase II, phase III and long-term extension studies of tofacitinib, 107 patients were found to develop malignancies [excluding non-melanoma skin cancer (NMSC), also known as keratinocyte carcinomas] (61, 62). The most common was lung cancer (n = 24), followed by breast cancer (n = 19), lymphomas (n = 10), and gastric cancer (n = 6). The overall incidence rate (IR) for all malignancies (excluding NMSC) in patients with RA treated with tofacitinib was 0.85 (95% CI, 0.70–1.02). Nevertheless, the incidences of all malignancies (excluding NMSC) were similar in the tofacitinib users compared with the general population (Standardized IR, 1.17; 95% CI, 0.96–1.41) (62). Consistent with this, a subsequent study following patients for 9.5 years of tofacitinib treatment documented no increased risk of malignancy in comparison to the reference population (63). Another study investigating the long-term safety of baricitinib in RA determined that incidences of malignancy (excluding NMSCs) were 0.8 (95% CI 0.4–1.5) per 100 patient-years for 2 mg baricitinib and 1.0 (95% CI 0.5–1.7) per 100 patient-years for 4 mg baricitinib, although there was no significant difference in the incidence of malignancy compared to the placebo group (64). Further long-term studies are required to appropriately determine the risk of developing malignancies when JAKi are used in AD patients. Occurrence of transient neutropenia and acne are additional important side effects to consider in the treatment with JAKi.
In conclusion, given its rapid symptom control combined with the reassuring safety profile, the use of abrocitinib, baricitinib, and upadacitinib can be considered as an important reliable systemic treatment option for adult patients with moderate-to-severe AD who are unresponsive to topical/skin-directed therapies. For abrocitinib, baricitinib, and upadacinitib, close observation of TEAEs is required as well as serial complete blood cell count with differential for neutrophil and platelet monitoring, CPK levels and lipid assessment. Hence, while needle-phobic patients may prefer an oral pill option, regular blood tests will be needed to monitor therapy. Additionally, as a clinical measure of drug efficacy, it has been hypothesized that acne as a TEAE may be due to the pharmacological effect of JAK inhibition.
Although gusacitinib has also demonstrated promising clinical outcomes in the treatment of moderate-to-severe AD, future large-scale phase III trials are required prior to considering their integration in current treatment guidelines. To the best of our knowledge, active clinical trials involving JAKi include phase III trials investigating the use of abrocitinib, baricitinib, and upadacitinib in pediatric populations. Additionally, a phase II clinical trial investigating the safety and efficacy of Jaktinib, a JAK1/2/3 inhibitor, in the treatment of moderate-to-severe AD is currently underway (ClinicalTrials.gov Identifier: NCT04612699).
JAKi represent a new therapeutic class to optimize AD treatment. As more clinical studies confirming the safety and efficacy of JAKi emerge, clinician education regarding this novel treatment will be important.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
ML, MB-R, and FG assessed study eligibility. ML and MB-R conducted data extraction and prepared figures. ML, MB-R, FG, MB, LF, MG, LG, SH, HH, IL, PL, DM, MW, JY, CL, and IVL reviewed included studies and the extracted data and wrote the paper. CL and IVL supervised the project. All authors contributed to the article and approved the submitted version.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The genesis of the paper was initiated at a meeting sponsored by a pharmaceutical company (Pfizer) and MB, LF, MG, LG, SH, HH, IL, PL, DM, MW, JY, CL, and IVL were paid to attend that meeting. This work was supported by unrestricted educational grant from Pfizer. IVL clinician-scientist salary is supported by the Fonds de Recherche du Quebec–Santé research grant #34753. No funding bodies or other organizations had any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
MB an advisory board member and speaker for AbbVie, Amgen, Leo Pharma, and Janssen; a speaker for Astellas and Merck; and an investigator for AbbVie, Astellas, Amgen, Leo Pharma, Novartis, Janssen, Sun Pharma, Lilly, Pfizer, and Celgene; LF an investigator for Pfizer, Amgen, Leo, and received honoraria as speaker and participant of advisory boards from the same companies. She is also a speaker for Pierre Fabre and Galderma; MG has been an investigator, speaker or advisory board member for Abbvie, Akros, Amgen, Arcutis, Boehringer Ingelheim, BMS, Celgene, Dermira, Dermavant, Galderma, GSK, Eli Lilly, Incyte, Janssen, Kyowa Kirin, Leo Pharma, Medimmune, Merck, Novartis, Pfizer, Regeneron, Sanofi Genzyme, Sun Pharmaceuticals, UCB, Valeant/Bausch; LG has been a consultant, advisory board member, speaker and clinical trials principal investigator for AbbVie, Eli Lilly, LaRoche Posay, Leo Pharma, Pfizer and Roche, and a consultant, advisory board member and speaker for Bausch Health, Johnson & Johnson, and Sanofi Aventis. SH has been a consultant and/or advisor and/or investigator for or received honoraria from Abbvie, Akros, Altius Healthcare, Amgen, Aralez, Arcutis, Bausch Health, Biopharma, BMS, Boehringer-Ingelheim, Celgene, Coherus, Concert Pharma, Cutanea, Dermira, Galderma, Glenmark, Incyte, Janssen, Leo, Lilly, Novartis, Pedia-Pharm, Pfizer, Regeneron, Sandoz, Sanofi, Sun Pharma, UCB; HH has been a consultant and/or advisor and/or investigator for or received honoraria from Abbvie, Amgen, Arcutis, Bausch Health, Boerhinger Ingelheim, Bristol Meyers Squibb, Celgene, Dermavant, Dermira, DS Biopharma, Eli Lilly, Galderma, GlaxoSmithKline, Incyte, Janssen, Leo Pharma, Medimmune, Merck, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharma, and UCB; PL has been a consultant and/or advisor and/or investigator for or received honoraria from Abbvie, Amgen, Aralez, Bausch Health, Boehringer-Ingelheim, Celgene, Dermira, Galderma, Janssen, Leo, Lilly, Novartis, Pfizer, Regeneron, Sanofi, Sun Pharma; DM has been a consultant and/or investigator and/or speaker for Abbvie, Celgene, Janssen, Leo Pharma, Lilly, Novartis, Pfizer, Regeneron, Sanofi, UCB; MW reports having received honoraria for ad board participation from Novartis, Sun Pharma and Pfizer; JY has been a speaker, consultant, and investigator for AbbVie, Allergan, Amgen, Arcutis, Astellas, Bausch Health, Boehringer Ingelheim, Bristol Meyers Squibb, Celgene, Centocor, Coherus, Dermavant, Dermira, Forward, Galderma, GlaxoSmithKline, Incyte, Janssen, Kyowa, Leo Pharma, Lilly, Medimmune, Merck, Novartis, Pfizer, Regeneron, Roche, Sandoz, Sanofi Genzyme, Sun Pharma, Takeda, UCB, and Xenon. CL was a consultant, speaker, and advisory board member for Amgen, Pfizer, AbbVie, Janssen, Novartis, and Celgene, and was an investigator for Amgen, Pfizer, AbbVie, Janssen, Lilly, Novartis, and Celgene. Dr Poulin was a speaker and advisory board member for AbbVie, Amgen, and Janssen, and was an investigator for AbbVie, Amgen, Celgene, Centocor, Lilly, Galderma, Incyte, Sun Pharma, Janssen, Leo Pharma, Merck, Novartis, Pfizer, and Roche; IVL received grant funding from Novartis, Merck, AbbVie, and Bristol Myers Squibb and honoraria from Janssen, Bausch, Galderma, Novartis, Pfizer, Sun Pharmaceuticals, Johnson & Johnson, and Actelion.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors acknowledge and thank RBC Consultants, for their invaluable assistance with preparing this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmed.2021.682547/full#supplementary-material
References
1. DaVeiga SP. Epidemiology of atopic dermatitis: a review. Allergy Asthma Proc. (2012) 33:227–34. doi: 10.2500/aap.2012.33.3569
2. Bartolucci S, Netchiporouk E, Litvinov IV. Recent therapeutic advances in pruritus management for atopic dermatitis patients: a welcome addition of asivatrep to our arsenal of future topical treatments. J Cutan Med Surg. (2019) 23:551–2. doi: 10.1177/1203475419860501
3. Netchiporouk E, Cohen BA. Recognizing and managing eczematous id reactions to molluscum contagiosum virus in children. Pediatrics. (2012) 129:e1072–5. doi: 10.1542/peds.2011-1054
4. Ashbaugh AG, Kwatra SG. Atopic dermatitis disease complications. Adv Exp Med Biol. (2017) 1027:47–55. doi: 10.1007/978-3-319-64804-0_5
5. Gooderham M, Lynde CW, Papp K, Bourcier M, Guenther L, Gulliver W, et al. Review of systemic treatment options for adult atopic dermatitis. J Cutan Med Surg. (2017) 21:31–9. doi: 10.1177/1203475416670364
6. Weidinger S, Novak N. Atopic dermatitis. Lancet. (2016) 387:1109–22. doi: 10.1016/S0140-6736(15)00149-X
7. Drucker AM, Wang AR, Li WQ, Sevetson E, Block JK, Qureshi AA. The burden of atopic dermatitis: summary of a report for the national eczema association. J Invest Dermatol. (2017) 137:26–30. doi: 10.1016/j.jid.2016.07.012
8. Furue K, Ito T, Tsuji G, Ulzii D, Vu YH, Kido-Nakahara M, et al. The IL-13-OVOL1-FLG axis in atopic dermatitis. Immunology. (2019) 158:281–6. doi: 10.1111/imm.13120
9. Giustizieri ML, Mascia F, Frezzolini A, De Pità O, Chinni LM, Giannetti A, et al. Keratinocytes from patients with atopic dermatitis and psoriasis show a distinct chemokine production profile in response to T cell-derived cytokines. J Allergy Clin Immunol. (2001) 107:871–7. doi: 10.1067/mai.2001.114707
10. Brunner PM, Guttman-Yassky E, Leung DY. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J Allergy Clin Immunol. (2017) 139:S65–76. doi: 10.1016/j.jaci.2017.01.011
11. Sidbury R, Tom WL, Bergman JN, Cooper KD, Silverman RA, Berger TG, et al. Guidelines of care for the management of atopic dermatitis: section 4. Prevention of disease flares and use of adjunctive therapies and approaches. J Am Acad Dermatol. (2014) 71:1218–33. doi: 10.1016/j.jaad.2014.08.038
12. Wong ITY, Tsuyuki RT, Cresswell-Melville A, Doiron P, Drucker AM. Guidelines for the management of atopic dermatitis (eczema) for pharmacists. Can Pharm J. (2017) 150:285–97. doi: 10.1177/1715163517710958
13. Lee JH, Son SW, Cho SH. A comprehensive review of the treatment of atopic eczema. Allergy Asthma Immunol Res. (2016) 8:181–90. doi: 10.4168/aair.2016.8.3.181
14. Torres AE, Lyons AB, Hamzavi IH, Lim HW. Role of phototherapy in the era of biologics. J Am Acad Dermatol. (2021) 84:479–85. doi: 10.1016/j.jaad.2020.04.095
15. Association NE. People With Atopic Dermatitis Want More and Better Treatment Options Novato, CA: National Eczema Association.(2016). Available online at: https://nationaleczema.org/in-your-words/
16. Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. (2016) 375:2335–48. doi: 10.1056/NEJMoa1610020
17. Khosravi H, Zhang S, Anderson AM, Ferris LK, Choudhary S, Patton T. Dupilumab drug survival, treatment failures, and insurance approval at a tertiary care center in the United States. J Am Acad Dermatol. (2020) 82:1023–4. doi: 10.1016/j.jaad.2019.12.034
18. de Wijs LEM, Bosma AL, Erler NS, Hollestein LM, Gerbens LAA, Middelkamp-Hup MA, et al. Effectiveness of dupilumab treatment in 95 patients with atopic dermatitis: daily practice data. Br J Dermatol. (2020) 182:418–26. doi: 10.1111/bjd.18179
19. Wang C, Kraus CN, Patel KG, Ganesan AK, Grando SA. Real-world experience of dupilumab treatment for atopic dermatitis in adults: a retrospective analysis of patients' records. Int J Dermatol. (2020) 59:253–6. doi: 10.1111/ijd.14573
20. Ratnarajah K, Le M, Muntyanu A, Mathieu S, Nigen S, Litvinov IV, et al. Inhibition of IL-13: a new pathway for atopic dermatitis. J Cutan Med Surg. (2020) 25:315–28. doi: 10.1177/1203475420982553
21. Wollenberg A, Howell MD, Guttman-Yassky E, Silverberg JI, Kell C, Ranade K, et al. Treatment of atopic dermatitis with tralokinumab, an anti–IL-13 mAb. J Allergy Clin Immunol. (2019) 143:135–41. doi: 10.1016/j.jaci.2018.05.029
22. Kabashima K, Furue M, Hanifin JM, Pulka G, Wollenberg A, Galus R, et al. Nemolizumab in patients with moderate-to-severe atopic dermatitis: randomized, phase II, long-term extension study. J Allergy Clin Immunol. (2018) 142:1121–30.e7. doi: 10.1016/j.jaci.2018.03.018
23. Simpson EL, Flohr C, Eichenfield LF, Bieber T, Sofen H, Taïeb A, et al. Efficacy and safety of lebrikizumab (an anti-IL-13 monoclonal antibody) in adults with moderate-to-severe atopic dermatitis inadequately controlled by topical corticosteroids: a randomized, placebo-controlled phase II trial (TREBLE). J Am Acad Dermatol. (2018) 78:863–71.e11. doi: 10.1016/j.jaad.2018.01.017
24. Smith D, Helgason H, Sulem P, Bjornsdottir US, Lim AC, Sveinbjornsson G, et al. A rare IL33 loss-of-function mutation reduces blood eosinophil counts and protects from asthma. PLoS genetics. (2017) 13:e1006659. doi: 10.1371/journal.pgen.1006659
25. Amano W, Nakajima S, Kunugi H, Numata Y, Kitoh A, Egawa G, et al. The Janus kinase inhibitor JTE-052 improves skin barrier function through suppressing signal transducer and activator of transcription 3 signaling. J Allergy Clin Immunol. (2015) 136:667–77.e7. doi: 10.1016/j.jaci.2015.03.051
26. Ghoreschi K, Laurence A, O'Shea JJ. Janus kinases in immune cell signaling. Immunol Rev. (2009) 228:273–87. doi: 10.1111/j.1600-065X.2008.00754.x
27. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
28. Solimani F, Meier K, Ghoreschi K. Emerging topical and systemic JAK inhibitors in dermatology. Front Immunol. (2019) 10:2847. doi: 10.3389/fimmu.2019.02847
29. Sterne JAC, Savović J, Page MJ, Elbers RG, Blencowe NS, Boutron I, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. (2019) 366:l4898. doi: 10.1136/bmj.l4898
30. Ma LL, Wang YY, Yang ZH, Huang D, Weng H, Zeng XT. Methodological quality (risk of bias) assessment tools for primary and secondary medical studies: what are they and which is better? Mil Med Res. (2020) 7:7. doi: 10.1186/s40779-020-00238-8
31. Higgins JPT, Savović J, Page MJ, Elbers RG, Sterne JAC. Assessing risk of bias in a randomized trial. In: Higgins JPT, Thomas J, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editors. Cochrane Handbook for Systematic Reviews of Interventions. Chichester: John Wiley and Sons (2019). p. 205–28. doi: 10.1002/9781119536604.ch8
32. Moher D, Liberati A, Tetzlaff J, Altman DG, The PG. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. (2009) 6:e1000097. doi: 10.1371/journal.pmed.1000097
33. Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. (2020) 396:255–66. doi: 10.1016/S0140-6736(20)30732-7
34. Silverberg JI, Simpson EL, Thyssen JP, Gooderham M, Chan G, Feeney C, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. (2020) 156:863–73. doi: 10.1001/jamadermatol.2020.1406
35. Simpson EL, Lacour JP, Spelman L, Galimberti R, Eichenfield LF, Bissonnette R, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol. (2020) 183:242–55. doi: 10.1111/bjd.18898
36. Reich K, Kabashima K, Peris K, Silverberg JI, Eichenfield LF, Bieber T, et al. Efficacy and safety of baricitinib combined with topical corticosteroids for treatment of moderate to severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. (2020) 156:1333–43. doi: 10.1001/jamadermatol.2020.3260
37. Guttman-Yassky E, Thaci D, Pangan AL, Hong HCH, Papp KA, Reich K, et al. Upadacitinib in adults with moderate to severe atopic dermatitis: 16-week results from a randomized, placebo-controlled trial. J Allergy Clin Immunol. (2020) 145:877–84. doi: 10.1016/j.jaci.2019.11.025
38. Bissonnette R, Maari C, Forman S, Bhatia N, Lee M, Fowler J, et al. The oral Janus kinase/spleen tyrosine kinase inhibitor ASN002 demonstrates efficacy and improves associated systemic inflammation in patients with moderate-to-severe atopic dermatitis: results from a randomized double-blind placebo-controlled study. Br J Dermatol. (2019) 181:733–42. doi: 10.1111/bjd.17932
39. Bieber T, Simpson EL, Silverberg JI, Thaçi D, Paul C, Pink AE, et al. Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. (2021) 384:1101-12. doi: 10.1056/NEJMoa2019380
40. Guttman-Yassky E, Teixeira HD, Simpson EL, Papp KA, Pangan AL, Blauvelt A, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. (2021) 397:2151-68. doi: 10.1016/S0140-6736(21)00588-2
41. Reich K, Teixeira HD, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2021) 397:2169-81. doi: 10.1016/S0140-6736(21)00589-4
42. Gooderham MJ, Forman SB, Bissonnette R, Beebe JS, Zhang W, Banfield C, et al. Efficacy and safety of oral janus kinase 1 inhibitor abrocitinib for patients with atopic dermatitis: a phase 2 randomized clinical trial. JAMA Dermatol. (2019) 155:1371–9. doi: 10.1001/jamadermatol.2019.2855
43. Guttman-Yassky E, Silverberg JI, Nemoto O, Forman SB, Wilke A, Prescilla R, et al. Baricitinib in adult patients with moderate-to-severe atopic dermatitis: a phase 2 parallel, double-blinded, randomized placebo-controlled multiple-dose study. J Am Acad Dermatol. (2019) 80:913–21.e9. doi: 10.1016/j.jaad.2018.01.018
44. Guttman-Yassky E, Beck LA, Anderson JK, Hu X, Gu Y, Teixeira HD, et al. Upadacitinib treatment withdrawal and retreatment in patients with moderate-to-severe atopic dermatitis from a phase 2b, randomized, controlled trial. J Am Acad Dermatol. (2019) 81(4 Suppl. 1):AB294. doi: 10.1016/j.jaad.2019.06.1265
45. Schmieder GJ, Draelos ZD, Pariser DM, Banfield C, Cox L, Hodge M, et al. Efficacy and safety of the Janus kinase 1 inhibitor PF-04965842 in patients with moderate-to-severe psoriasis: phase II, randomized, double-blind, placebo-controlled study. Br J Dermatol. (2018) 179:54–62. doi: 10.1111/bjd.16004
46. Fridman JS, Scherle PA, Collins R, Burn TC, Li Y, Li J, et al. Selective inhibition of JAK1 and JAK2 is efficacious in rodent models of arthritis: preclinical characterization of INCB028050. J Immunol. (2010) 184:5298–307. doi: 10.4049/jimmunol.0902819
47. Papp KA, Krueger JG, Feldman SR, Langley RG, Thaci D, Torii H, et al. Tofacitinib, an oral Janus kinase inhibitor, for the treatment of chronic plaque psoriasis: long-term efficacy and safety results from 2 randomized phase-III studies and 1 open-label long-term extension study. J Am Acad Dermatol. (2016) 74:841–50. doi: 10.1016/j.jaad.2016.01.013
48. Gladman D, Rigby W, Azevedo VF, Behrens F, Blanco R, Kaszuba A, et al. Tofacitinib for psoriatic arthritis in patients with an inadequate response to TNF inhibitors. N Engl J Med. (2017) 377:1525–36. doi: 10.1056/NEJMoa1615977
49. Jorgensen SC, Tse CL, Burry L, Dresser LD. Baricitinib: a review of pharmacology, safety, and emerging clinical experience in COVID-19. Pharmacotherapy. (2020) 40:843–56. doi: 10.1002/phar.2438
50. Sandborn WJ, Ghosh S, Panes J, Schreiber S, D'Haens G, Tanida S, et al. Efficacy of upadacitinib in a randomized trial of patients with active ulcerative colitis. Gastroenterology. (2020) 158:2139–49.e14. doi: 10.1053/j.gastro.2020.02.030
51. van der Heijde D, Song I-H, Pangan AL, Deodhar A, Van den Bosch F, Maksymowych WP, et al. Efficacy and safety of upadacitinib in patients with active ankylosing spondylitis (SELECT-AXIS 1): a multicentre, randomised, double-blind, placebo-controlled, phase 2/3 trial. The Lancet. (2019) 394:2108–17. doi: 10.1016/S0140-6736(19)32534-6
52. Fleischmann R, Pangan AL, Song IH, Mysler E, Bessette L, Peterfy C, et al. Upadacitinib versus placebo or adalimumab in patients with rheumatoid arthritis and an inadequate response to methotrexate: results of a phase III, double-blind, randomized controlled trial. Arthritis Rheumatol. (2019) 71:1788–800. doi: 10.1002/art.41032
53. Anjara P, Jiang M, Mundae M. Symptomatic elevation creatine kinase following treatment of rheumatoid arthritis with baricitinib. Clin Rheumatol. (2020) 39:613–4. doi: 10.1007/s10067-019-04833-6
54. Wolk R, Armstrong EJ, Hansen PR, Thiers B, Lan S, Tallman AM, et al. Effect of tofacitinib on lipid levels and lipid-related parameters in patients with moderate to severe psoriasis. J Clin Lipidol. (2017) 11:1243–56. doi: 10.1016/j.jacl.2017.06.012
55. Qiu C, Zhao X, She L, Shi Z, Deng Z, Tan L, et al. Baricitinib induces LDL-C and HDL-C increases in rheumatoid arthritis: a meta-analysis of randomized controlled trials. Lipids Health Dis. (2019) 18:54. doi: 10.1186/s12944-019-0994-7
56. Taylor PC, Kremer JM, Emery P, Zuckerman SH, Ruotolo G, Zhong J, et al. Lipid profile and effect of statin treatment in pooled phase II and phase III baricitinib studies. Ann Rheum Dis. (2018) 77:988–95. doi: 10.1136/annrheumdis-2017-212461
57. Ibrahim F, Scott DL. Thromboembolism and Janus kinase inhibitors. Drug Safety. (2020) 43:831–3. doi: 10.1007/s40264-020-00973-w
58. European Medicines Agency. Oluminant: Summary of Product Characteristics. (2017). Available online at: https://www.ema.europa.eu/en/medicines/human/EPAR/olumiant
59. US Food and Drug Administration. Safety trial finds risk of blood clots in the lungs and death with higher dose of tofacitinib (Xeljanz, Xeljanz XR) in rheumatoid arthritis patients; FDA to investigate. Saf. Announc. (2019). Available online at: https://www.fda.gov/drugs/drug-safety-and-availability/safety-trial-finds-risk-bloodclots-lungs-and-death-higher-dose-tofacitinib-xeljanz-xeljanz-xr
60. Olivera PA, Lasa JS, Bonovas S, Danese S, Peyrin-Biroulet L. Safety of Janus kinase inhibitors in patients with inflammatory bowel diseases or other immune-mediated diseases: a systematic review and meta-analysis. Gastroenterology. (2020) 158:1554–73. doi: 10.1053/j.gastro.2020.01.001
61. Harigai M. Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology. (2019) 58(Suppl. 1):i34–42. doi: 10.1093/rheumatology/key287
62. Sivaraman P, Cohen SB. Malignancy and Janus kinase inhibition. Rheum Dis Clin North Am. (2017) 43:79–93. doi: 10.1016/j.rdc.2016.09.008
63. Wollenhaupt J, Lee EB, Curtis JR, Silverfield J, Terry K, Soma K, et al. Safety and efficacy of tofacitinib for up to 9.5 years in the treatment of rheumatoid arthritis: final results of a global, open-label, long-term extension study. Arthritis Res Ther. (2019) 21:89. doi: 10.1186/s13075-019-1866-2
64. Genovese MC, Smolen JS, Takeuchi T, Burmester G, Brinker D, Rooney TP, et al. Safety profile of baricitinib for the treatment of rheumatoid arthritis over a median of 3 years of treatment: an updated integrated safety analysis. Lancet Rheumatol. (2020) 2:e347–57. doi: 10.1016/S2665-9913(20)30032-1
Keywords: JAK inhibitor, janus kinase, atopic dermatitis, eczema, abrocitinib, baricitinib, gusacitinib, upadacitinib
Citation: Le M, Berman-Rosa M, Ghazawi FM, Bourcier M, Fiorillo L, Gooderham M, Guenther L, Hanna S, Hong HC-H, Landells I, Lansang P, Marcoux D, Wiseman MC, Yeung J, Lynde C and Litvinov IV (2021) Systematic Review on the Efficacy and Safety of Oral Janus Kinase Inhibitors for the Treatment of Atopic Dermatitis. Front. Med. 8:682547. doi: 10.3389/fmed.2021.682547
Received: 18 March 2021; Accepted: 21 June 2021;
Published: 01 September 2021.
Edited by:
Oleg E. Akilov, University of Pittsburgh, United StatesReviewed by:
Andreas Wollenberg, LMU Munich University Hospital, GermanyRobert Stewart Dawe, NHS Tayside, United Kingdom
Copyright © 2021 Le, Berman-Rosa, Ghazawi, Bourcier, Fiorillo, Gooderham, Guenther, Hanna, Hong, Landells, Lansang, Marcoux, Wiseman, Yeung, Lynde and Litvinov. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ivan V. Litvinov, ivan.litvinov@mcgill.ca; Charles Lynde, derma@lynderma.com