Biomarkers in Progressive Fibrosing Interstitial Lung Disease: Optimizing Diagnosis, Prognosis, and Treatment Response
 Willis S. Bowman
Willis S. Bowman Gabrielle A. Echt
Gabrielle A. Echt Justin M. Oldham
Justin M. Oldham- Division of Pulmonary, Critical Care, and Sleep Medicine, University of California, Davis, Davis, CA, United States
Interstitial lung disease (ILD) comprises a heterogenous group of diffuse lung disorders that commonly result in irreversible pulmonary fibrosis. While idiopathic pulmonary fibrosis (IPF) is the prototypical progressive fibrosing ILD (PF-ILD), a high proportion of patients with other ILD subtypes develop a PF-ILD phenotype. Evidence exists for shared pathobiology leading to progressive fibrosis, suggesting that biomarkers of disease activity may prove informative across the wide spectrum of ILDs. Biomarker investigation to date has identified a number of molecular markers that predict relevant ILD endpoints, including disease presence, prognosis, and/or treatment response. In this review, we provide an overview of potentially informative biomarkers in patients with ILD, including those suggestive of a PF-ILD phenotype. We highlight the recent genomic, transcriptomic, and proteomic investigations that identified these biomarkers and discuss the body compartments in which they are found, including the peripheral blood, airway, and lung parenchyma. Finally, we identify critical gaps in knowledge within the field of ILD biomarker research and propose steps to advance the field toward biomarker implementation.
Introduction
Interstitial lung disease (ILD) comprises a heterogeneous group of diffuse parenchymal lung processes characterized by variable degrees of inflammation and fibrosis. While etiology varies by ILD subtype, studies suggest that pathobiology leading to pulmonary fibrosis may be shared across subtypes (1, 2). Idiopathic pulmonary fibrosis (IPF) is among the most common forms of fibrotic-predominant ILD and is characterized by progressive parenchymal fibrosis leading to clinical deterioration and high mortality (3). Other common forms of ILD, including connective tissue-associated ILD (CTD-ILD), chronic hypersensitivity pneumonitis (CHP), and unclassifiable ILD (uILD) manifest variable patterns of inflammation and fibrosis and can approximate the natural history of IPF when progressive (2, 4). Immunosuppressive therapy is thought to provide benefit for patients with inflammatory ILD (5, 6), but was found to cause harm to patients with IPF (7), underscoring the importance of an accurate diagnosis. Patients with IPF are now treated with antifibrotic therapy, which slowed disease progression in several randomized controlled trials (8–11). More recently, anti-fibrotic therapy was shown to provide similar benefit in patients with CTD-ILD, CHP, uILD, and others with progressive fibrosing ILD (PF-ILD) phenotype (12, 13).
Despite advances in the treatment of PF-ILD, predicting a progressive phenotype remains difficult. Biomarkers have advanced our understanding of IPF and are now beginning to inform diagnosis, prognosis, and treatment in patients with ILD, including those with PF-ILD. In this review we provide an overview of the current state of ILD biomarker research, including relevant biomarkers to help discriminate ILDs, prognosticate disease course, and assess treatment response. Finally, we highlight unmet needs in ILD biomarker research and discuss strategies for biomarker implementation.
The PF-ILD Phenotype
A number of clinical criteria for identifying a PF-ILD phenotype have been proposed. While few have yet been validated, they generally utilize longitudinal measures of progressive disease, including lung function decline, worsening respiratory symptoms, and increasing extent of radiologic fibrosis (12–15). Defining PF-ILD by presence of these features stems largely from their known association with worse outcomes in ILD. Longitudinal decline in forced vital capacity (FVC) is perhaps the best near-term predictor of subsequent mortality. While a categorical FVC decline of ≥10% has been associated with an increased mortality risk in patients with IPF (16–19), CHP (20–22), and CTD-ILD (23), even modest FVC decline of≥5% decline has been linked to worse outcome in IPF (16). Baseline extent of fibrosis on high-resolution computed tomography (HRCT) has been linked to prognosis in different ILD subtypes (24, 25), even in the absence of FVC decline (26), and suggests that longitudinal increase in fibrosis extent is a good measure of progressive ILD (25, 27, 28). Symptomatic worsening, either alone or in combination with FVC decline or fibrotic progression on HRCT also suggests ILD worsening, but has yet been to validated as a reliable predictor of outcome.
Biomarker Acquisition
A biomarker is defined as an indicator of normal biological processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions (29). Sources of biomarkers that may inform diagnosis, outcomes, and treatment response in ILD include the peripheral blood, airway, and lung parenchyma. Peripheral blood is easily obtained, and acquisition requires little training beyond phlebotomy. While airway biomarkers may be obtained non-invasively via exhaled breath, most studies to date have employed bronchoscopy to obtain these biomarkers via bronchoalveolar lavage. Obtaining parenchymal biomarkers requires increasingly invasive bronchoscopic approaches depending on specimen size or by surgical lung biopsy, usually by way of video-assisted thoracoscopic surgery. Novel functional and quantitative imaging techniques are also emerging as non-invasive parenchymal biomarkers (30–32).
While peripheral blood is easy to obtain in cross-section and serially over time, biomarkers from this compartment may not reflect pathobiologic processes in the lung. Conversely, while those obtained from the airway or lung parenchyma may better reflect the parenchymal pathobiology, invasive techniques are required generally for acquisition and limit the widespread use of these modalities along with serial biospecimen acquisition. Radiologic biomarkers do allow for serial acquisition, but radiation exposure remains a consideration, especially among younger individuals with ILD. Biomarkers within each compartment of interest can be further classified according to the endpoints with which they are associated, including disease discrimination, prognosis, and treatment response. Below we highlight key biomarkers within each compartment and the endpoints they help predict in patients with ILD.
Peripheral Blood Biomarkers
Diagnostic
A large number of plasma and serum protein biomarkers discriminate ILD from healthy controls, ILD within in a disease state, and between ILD subtypes (Table 1) (33–69, 71–104). Despite the considerable work performed in this arena, test performance for individual biomarkers has generally been insufficient to justify their incorporation into clinical practice. Recent investigations have demonstrated increased potential when modeling plasma biomarkers in aggregate. Doyle et al. demonstrated matrix metalloproteinase 7 (MMP-7), pulmonary and activation-related chemokine (PARC), and surfactant-protein D (SP-D) effectively predicted ILD in patients with rheumatoid arthritis (RA) when modeled with clinical characteristics (84). White and colleagues then employed a similar approach, using plasma MMP-7, SP-D, and osteopontin concentration to derive and validate a protein prediction score to distinguish IPF from healthy controls and most alternative ILDs (85). One group for which this scoring system performed poorly was RA-ILD, which shares morphologic features with IPF. Recently, Sanders et al. showed that biomarkers hold promise for identifying early ILD, termed interstitial lung abnormalities (ILAs). These authors found that GDF15 and four other plasma biomarkers were increased in community dwelling adults with ILAs (48), with GDF15 validated in an independent cohort. These findings suggest that plasma biomarkers may eventually allow for non-invasive ILD screening (105).

Table 1. Diagnostic ILD plasma/serum biomarkers.
Genomic investigation of DNA acquired from peripheral blood has identified several common gene variants associated with ILD. The variant with strongest effect is a polymorphism in the promoter region of MUC5B, which encodes a mucin producing gene critical for airway host defense (106). The presence of this variant increases the risk of developing IPF by 5-fold (107–109) and was recently shown to increase the risk of developing rheumatoid arthritis (RA)-associated ILD, especially with concurrent usual interstitial pneumonia (UIP) pattern (110, 111). Interestingly, the MUC5B promoter variant does not appear to increase risk for ILD due to systemic sclerosis or anti-synthetase syndrome (112, 113). A number of other variants have been linked to IPF susceptibility but with smaller effect association (109, 114, 115). While these studies were informative, it is unlikely that common gene variants, including the MUC5B promoter variant, will allow for cost-effective ILD screening, as such variants are found in a large minority of unaffected individuals.
Prognostic
In addition to discriminating ILD presence, many of the biomarkers described above have also been linked to prognosis, including survival, near-term lung function decline and ILD exacerbation (Table 2) (116–153). Plasma biomarkers most commonly linked to differential survival include chemokine (C-C motif) ligand 18 (CCL18) (44, 126, 127, 129, 130), Krebs von den Lungen 6 (KL-6) (131, 133, 138, 142), chitinase-3-like protein 1 (YKL-40) (101, 104, 117), cancer antigen 125 (CA-125) (117–119), and MMP-7, (117, 121, 145, 153) which have been shown to predict this endpoint across diverse forms of ILD. Our group recently showed that several of these biomarkers predicted differential survival in both antifibrotic treated and untreated patients with IPF, but at higher categorical thresholds in anti-fibrotic treated patients (117).
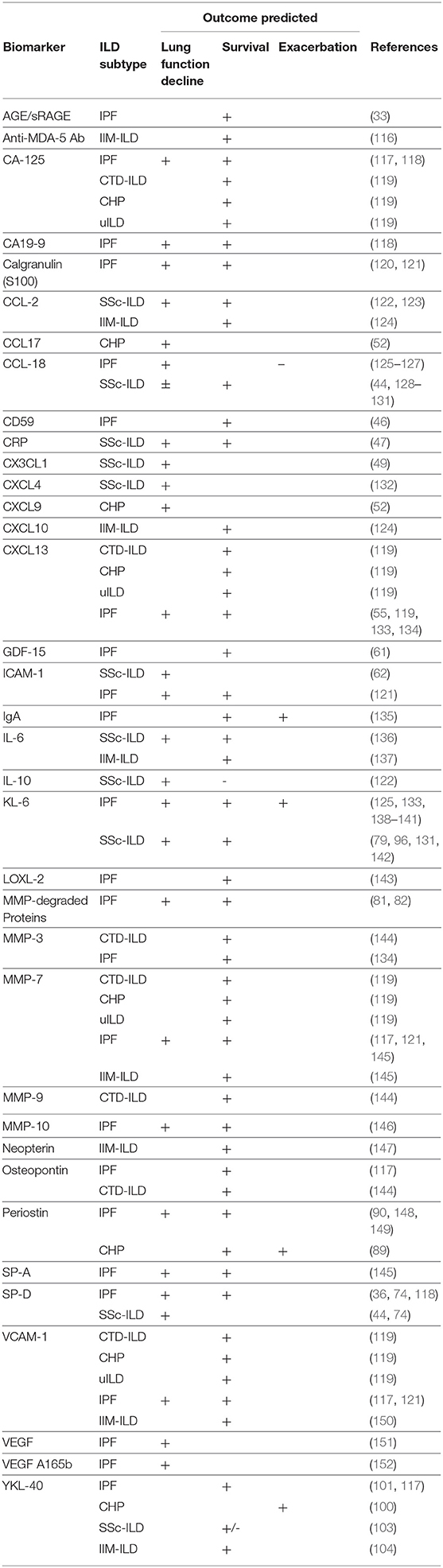
Table 2. Prognostic ILD plasma/serum biomarkers.
While most biomarkers are measured in cross-section, Maher and colleagues showed longitudinal change in plasma CA-125 concentration to predict subsequent mortality in a prospectively recruited IPF cohort (118). Neoepitopes of protein fragments created during extracellular matrix turnover may also prove to be valuable biomarkers. Several have been shown to predict subsequent IPF survival when modeled in cross-section and over time (81, 82). Additional novel plasma biomarkers of ILD survival are expected in the coming years as high-throughput proteomic platforms are increasingly utilized (154).
Other blood-based biomarkers also hold potential. Monocyte count, obtained as part of a complete blood count, was recently shown to predict increased mortality across multiple IPF cohorts (155). Kreuter et al. also showed increasing monocyte count to predict near-term hospitalization and IPF progression, as measured by death, decreasing walk distance or ≥10% categorical decline in FVC (156). Because FVC decline is among the earliest objective indicators of a progressive phenotype and change in FVC among the most commonly utilized primary endpoint in clinical trials, biomarkers predictive of FVC decline are of particular importance for identifying early progression and enriching clinical trial cohorts. CCL18 (126), MMP7 (157), SP-D (74, 158), and interleukin-6 (IL-6) (136) have each demonstrated potential in predicting near-term FVC decline in patients with ILD, though none with sufficient risk explanation to justify clinical implementation to date.
Genomic biomarkers have also been linked to differential ILD survival. The MUC5B promoter SNP, despite increasing IPF risk (108), was paradoxically associated with reduced mortality in IPF (159). This observation may be been driven by index event bias (160), leaving it unclear what role MUC5B plays in ILD progression. The same polymorphism has also been associated with increased mortality risk in patients with CHP (161). Short leukocyte telomere length has been shown to predict survival across numerous ILD cohorts and subtypes (161–163) and patients with rare mutations in telomerase encoding genes, including TERT, TERC, PARN, and RTEL1 were shown to display survival similar to IPF irrespective of the clinical ILD diagnosis (164). Recent studies have also suggested circulating mitochondrial DNA may be a relevant biomarker for predicting IPF outcomes (165, 166). Transcriptomic biomarkers also appear informative, as a 52-gene signature has been shown to reliably predict IPF survival across numerous cohorts in the US and Europe (167, 168).
Treatment Response
Compared to the biomarkers of diagnostic and prognostic utility in ILD, there is less evidence supporting the role of biomarkers to predict or monitor treatment response in patients with ILD. Serial measurements of KL-6, SP-D, IL-6, CXCL-4, and C-reactive protein (CRP) among others have correlated with lung function changes following immunosuppressive treatment in SSc-ILD (51, 169), RA-ILD (170), and IIM-ILD (171). Assassi and colleagues recently developed a prediction model for response to cyclophosphamide and mycophenolate in SSc-ILD using interferon-induced serum proteins (172). Ikeda et al. showed plasma SP-D concentration may predict favorable response to pirfenidone (173) in patients with IPF, though Neighbors and colleagues showed consistent pirfenidone treatment effect irrespective of plasma concentration for a panel of prospectively collected candidate biomarkers (126).
Genomic biomarkers have also suggested potential for predicting differential treatment response. Two post-hoc analyses of the PANTHER-IPF trial have been of particular interest. The first showed a common gene polymorphism in TOLLIP to predict differential progression in patients treated with N-acetylcysteine (174). The next showed short leukocyte telomere length was associated with worse outcome in patients treated with immunosuppressant therapy (175). The former provided the rationale for the recently funded IPF PRECISIONS trial (NCT 04300920) which will test this observation prospectively, while the latter has raised the question of whether immunosuppressant therapy should be used for other forms of ILD in the setting of short telomere length. Adegunsoye et al. recently demonstrated similar outcomes among CHP patients with short telomere length treated with mycophenolate mofetil, though sample size was small (176). At present, telomere length does not appear to influence response to anti-fibrotic therapy in patients with IPF (177).
Airway Biomarkers
Diagnostic
Bronchoalveolar lavage (BAL) has a long and controversial history in the evaluation of ILD (178, 179). Its primary use remains discriminating CHP from other forms of ILD, which is supported by elevated lymphocyte count (180). Accordingly, BAL with cellular analysis is now formally recommended to assist in the diagnosis of CHP (181) and has received a conditional recommendation for excluding other potential ILD causes when diagnosing IPF (182). BAL protein biomarkers may help differentiate ILD subtypes, including organizing pneumonia from UIP in patients with RA-ILD (183), IPF from other forms of ILD (93, 184–186), as well as CHP from IPF (100). Exhaled breath analysis of volatile organic compounds is an evolving, but promising modality for discriminating ILD subtypes that may 1 day provide a non-invasive alternative to BAL (187).
Prognostic
Airway biomarkers of ILD outcomes have been identified, but studies are limited relative to those performed in peripheral blood. Some studies have assessed whether BAL cellular makeup can predict outcome in patients with different ILD subtypes, but results have had mixed (188–191). BAL protein biomarkers, including YKL-40, IL-15, IL-2, and TNF have been linked to differential survival in various ILD subtypes (101, 150, 192), but these studies have been generally limited by small sample sizes. Airway biomarkers of near-term FVC function decline are also limited, but BAL interferon gamma and transforming growth factor beta may predict progressive RA-ILD (193). Norman et al. recently showed that a BAL fluid proteomic signature may predict near-term IPF progression, with high concentrations of immune-regulatory proteins being associated with slower progression (194).
Parenchymal Lung Biomarkers
Diagnostic
Transcriptomic analysis of lung tissue has emerged as an exciting arena of biomarker research. Furusawa et al. recently employed this methodology to identify gene signatures unique to patients with IPF and CHP that may allow for better diagnostic discrimination if this can be implemented using less invasive measures (195). Transcriptomic analysis of lung tissue obtained by transbronchial biopsy has resulted in the first commercially available ILD biomarker. The Envisia® genomic classifier (Veracyte, South San Francisco, CA) predicts the presence of histologic usual interstitial pneumonia using a proprietary gene expression-based signature (196). This tool has been shown to predict histologic UIP with good test performance and increases diagnostic confidence for IPF when positive (197, 198).
Several unanswered questions need to be addressed before widespread use of Envisia in the ILD evaluation. First, because UIP can be observed in other ILD subtypes, including RA-ILD, SSc-ILD, asbestosis, and CHP (2, 199–202), it remains unclear how well this molecular diagnostic tool discriminates UIP due to IPF from UIP seen in these other conditions. Next, the radiologic pattern for which this tool is most useful remains unclear. Because those with probable UIP on HRCT can meet IPF criteria in the appropriate clinical setting (203, 204), the utility of genomic UIP in these patients is unclear. Additionally, because those with an alternative diagnosis pattern on HRCT can fail to meet IPF criteria even in the setting of histologic UIP (182), the meaning of genomic UIP in such patients remains unclear. Finally, it remains unclear whether the phenotype identified by this molecular diagnostic tool approximates that of IPF in other ILD subtypes. Demonstrating that genomic UIP follows an IPF-like progressive phenotype will be important to support its clinical utility.
Prognostic
Few parenchymal lung biomarkers have been identified to predict outcomes in patients with ILD. Telomere length in type II alveolar cells is a notable exception that predicts decreased survival in patients with IPF (205). However, telomere length was recently shown to correlate across human tissues (206), suggesting that telomere length measurement in peripheral blood provides a safer and easier alternative to parenchymal-based measures.
Unmet Needs in ILD Biomarker Research
While the last two decades have seen impressive progress made in biomarker discovery, a number of unmet needs remain. First, as PF-ILD evolves as a phenotype, the identification and implementation of biomarkers that predict PF-ILD will be critical to our success as a community. Proposed criteria for identifying patients with PF-ILD rely on objective markers of ILD progression to manifest. Predicting progression before it occurs and initiating appropriate therapy remains our best chance to prevent irreversible fibrosis from developing. Biomarkers could also mitigate the uncertainty surrounding prognosis, CT surveillance, and need for early therapy in the substantial number of asymptomatic older adults with incidentally discovered ILAs. Next, while survival is perhaps the most important ILD outcome, biomarkers of survival are less likely to influence near-term treatment decisions or clinical trial design. Because change in FVC is commonly used to assess treatment response, biomarkers predictive of near-term change in FVC are more likely to be informative and therefore implemented. Such biomarkers also have high potential to enrich clinical trial cohorts with patients most likely to experience near-term FVC decline, thereby reducing sample sizes needed to detect treatment effect. Finally, our ability to predict which patients will respond favorably to specific ILD therapies remains limited. Current measures of ILD disease progression may reflect worsening inflammation or progressive fibrosis but cannot differentiate between the two. The means to reliably discriminate inflammatory from fibrotic lung injury would allow us to predict favorable response to immunosuppression or antifibrotic treatments that could halt or potentially reverse loss of lung function. For precision medicine to become a reality, we must have viable biomarkers to guide this approach.
We and others have demonstrated the ability of aggregated biomarkers to augment risk explanation when compared to biomarkers modeled in isolation. Nearly all validated clinical prediction models for predicting ILD outcomes utilize several clinical parameters. A similar approach is likely necessary with biomarker modeling. Multi-dimensional prediction models that incorporate clinical and molecular data may be better yet. With the emergence of high-throughput genomic, transcriptomic, and proteomic platforms, a number of novel and informative ILD biomarkers are likely to emerge over the next decade. A mechanism to quickly and seamlessly incorporate these novel biomarkers into existing prediction tools will be critical for moving the field forward.
Conclusion
A host of biomarkers drawn from peripheral blood, airway, and parenchymal compartments have proven informative in patients with ILD. These include genomic, transcriptomic, and proteomic determinants of ILD presence, prognosis, and treatment response. While most studies have used IPF as the prototypical PF-ILD, a rapid expansion of this research to include other ILD subtypes meeting validated criteria for PF-ILD is expected in the coming years. Such investigations have high potential to identify biomarkers that transcend clinical ILD diagnosis and instead identify a homogeneous PF-ILD endotype.
Author Contributions
WB, JO, and GE: literature review. WB and JO: manuscript preparation. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by T32HL007013 to WB and K23HL138190 to JO.
Conflict of Interest
JO has received consultancy or speaker fees from Boehringer Ingelheim and Genentech, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Vasakova M, Poletti V. Fibrosing interstitial lung diseases involve different pathogenic pathways with similar outcomes. Sarcoidosis Vasc Diffuse Lung Dis. (2015) 32:246–50.
2. Kolb M, Vasakova M. The natural history of progressive fibrosing interstitial lung diseases. Respir Res. (2019) 20:57. doi: 10.1186/s12931-019-1022-1
3. Nathan SD, Shlobin OA, Weir N, Ahmad S, Kaldjob JM, Battle E, et al. Long-term course and prognosis of idiopathic pulmonary fibrosis in the new millennium. Chest. (2011) 140:221–9. doi: 10.1378/chest.10-2572
4. Jacob J, Bartholmai BJ, Egashira R, Brun AL, Rajagopalan S, Karwoski R, et al. Chronic hypersensitivity pneumonitis: identification of key prognostic determinants using automated CT analysis. BMC Pulm Med. (2017) 17:81. doi: 10.1186/s12890-017-0418-2
5. Tashkin DP, Elashoff R, Clements PJ, Goldin J, Roth MD, Furst DE, et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med. (2006) 354:2655–66. doi: 10.1056/NEJMoa055120
6. Tashkin DP, Roth MD, Clements PJ, Furst DE, Khanna D, Kleerup EC, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. (2016) 4:708–19. doi: 10.1016/S2213-2600(16)30152-7
7. Idiopathic Pulmonary Fibrosis Clinical Research Network, Raghu G, Anstrom KJ, King TE Jr, Lasky JA, Martinez FJ. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N Engl J Med. (2012) 366:1968–77. doi: 10.1056/NEJMoa1113354
8. Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, Costabel U, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2071–82. doi: 10.1056/NEJMoa1402584
9. King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, Glassberg MK, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. (2014) 370:2083–92. doi: 10.1056/NEJMoa1402582
10. Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, Kardatzke D, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. (2011) 377:1760–9. doi: 10.1016/S0140-6736(11)60405-4
11. Richeldi L, Costabel U, Selman M, Kim DS, Hansell DM, Nicholson AG, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. (2011) 365:1079–87. doi: 10.1056/NEJMoa1103690
12. Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med. (2019) 381:1718–27. doi: 10.1056/NEJMoa1908681
13. Maher TM, Corte TJ, Fischer A, Kreuter M, Lederer DJ, Molina-Molina M, et al. Pirfenidone in patients with unclassifiable progressive fibrosing interstitial lung disease: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet Respir Med. (2020) 8:147–57. doi: 10.1016/S2213-2600(19)30341-8
14. Behr J, Neuser P, Prasse A, Kreuter M, Rabe K, Schade-Brittinger C, et al. Exploring efficacy and safety of oral Pirfenidone for progressive, non-IPF lung fibrosis (RELIEF) - a randomized, double-blind, placebo-controlled, parallel group, multi-center, phase II trial. BMC Pulm Med. (2017) 17:122. doi: 10.1186/s12890-017-0462-y
15. George PM, Spagnolo P, Kreuter M, Altinisik G, Bonifazi M, Martinez FJ, et al. Progressive fibrosing interstitial lung disease: clinical uncertainties, consensus recommendations, and research priorities. Lancet Respir Med. (2020) 8:925–34. doi: 10.1016/S2213-2600(20)30355-6
16. Zappala CJ, Latsi PI, Nicholson AG, Colby TV, Cramer D, Renzoni EA, et al. Marginal decline in forced vital capacity is associated with a poor outcome in idiopathic pulmonary fibrosis. Eur Respir J. (2010) 35:830–6. doi: 10.1183/09031936.00155108
17. Reichmann WM, Yu YF, Macaulay D, Wu EQ, Nathan SD. Change in forced vital capacity and associated subsequent outcomes in patients with newly diagnosed idiopathic pulmonary fibrosis. BMC Pulm Med. (2015) 15:167. doi: 10.1186/s12890-015-0161-5
18. du Bois RM, Weycker D, Albera C, Bradford WZ, Costabel U, Kartashov A, et al. Forced vital capacity in patients with idiopathic pulmonary fibrosis: test properties and minimal clinically important difference. Am J Respir Crit Care Med. (2011) 184:1382–9. doi: 10.1164/rccm.201105-0840OC
19. Collard HR, King TE Jr, Bartelson BB, Vourlekis JS, Schwarz MI, Brown KK. Changes in clinical and physiologic variables predict survival in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2003) 168:538–42. doi: 10.1164/rccm.200211-1311OC
20. Goh NS, Hoyles RK, Denton CP, Hansell DM, Renzoni EA, Maher TM, et al. Short-term pulmonary function trends are predictive of mortality in interstitial lung disease associated with systemic sclerosis. Arthritis Rheumatol. (2017) 69:1670–8. doi: 10.1002/art.40130
21. Solomon JJ, Chung JH, Cosgrove GP, Demoruelle MK, Fernandez-Perez ER, Fischer A, et al. Predictors of mortality in rheumatoid arthritis-associated interstitial lung disease. Eur Respir J. (2016) 47:588–96. doi: 10.1183/13993003.00357-2015
22. Volkmann ER, Tashkin DP, Sim M, Li N, Goldmuntz E, Keyes-Elstein L, et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann Rheum Dis. (2019) 78:122–30. doi: 10.1136/annrheumdis-2018-213708
23. Gimenez A, Storrer K, Kuranishi L, Soares MR, Ferreira RG, Pereira CAC. Change in FVC and survival in chronic fibrotic hypersensitivity pneumonitis. Thorax. (2018) 73:391–2. doi: 10.1136/thoraxjnl-2017-210035
24. Walsh SL, Sverzellati N, Devaraj A, Wells AU, Hansell DM. Chronic hypersensitivity pneumonitis: high resolution computed tomography patterns and pulmonary function indices as prognostic determinants. Eur Radiol. (2012) 22:1672–9. doi: 10.1007/s00330-012-2427-0
25. Lee SM, Seo JB, Oh SY, Kim TH, Song JW, Lee SM, et al. Prediction of survival by texture-based automated quantitative assessment of regional disease patterns on CT in idiopathic pulmonary fibrosis. Eur Radiol. (2018) 28:1293–300. doi: 10.1007/s00330-017-5028-0
26. Oda K, Ishimoto H, Yatera K, Naito K, Ogoshi T, Yamasaki K, et al. High-resolution CT scoring system-based grading scale predicts the clinical outcomes in patients with idiopathic pulmonary fibrosis. Respir Res. (2014) 15:10. doi: 10.1186/1465-9921-15-10
27. Hwang JH, Misumi S, Curran-Everett D, Brown KK, Sahin H, Lynch DA. Longitudinal follow-up of fibrosing interstitial pneumonia: relationship between physiologic testing, computed tomography changes, and survival rate. J Thorac Imaging. (2011) 26:209–17. doi: 10.1097/RTI.0b013e3181e35823
28. Jacob J, Bartholmai BJ, van Moorsel CHM, Rajagopalan S, Devaraj A, van Es HW, et al. Longitudinal prediction of outcome in idiopathic pulmonary fibrosis using automated CT analysis. Eur Respir J. (2019) 54:1802341. doi: 10.1183/13993003.02341-2018
29. Wu AC, Kiley JP, Noel PJ, Amur S, Burchard EG, Clancy JP, et al. Current status and future opportunities in lung precision medicine research with a focus on biomarkers. an American Thoracic Society/National Heart, Lung, and Blood Institute Research Statement. Am J Respir Crit Care Med. (2018) 198:e116–36. doi: 10.1164/rccm.201810-1895ST
30. Walsh SLF, Calandriello L, Silva M, Sverzellati N. Deep learning for classifying fibrotic lung disease on high-resolution computed tomography: a case-cohort study. Lancet Respir Med. (2018) 6:837–45. doi: 10.1016/S2213-2600(18)30286-8
31. Jacob J, Bartholmai BJ, Rajagopalan S, van Moorsel CHM, van Es HW, van Beek FT, et al. Predicting outcomes in idiopathic pulmonary fibrosis using automated computed tomographic analysis. Am J Respir Crit Care Med. (2018) 198:767–76. doi: 10.1164/rccm.201711-2174OC
32. Jacob J, Bartholmai BJ, Rajagopalan S, Brun AL, Egashira R, Karwoski R, et al. Evaluation of computer-based computer tomography stratification against outcome models in connective tissue disease-related interstitial lung disease: a patient outcome study. BMC Med. (2016) 14:190. doi: 10.1186/s12916-016-0739-7
33. Machahua C, Montes-Worboys A, Planas-Cerezales L, Buendia-Flores R, Molina-Molina M, Vicens-Zygmunt V. Serum AGE/RAGEs as potential biomarker in idiopathic pulmonary fibrosis. Respir Res. (2018) 19:215. doi: 10.1186/s12931-018-0924-7
34. d'Alessandro M, Bergantini L, Cameli P, Lanzarone N, Perillo F, Perrone A, et al. BAL and serum multiplex lipid profiling in idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Life Sci. (2020) 256:117995. doi: 10.1016/j.lfs.2020.117995
35. Wang T, Zheng XJ, Ji YL, Liang ZA, Liang BM. Tumour markers in rheumatoid arthritis-associated interstitial lung disease. Clin Exp Rheumatol. (2016) 34:587–91.
36. Doubkova M, Karpisek M, Mazoch J, Skrickova J, Doubek M. Prognostic significance of surfactant protein A, surfactant protein D, Clara cell protein 16, S100 protein, trefoil factor 3, and prostatic secretory protein 94 in idiopathic pulmonary fibrosis, sarcoidosis, and chronic pulmonary obstructive disease. Sarcoidosis Vasc Diffuse Lung Dis. (2016) 33:224–34.
37. Buendia-Roldan I, Ruiz V, Sierra P, Montes E, Ramirez R, Vega A, et al. Increased expression of CC16 in patients with idiopathic pulmonary fibrosis. PLoS ONE. (2016) 11:e0168552. doi: 10.1371/journal.pone.0168552
38. Hasegawa M, Fujimoto M, Hamaguchi Y, Matsushita T, Inoue K, Sato S, et al. Use of serum clara cell 16-kDa (CC16) levels as a potential indicator of active pulmonary fibrosis in systemic sclerosis. J Rheumatol. (2011) 38:877–84. doi: 10.3899/jrheum.100591
39. Ohnishi H, Yokoyama A, Kondo K, Hamada H, Abe M, Nishimura K, et al. Comparative study of KL-6, surfactant protein-A, surfactant protein-D, and monocyte chemoattractant protein-1 as serum markers for interstitial lung diseases. Am J Respir Crit Care Med. (2002) 165:378–81. doi: 10.1164/ajrccm.165.3.2107134
40. Hasegawa M, Sato S, Takehara K. Augmented production of chemokines (monocyte chemotactic protein-1 (MCP-1), macrophage inflammatory protein-1alpha (MIP-1alpha) and MIP-1beta) in patients with systemic sclerosis: MCP-1 and MIP-1alpha may be involved in the development of pulmonary fibrosis. Clin Exp Immunol. (1999) 117:159–65. doi: 10.1046/j.1365-2249.1999.00929.x
41. Rosas IO, Richards TJ, Konishi K, Zhang Y, Gibson K, Lokshin AE, et al. MMP1 and MMP7 as potential peripheral blood biomarkers in idiopathic pulmonary fibrosis. PLoS Med. (2008) 5:e93. doi: 10.1371/journal.pmed.0050093
42. Watanabe M, Horimasu Y, Iwamoto H, Yamaguchi K, Sakamoto S, Masuda T, et al. C-C motif chemokine ligand 15 may be a useful biomarker for predicting the prognosis of patients with chronic hypersensitivity pneumonitis. Respiration. (2019) 98:212–20. doi: 10.1159/000500576
43. Raghu G, Richeldi L, Jagerschmidt A, Martin V, Subramaniam A, Ozoux ML, et al. Idiopathic pulmonary fibrosis: prospective, case-controlled study of natural history and circulating biomarkers. Chest. (2018) 154:1359–70. doi: 10.1016/j.chest.2018.08.1083
44. Elhai M, Hoffmann-Vold AM, Avouac J, Pezet S, Cauvet A, Leblond A, et al. Performance of candidate serum biomarkers for systemic sclerosis-associated interstitial lung disease. Arthritis Rheumatol. (2019) 71:972–82. doi: 10.1002/art.40815
45. Prasse A, Pechkovsky DV, Toews GB, Schafer M, Eggeling S, Ludwig C, et al. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum. (2007) 56:1685–93. doi: 10.1002/art.22559
46. Cameli P, Bergantini L, D'Alessandro M, Vietri L, Cameli M, Sestini P, et al. Serum CD59: a novel biomarker of idiopathic pulmonary fibrosis? Panminerva Med. (2020). doi: 10.23736/S0031-0808.20.03893-8. [Epub ahead of print].
47. Liu X, Mayes MD, Pedroza C, Draeger HT, Gonzalez EB, Harper BE, et al. Does C-reactive protein predict the long-term progression of interstitial lung disease and survival in patients with early systemic sclerosis? Arthritis Care Res. (2013) 65:1375–80. doi: 10.1002/acr.21968
48. Sanders JL, Putman RK, Dupuis J, Xu H, Murabito JM, Araki T, et al. The association of aging biomarkers, interstitial lung abnormalities, and mortality. Am J Respir Crit Care Med. (2020). doi: 10.1164/rccm.202007-2993OC. [Epub ahead of print].
49. Hoffmann-Vold AM, Weigt SS, Palchevskiy V, Volkmann E, Saggar R, Li N, et al. Augmented concentrations of CX3CL1 are associated with interstitial lung disease in systemic sclerosis. PLoS ONE. (2018) 13:e0206545. doi: 10.1371/journal.pone.0206545
50. Liang M, Jiang Z, Huang Q, Liu L, Xue Y, Zhu X, et al. Clinical association of chemokine (C-X-C motif) Ligand 1 (CXCL1) with interstitial pneumonia with Autoimmune Features (IPAF). Sci Rep. (2016) 6:38949. doi: 10.1038/srep38949
51. Volkmann ER, Tashkin DP, Roth MD, Clements PJ, Khanna D, Furst DE, et al. Changes in plasma CXCL4 levels are associated with improvements in lung function in patients receiving immunosuppressive therapy for systemic sclerosis-related interstitial lung disease. Arthritis Res Ther. (2016) 18:305. doi: 10.1186/s13075-016-1203-y
52. Nukui Y, Yamana T, Masuo M, Tateishi T, Kishino M, Tateishi U, et al. Serum CXCL9 and CCL17 as biomarkers of declining pulmonary function in chronic bird-related hypersensitivity pneumonitis. PLoS ONE. (2019) 14:e0220462. doi: 10.1371/journal.pone.0220462
53. Chen J, Doyle TJ, Liu Y, Aggarwal R, Wang X, Shi Y, et al. Biomarkers of rheumatoid arthritis-associated interstitial lung disease. Arthritis Rheumatol. (2015) 67:28–38. doi: 10.1002/art.38904
54. Antonelli A, Ferri C, Fallahi P, Ferrari SM, Giuggioli D, Colaci M, et al. CXCL10 (alpha) and CCL2 (beta) chemokines in systemic sclerosis–a longitudinal study. Rheumatology. (2008) 47:45–9. doi: 10.1093/rheumatology/kem313
55. Vuga LJ, Tedrow JR, Pandit KV, Tan J, Kass DJ, Xue J, et al. C-X-C motif chemokine 13 (CXCL13) is a prognostic biomarker of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2014) 189:966–74. doi: 10.1164/rccm.201309-1592OC
56. Kumanovics G, Minier T, Radics J, Palinkas L, Berki T, Czirjak L. Comprehensive investigation of novel serum markers of pulmonary fibrosis associated with systemic sclerosis and dermato/polymyositis. Clin Exp Rheumatol. (2008) 26:414–20.
57. Ates A, Kinikli G, Turgay M, Duman M. Serum-soluble selectin levels in patients with rheumatoid arthritis and systemic sclerosis. Scand J Immunol. (2004) 59:315–20. doi: 10.1111/j.0300-9475.2004.01389.x
58. Lambrecht S, Smith V, De Wilde K, Coudenys J, Decuman S, Deforce D, et al. Growth differentiation factor 15, a marker of lung involvement in systemic sclerosis, is involved in fibrosis development but is not indispensable for fibrosis development. Arthritis Rheumatol. (2014) 66:418–27. doi: 10.1002/art.38241
59. Gamal SM, Elgengehy FT, Kamal A, El Bakry SA, Shabaan E, Elgendy A, et al. Growth Differentiation Factor-15 (GDF-15) level and relation to clinical manifestations in egyptian systemic sclerosis patients: preliminary data. Immunol Invest. (2017) 46:703–13. doi: 10.1080/08820139.2017.1360340
60. Yanaba K, Asano Y, Tada Y, Sugaya M, Kadono T, Sato S. Clinical significance of serum growth differentiation factor-15 levels in systemic sclerosis: association with disease severity. Mod Rheumatol. (2012) 22:668–75. doi: 10.3109/s10165-011-0568-7
61. Zhang Y, Jiang M, Nouraie M, Roth MG, Tabib T, Winters S, et al. GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2019) 317:L510L21. doi: 10.1152/ajplung.00062.2019
62. Hasegawa M, Asano Y, Endo H, Fujimoto M, Goto D, Ihn H, et al. Serum adhesion molecule levels as prognostic markers in patients with early systemic sclerosis: a multicentre, prospective, observational study. PLoS ONE. (2014) 9:e88150. doi: 10.1371/journal.pone.0088150
63. Guiot J, Bondue B, Henket M, Corhay JL, Louis R. Raised serum levels of IGFBP-1 and IGFBP-2 in idiopathic pulmonary fibrosis. BMC Pulm Med. (2016) 16:86. doi: 10.1186/s12890-016-0249-6
64. Khadilkar PV, Khopkar US, Nadkar MY, Rajadhyaksha AG, Chougule DA, Deshpande SD, et al. Fibrotic cytokine interplay in evaluation of disease activity in treatment naive systemic sclerosis patients from Western India. J Assoc Physicians India. (2019) 67:26–30.
65. Abdel-Magied RA, Kamel SR, Said AF, Ali HM, Abdel Gawad EA, Moussa MM. Serum interleukin-6 in systemic sclerosis and its correlation with disease parameters and cardiopulmonary involvement. Sarcoidosis Vasc Diffuse Lung Dis. (2016) 33:321–30. doi: 10.1016/j.ejcdt.2016.02.002
66. Tsoutsou PG, Gourgoulianis KI, Petinaki E, Germenis A, Tsoutsou AG, Mpaka M, et al. Cytokine levels in the sera of patients with idiopathic pulmonary fibrosis. Respir Med. (2006) 100:938–45. doi: 10.1016/j.rmed.2005.06.016
67. Ishikawa Y, Iwata S, Hanami K, Nawata A, Zhang M, Yamagata K, et al. Relevance of interferon-gamma in pathogenesis of life-threatening rapidly progressive interstitial lung disease in patients with dermatomyositis. Arthritis Res Ther. (2018) 20:240. doi: 10.1186/s13075-018-1737-2
68. Yanaba K, Yoshizaki A, Asano Y, Kadono T, Sato S. Serum IL-33 levels are raised in patients with systemic sclerosis: association with extent of skin sclerosis and severity of pulmonary fibrosis. Clin Rheumatol. (2011) 30:825–30. doi: 10.1007/s10067-011-1686-5
69. Tang J, Lei L, Pan J, Zhao C, Wen J. Higher levels of serum interleukin-35 are associated with the severity of pulmonary fibrosis and Th2 responses in patients with systemic sclerosis. Rheumatol Int. (2018) 38:1511–9. doi: 10.1007/s00296-018-4071-8
70. Zheng M, Lou A, Zhang H, Zhu S, Yang M, Lai W. Serum KL-6, CA19-9, CA125 and CEA are diagnostic biomarkers for rheumatoid arthritis-associated interstitial lung disease in the chinese population. Rheumatol Ther. (2021) 8:517–27. doi: 10.1007/s40744-021-00288-x
71. Kobayashi J, Kitamura S. KL-6: a serum marker for interstitial pneumonia. Chest. (1995) 108:311–5. doi: 10.1378/chest.108.2.311
72. Takahashi T, Munakata M, Ohtsuka Y, Satoh-Kamachi A, Sato R, Homma Y, et al. Serum KL-6 concentrations in dairy farmers. Chest. (2000) 118:445–50. doi: 10.1378/chest.118.2.445
73. Ji Y, Bourke SJ, Spears M, Wain LV, Boyd G, Lynch PP, et al. Krebs von den Lungen-6 (KL-6) is a pathophysiological biomarker of early-stage acute hypersensitivity pneumonitis among pigeon fanciers. Clin Exp Allergy. (2020) 50:1391–9. doi: 10.1111/cea.13744
74. Kennedy B, Branagan P, Moloney F, Haroon M, O'Connell OJ, O'Connor TM, et al. Biomarkers to identify ILD and predict lung function decline in scleroderma lung disease or idiopathic pulmonary fibrosis. Sarcoidosis Vasc Diffuse Lung Dis. (2015) 32:228–36.
75. Samukawa T, Hamada T, Uto H, Yanagi M, Tsukuya G, Nosaki T, et al. The elevation of serum napsin A in idiopathic pulmonary fibrosis, compared with KL-6, surfactant protein-A and surfactant protein-D. BMC Pulm Med. (2012) 12:55. doi: 10.1186/1471-2466-12-55
76. Hant FN, Ludwicka-Bradley A, Wang HJ, Li N, Elashoff R, Tashkin DP, et al. Surfactant protein D and KL-6 as serum biomarkers of interstitial lung disease in patients with scleroderma. J Rheumatol. (2009) 36:773–80. doi: 10.3899/jrheum.080633
77. Yanaba K, Hasegawa M, Takehara K, Sato S. Comparative study of serum surfactant protein-D and KL-6 concentrations in patients with systemic sclerosis as markers for monitoring the activity of pulmonary fibrosis. J Rheumatol. (2004) 31:1112–20.
78. Sato S, Nagaoka T, Hasegawa M, Nishijima C, Takehara K. Elevated serum KL-6 levels in patients with systemic sclerosis: association with the severity of pulmonary fibrosis. Dermatology. (2000) 200:196–201. doi: 10.1159/000018382
79. Yanaba K, Hasegawa M, Hamaguchi Y, Fujimoto M, Takehara K, Sato S. Longitudinal analysis of serum KL-6 levels in patients with systemic sclerosis: association with the activity of pulmonary fibrosis. Clin Exp Rheumatol. (2003) 21:429–36.
80. Lee JS, Lee EY, Ha YJ, Kang EH, Lee YJ, Song YW. Serum KL-6 levels reflect the severity of interstitial lung disease associated with connective tissue disease. Arthritis Res Ther. (2019) 21:58. doi: 10.1186/s13075-019-1835-9
81. Jenkins RG, Simpson JK, Saini G, Bentley JH, Russell AM, Braybrooke R, et al. Longitudinal change in collagen degradation biomarkers in idiopathic pulmonary fibrosis: an analysis from the prospective, multicentre PROFILE study. Lancet Respir Med. (2015) 3:462–72. doi: 10.1016/S2213-2600(15)00048-X
82. Organ LA, Duggan AR, Oballa E, Taggart SC, Simpson JK, Kang'ombe AR, et al. Biomarkers of collagen synthesis predict progression in the PROFILE idiopathic pulmonary fibrosis cohort. Respir Res. (2019) 20:148. doi: 10.1186/s12931-019-1118-7
83. Young-Min SA, Beeton C, Laughton R, Plumpton T, Bartram S, Murphy G, et al. Serum TIMP-1, TIMP-2, and MMP-1 in patients with systemic sclerosis, primary Raynaud's phenomenon, and in normal controls. Ann Rheum Dis. (2001) 60:846–851.
84. Doyle TJ, Patel AS, Hatabu H, Nishino M, Wu G, Osorio JC, et al. Detection of rheumatoid arthritis-interstitial lung disease is enhanced by serum biomarkers. Am J Respir Crit Care Med. (2015) 191:1403–12. doi: 10.1164/rccm.201411-1950OC
85. White ES, Xia M, Murray S, Dyal R, Flaherty CM, Flaherty KR, et al. Plasma surfactant Protein-D, matrix Metalloproteinase-7, and osteopontin index distinguishes idiopathic pulmonary fibrosis from other idiopathic interstitial pneumonias. Am J Respir Crit Care Med. (2016) 194:1242–51. doi: 10.1164/rccm.201505-0862OC
86. Moinzadeh P, Krieg T, Hellmich M, Brinckmann J, Neumann E, Muller-Ladner U, et al. Elevated MMP-7 levels in patients with systemic sclerosis: correlation with pulmonary involvement. Exp Dermatol. (2011) 20:770–3. doi: 10.1111/j.1600-0625.2011.01321.x
87. Manetti M, Guiducci S, Romano E, Bellando-Randone S, Conforti ML, Ibba-Manneschi L, et al. Increased serum levels and tissue expression of matrix metalloproteinase-12 in patients with systemic sclerosis: correlation with severity of skin and pulmonary fibrosis and vascular damage. Ann Rheum Dis. (2012) 71:1064–72. doi: 10.1136/annrheumdis-2011-200837
88. Guiot J, Struman I, Chavez V, Henket M, Herzog M, Scoubeau K, et al. Altered epigenetic features in circulating nucleosomes in idiopathic pulmonary fibrosis. Clin Epigenetics. (2017) 9:84. doi: 10.1186/s13148-017-0383-x
89. Nukui Y, Miyazaki Y, Masuo M, Okamoto T, Furusawa H, Tateishi T, et al. Periostin as a predictor of prognosis in chronic bird-related hypersensitivity pneumonitis. Allergol Int. (2019) 68:363–369. doi: 10.1016/j.alit.2019.02.007
90. Ohta S, Okamoto M, Fujimoto K, Sakamoto N, Takahashi K, Yamamoto H, et al. The usefulness of monomeric periostin as a biomarker for idiopathic pulmonary fibrosis. PLoS ONE. (2017) 12:e0174547. doi: 10.1371/journal.pone.0174547
91. Kuroki Y, Tsutahara S, Shijubo N, Takahashi H, Shiratori M, Hattori A, et al. Elevated levels of lung surfactant protein A in sera from patients with idiopathic pulmonary fibrosis and pulmonary alveolar proteinosis. Am Rev Respir Dis. (1993) 147:723–9. doi: 10.1164/ajrccm/147.3.723
92. Honda Y, Kuroki Y, Shijubo N, Fujishima T, Takahashi H, Hosoda K, et al. Aberrant appearance of lung surfactant protein A in sera of patients with idiopathic pulmonary fibrosis and its clinical significance. Respiration. (1995) 62:64–9. doi: 10.1159/000196393
93. Ishii H, Mukae H, Kadota J, Kaida H, Nagata T, Abe K, et al. High serum concentrations of surfactant protein A in usual interstitial pneumonia compared with non-specific interstitial pneumonia. Thorax. (2003) 58:52–7. doi: 10.1136/thorax.58.1.52
94. Takahashi H, Kuroki Y, Tanaka H, Saito T, Kurokawa K, Chiba H, et al. Serum levels of surfactant proteins A and D are useful biomarkers for interstitial lung disease in patients with progressive systemic sclerosis. Am J Respir Crit Care Med. (2000) 162:258–63. doi: 10.1164/ajrccm.162.1.9903014
95. Honda Y, Kuroki Y, Matsuura E, Nagae H, Takahashi H, Akino T, et al. Pulmonary surfactant protein D in sera and bronchoalveolar lavage fluids. Am J Respir Crit Care Med. (1995) 152:1860–6. doi: 10.1164/ajrccm.152.6.8520747
96. Asano Y, Ihn H, Yamane K, Yazawa N, Kubo M, Fujimoto M, et al. Clinical significance of surfactant protein D as a serum marker for evaluating pulmonary fibrosis in patients with systemic sclerosis. Arthritis Rheum. (2001) 44:1363–9. doi: 10.1002/1529-0131(200106)44:6<1363::AID-ART229>3.0.CO;2-5
97. Maeda M, Ichiki Y, Aoyama Y, Kitajima Y. Surfactant protein D (SP-D) and systemic scleroderma (SSc). J Dermatol. (2001) 28:467–74. doi: 10.1111/j.1346-8138.2001.tb00013.x
98. Kikuchi K, Kubo M, Sato S, Fujimoto M, Tamaki K. Serum tissue inhibitor of metalloproteinases in patients with systemic sclerosis. J Am Acad Dermatol. (1995) 33:973–8. doi: 10.1016/0190-9622(95)90289-9
99. Yu M, Guo Y, Zhang P, Xue J, Yang J, Cai Q, et al. Increased circulating Wnt5a protein in patients with rheumatoid arthritis-associated interstitial pneumonia (RA-ILD). Immunobiology. (2019) 224:551–9. doi: 10.1016/j.imbio.2019.04.006-
100. Long X, He X, Ohshimo S, Griese M, Sarria R, Guzman J, et al. Serum YKL-40 as predictor of outcome in hypersensitivity pneumonitis. Eur Respir J. (2017) 49:1501924. doi: 10.1183/13993003.01924-2015
101. Korthagen NM, van Moorsel CH, Barlo NP, Ruven HJ, Kruit A, Heron M, et al. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir Med. (2011) 105:106–13. doi: 10.1016/j.rmed.2010.09.012
102. Korthagen NM, van Moorsel CH, Zanen P, Ruven HJ, Grutters JC. Evaluation of circulating YKL-40 levels in idiopathic interstitial pneumonias. Lung. (2014) 192:975–80. doi: 10.1007/s00408-014-9647-9
103. Nordenbaek C, Johansen JS, Halberg P, Wiik A, Garbarsch C, Ullman S, et al. High serum levels of YKL-40 in patients with systemic sclerosis are associated with pulmonary involvement. Scand J Rheumatol. (2005) 34:293–7. doi: 10.1080/03009740510018598
104. Hozumi H, Fujisawa T, Enomoto N, Nakashima R, Enomoto Y, Suzuki Y, et al. Clinical Utility of YKL-40 in Polymyositis/dermatomyositis-associated Interstitial Lung Disease. J Rheumatol. (2017) 44:1394–401. doi: 10.3899/jrheum.170373
105. Oldham JM. Interstitial lung abnormalities and aging biomarkers: a mediation. Am J Respir Crit Care Med. (2020). doi: 10.1164/rccm.202011-4046ED. [Epub ahead of print].
106. Roy MG, Livraghi-Butrico A, Fletcher AA, McElwee MM, Evans SE, Boerner RM, et al. Muc5b is required for airway defence. Nature. (2014) 505:412–6. doi: 10.1038/nature12807
107. Hunninghake GM, Hatabu H, Okajima Y, Gao W, Dupuis J, Latourelle JC, et al. MUC5B promoter polymorphism and interstitial lung abnormalities. N Engl J Med. (2013) 368:2192–200. doi: 10.1056/NEJMoa1216076
108. Fingerlin TE, Murphy E, Zhang W, Peljto AL, Brown KK, Steele MP, et al. Genome-wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet. (2013) 45:613–20. doi: 10.1038/ng.2609
109. Allen RJ, Guillen-Guio B, Oldham JM, Ma SF, Dressen A, Paynton ML, et al. Genome-wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2020) 201:564–74. doi: 10.1164/rccm.201905-1017OC
110. Juge PA, Lee JS, Ebstein E, Furukawa H, Dobrinskikh E, Gazal S, et al. MUC5B promoter variant and rheumatoid arthritis with interstitial lung disease. N Engl J Med. (2018) 379:2209–19.
111. Wang N, Zhang Q, Jing X, Guo J, Huang H, Xu Z. The association between MUC5B mutations and clinical outcome in patients with rheumatoid arthritis-associated interstitial lung disease: a retrospective exploratory study in China. Med Sci Monit. (2020) 26:e920137. doi: 10.12659/MSM.920137
112. Lopez-Mejias R, Remuzgo-Martinez S, Genre F, Pulito-Cueto V, Rozas SMF, Llorca J, et al. Influence of MUC5B gene on antisynthetase syndrome. Sci Rep. (2020) 10:1415. doi: 10.1038/s41598-020-58400-0
113. Johnson C, Rosen P, Lloyd T, Horton M, Christopher-Stine L, Oddis CV, et al. Exploration of the MUC5B promoter variant and ILD risk in patients with autoimmune myositis. Respir Med. (2017) 130:52–54. doi: 10.1016/j.rmed.2017.07.010
114. Allen RJ, Porte J, Braybrooke R, Flores C, Fingerlin TE, Oldham JM, et al. Genetic variants associated with susceptibility to idiopathic pulmonary fibrosis in people of European ancestry: a genome-wide association study. Lancet Respir Med. (2017) 5:869–80. doi: 10.1016/S2213-2600(17)30387-9
115. Noth I, Zhang Y, Ma SF, Flores C, Barber M, Huang Y, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med. (2013) 1:309–17. doi: 10.1016/S2213-2600(13)70045-6
116. Gono T, Sato S, Kawaguchi Y, Kuwana M, Hanaoka M, Katsumata Y, et al. Anti-MDA5 antibody, ferritin and IL-18 are useful for the evaluation of response to treatment in interstitial lung disease with anti-MDA5 antibody-positive dermatomyositis. Rheumatology. (2012) 51:1563–70. doi: 10.1093/rheumatology/kes102
117. Adegunsoye A, Alqalyoobi S, Linderholm A, Bowman WS, Lee CT, Pugashetti JV, et al. Circulating plasma biomarkers of survival in antifibrotic-treated patients with idiopathic pulmonary fibrosis. Chest. (2020) 158:1526–34. doi: 10.1016/j.chest.2020.04.066
118. Maher TM, Oballa E, Simpson JK, Porte J, Habgood A, Fahy WA, et al. An epithelial biomarker signature for idiopathic pulmonary fibrosis: an analysis from the multicentre PROFILE cohort study. Lancet Respir Med. (2017) 5:946–55. doi: 10.1016/S2213-2600(17)30430-7
119. Alqalyoobi S, Adegunsoye A, Linderholm A, Hrusch C, Cutting C, Ma SF, et al. Circulating plasma biomarkers of progressive interstitial lung disease. Am J Respir Crit Care Med. (2020) 201:250–3. doi: 10.1164/rccm.201907-1343LE
120. Akiyama N, Hozumi H, Isayama T, Okada J, Sugiura K, Yasui H, et al. Clinical significance of serum S100 calcium-binding protein A4 in idiopathic pulmonary fibrosis. Respirology. (2019) 25:743–9. doi: 10.1111/resp.13707
121. Richards TJ, Kaminski N, Baribaud F, Flavin S, Brodmerkel C, Horowitz D, et al. Peripheral blood proteins predict mortality in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2012) 185:67–76. doi: 10.1164/rccm.201101-0058OC
122. Wu M, Baron M, Pedroza C, Salazar GA, Ying J, Charles J, et al. CCL2 in the circulation predicts long-term progression of interstitial lung disease in patients with early systemic sclerosis: data from two independent cohorts. Arthritis Rheumatol. (2017) 69:1871–8. doi: 10.1002/art.40171
123. Hasegawa M, Fujimoto M, Matsushita T, Hamaguchi Y, Takehara K, Sato S. Serum chemokine and cytokine levels as indicators of disease activity in patients with systemic sclerosis. Clin Rheumatol. (2011) 30:231–7. doi: 10.1007/s10067-010-1610-4
124. Oda K, Kotani T, Takeuchi T, Ishida T, Shoda T, Isoda K, et al. Chemokine profiles of interstitial pneumonia in patients with dermatomyositis: a case control study. Sci Rep. (2017) 7:1635. doi: 10.1038/s41598-017-01685-5
125. Ohshimo S, Ishikawa N, Horimasu Y, Hattori N, Hirohashi N, Tanigawa K, et al. Baseline KL-6 predicts increased risk for acute exacerbation of idiopathic pulmonary fibrosis. Respir Med. (2014) 108:1031–9. doi: 10.1016/j.rmed.2014.04.009
126. Neighbors M, Cabanski CR, Ramalingam TR, Sheng XR, Tew GW, Gu C, et al. Prognostic and predictive biomarkers for patients with idiopathic pulmonary fibrosis treated with pirfenidone: post-hoc assessment of the CAPACITY and ASCEND trials. Lancet Respir Med. (2018) 6:615–26. doi: 10.1016/S2213-2600(18)30185-1
127. Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, et al. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2009) 179:717–23. doi: 10.1164/rccm.200808-1201OC
128. Elhaj M, Charles J, Pedroza C, Liu X, Zhou X, Estrada YMRM, et al. Can serum surfactant protein D or CC-chemokine ligand 18 predict outcome of interstitial lung disease in patients with early systemic sclerosis? J Rheumatol. (2013) 40:1114–20. doi: 10.3899/jrheum.120997
129. Tiev KP, Hua-Huy T, Kettaneh A, Gain M, Duong-Quy S, Toledano C, et al. Serum CC chemokine ligand-18 predicts lung disease worsening in systemic sclerosis. Eur Respir J. (2011) 38:1355–60. doi: 10.1183/09031936.00004711
130. Hoffmann-Vold AM, Tennoe AH, Garen T, Midtvedt O, Abraityte A, Aalokken TM, et al. High level of chemokine ccl18 is associated with pulmonary function deterioration, lung fibrosis progression, and reduced survival in systemic sclerosis. Chest. (2016) 150:299–306. doi: 10.1016/j.chest.2016.03.004
131. Salazar GA, Kuwana M, Wu M, Estrada YMRM, Ying J, Charles J, et al. KL-6 but not CCL-18 Is a predictor of early progression in systemic sclerosis-related interstitial lung disease. J Rheumatol. (2018) 45:1153–8. doi: 10.3899/jrheum.170518
132. van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. (2014) 370:433–43. doi: 10.1056/NEJMoa1114576
133. Guo L, Yang Y, Liu F, Jiang C, Yang Y, Pu H, et al. Clinical research on prognostic evaluation of subjects with IPF by peripheral blood biomarkers, quantitative imaging characteristics and pulmonary function parameters. Arch Bronconeumol. (2019) 56:365–72. doi: 10.1016/j.arbr.2019.08.019
134. DePianto DJ, Chandriani S, Abbas AR, Jia G, N'Diaye EN, Caplazi P, et al. Heterogeneous gene expression signatures correspond to distinct lung pathologies and biomarkers of disease severity in idiopathic pulmonary fibrosis. Thorax. (2015) 70:48–56. doi: 10.1136/thoraxjnl-2013-204596
135. Ten Klooster L, van Moorsel CH, Kwakkel-van Erp JM, van Velzen-Blad H, Grutters JC. Immunoglobulin A in serum: an old acquaintance as a new prognostic biomarker in idiopathic pulmonary fibrosis. Clin Exp Immunol. (2015) 181:357–61. doi: 10.1111/cei.12636
136. De Lauretis A, Sestini P, Pantelidis P, Hoyles R, Hansell DM, Goh NS, et al. Serum interleukin 6 is predictive of early functional decline and mortality in interstitial lung disease associated with systemic sclerosis. J Rheumatol. (2013) 40:435–46. doi: 10.3899/jrheum.120725
137. Nara M, Komatsuda A, Omokawa A, Togashi M, Okuyama S, Sawada K, et al. Serum interleukin 6 levels as a useful prognostic predictor of clinically amyopathic dermatomyositis with rapidly progressive interstitial lung disease. Mod Rheumatol. (2014) 24:633–6. doi: 10.3109/14397595.2013.844390
138. Wakamatsu K, Nagata N, Kumazoe H, Oda K, Ishimoto H, Yoshimi M, et al. Prognostic value of serial serum KL-6 measurements in patients with idiopathic pulmonary fibrosis. Respir Investig. (2017) 55:16–23. doi: 10.1016/j.resinv.2016.09.003
139. Yokoyama A, Kohno N, Hamada H, Sakatani M, Ueda E, Kondo K, et al. Circulating KL-6 predicts the outcome of rapidly progressive idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (1998) 158:1680–4. doi: 10.1164/ajrccm.158.5.9803115
140. Yokoyama A, Kondo K, Nakajima M, Matsushima T, Takahashi T, Nishimura M, et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology. (2006) 11:164–8. doi: 10.1111/j.1440-1843.2006.00834.x
141. Satoh H, Kurishima K, Ishikawa H, Ohtsuka M. Increased levels of KL-6 and subsequent mortality in patients with interstitial lung diseases. J Intern Med. (2006) 260:429–34. doi: 10.1111/j.1365-2796.2006.01704.x
142. Kuwana M, Shirai Y, Takeuchi T. Elevated serum krebs von den Lungen-6 in early disease predicts subsequent deterioration of pulmonary function in patients with systemic sclerosis and interstitial lung disease. J Rheumatol. (2016) 43:1825–31. doi: 10.3899/jrheum.160339
143. Chien JW, Richards TJ, Gibson KF, Zhang Y, Lindell KO, Shao L, et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J. (2014) 43:1430–438. doi: 10.1183/09031936.00141013
144. Oka S, Furukawa H, Shimada K, Hayakawa H, Fukui N, Tsuchiya N, et al. Serum biomarker analysis of collagen disease patients with acute-onset diffuse interstitial lung disease. BMC Immunol. (2013) 14:9. doi: 10.1186/1471-2172-14-9
145. Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest. (2013) 143:1422–9. doi: 10.1378/chest.11-2735
146. Sokai A, Handa T, Tanizawa K, Oga T, Uno K, Tsuruyama T, et al. Matrix metalloproteinase-10: a novel biomarker for idiopathic pulmonary fibrosis. Respir Res. (2015) 16:120. doi: 10.1186/s12931-015-0280-9
147. Peng QL, Zhang YM, Liang L, Liu X, Ye LF, Yang HB, et al. A high level of serum neopterin is associated with rapidly progressive interstitial lung disease and reduced survival in dermatomyositis. Clin Exp Immunol. (2020) 199:314–25. doi: 10.1111/cei.13404
148. Tajiri M, Okamoto M, Fujimoto K, Johkoh T, Ono J, Tominaga M, et al. Serum level of periostin can predict long-term outcome of idiopathic pulmonary fibrosis. Respir Investig. (2015) 53:73–81. doi: 10.1016/j.resinv.2014.12.003
149. Naik PK, Bozyk PD, Bentley JK, Popova AP, Birch CM, Wilke CA, et al. Periostin promotes fibrosis and predicts progression in patients with idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L1046–56. doi: 10.1152/ajplung.00139.2012
150. Shimizu T, Koga T, Furukawa K, Horai Y, Fujikawa K, Okada A, et al. IL-15 is a biomarker involved in the development of rapidly progressive interstitial lung disease complicated with polymyositis/dermatomyositis. J Intern Med. (2020) 289:206–20 doi: 10.1111/joim.13154
151. Ando M, Miyazaki E, Ito T, Hiroshige S, Nureki SI, Ueno T, et al. Significance of serum vascular endothelial growth factor level in patients with idiopathic pulmonary fibrosis. Lung. (2010) 188:247–52. doi: 10.1007/s00408-009-9223-x
152. Barratt SL, Blythe T, Jarrett C, Ourradi K, Shelley-Fraser G, Day MJ, et al. Differential expression of VEGF-Axxx isoforms is critical for development of pulmonary fibrosis. Am J Respir Crit Care Med. (2017) 196:479–93. doi: 10.1164/rccm.201603-0568OC
153. Nakatsuka Y, Handa T, Nakashima R, Tanizawa K, Kubo T, Murase Y, et al. Serum matrix metalloproteinase levels in polymyositis/dermatomyositis patients with interstitial lung disease. Rheumatology. (2019) 58:1465–73. doi: 10.1093/rheumatology/kez065
154. Ashley SL, Xia M, Murray S, O'Dwyer DN, Grant E, White ES, et al. Six-SOMAmer index relating to immune, protease and angiogenic functions predicts progression in IPF. PLoS ONE. (2016) 11:e0159878. doi: 10.1371/journal.pone.0159878
155. Scott MKD, Quinn K, Li Q, Carroll R, Warsinske H, Vallania F, et al. Increased monocyte count as a cellular biomarker for poor outcomes in fibrotic diseases: a retrospective, multicentre cohort study. Lancet Respir Med. (2019) 7:497–508. doi: 10.1016/S2213-2600(18)30508-3
156. Kreuter M, Lee JS, Tzouvelekis A, Oldham JM, Molyneaux PL, Weycker D, et al. Monocyte count as a prognostic biomarker in patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2021). doi: 10.1164/rccm.202003-0669OC. [Epub ahead of print].
157. Bauer Y, White ES, de Bernard S, Cornelisse P, Leconte I, Morganti A, et al. MMP-7 is a predictive biomarker of disease progression in patients with idiopathic pulmonary fibrosis. ERJ Open Res. (2017) 3:00074–2016. doi: 10.1183/23120541.00074-2016
158. Ikeda K, Shiratori M, Chiba H, Nishikiori H, Yokoo K, Saito A, et al. Serum surfactant protein D predicts the outcome of patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir Med. (2017) 131:184–91. doi: 10.1016/j.rmed.2017.08.021
159. Peljto AL, Zhang Y, Fingerlin TE, Ma SF, Garcia JG, Richards TJ, et al. Association between the MUC5B promoter polymorphism and survival in patients with idiopathic pulmonary fibrosis. JAMA. (2013) 309:2232–9. doi: 10.1001/jama.2013.5827
160. Dudbridge F, Allen RJ, Sheehan NA, Schmidt AF, Lee JC, Jenkins RG, et al. Adjustment for index event bias in genome-wide association studies of subsequent events. Nat Commun. (2019) 10:1561. doi: 10.1038/s41467-019-09381-w
161. Ley B, Newton CA, Arnould I, Elicker BM, Henry TS, Vittinghoff E, et al. The MUC5B promoter polymorphism and telomere length in patients with chronic hypersensitivity pneumonitis: an observational cohort-control study. Lancet Respir Med. (2017) 5:639–47. doi: 10.1016/S2213-2600(17)30216-3
162. Stuart BD, Lee JS, Kozlitina J, Noth I, Devine MS, Glazer CS, et al. Effect of telomere length on survival in patients with idiopathic pulmonary fibrosis: an observational cohort study with independent validation. Lancet Respir Med. (2014) 2:557–65. doi: 10.1016/S2213-2600(14)70124-9
163. Newton CA, Oldham JM, Ley B, Anand V, Adegunsoye A, Liu G, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J. (2019) 53:1801641. doi: 10.1183/13993003.01641-2018
164. Newton CA, Batra K, Torrealba J, Kozlitina J, Glazer CS, Aravena C, et al. Telomere-related lung fibrosis is diagnostically heterogeneous but uniformly progressive. Eur Respir J. (2016) 48:1710–20. doi: 10.1183/13993003.00308-2016
165. Ryu C, Sun H, Gulati M, Herazo-Maya JD, Chen Y, Osafo-Addo A, et al. Extracellular mitochondrial DNA is generated by fibroblasts and predicts death in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2017) 196:1571–81. doi: 10.1164/rccm.201612-2480OC
166. Sakamoto K, Furukawa T, Yamano Y, Kataoka K, Teramachi R, Walia A, et al. Serum mitochondrial DNA predicts the risk of acute exacerbation and progression of IPF. Eur Respir J. (2020) 57:2001346. doi: 10.1183/13993003.01346-2020
167. Herazo-Maya JD, Noth I, Duncan SR, Kim S, Ma SF, Tseng GC, et al. Peripheral blood mononuclear cell gene expression profiles predict poor outcome in idiopathic pulmonary fibrosis. Sci Transl Med. (2013) 5:205ra136. doi: 10.1126/scitranslmed.3005964
168. Herazo-Maya JD, Sun J, Molyneaux PL, Li Q, Villalba JA, Tzouvelekis A, et al. Validation of a 52-gene risk profile for outcome prediction in patients with idiopathic pulmonary fibrosis: an international, multicentre, cohort study. Lancet Respir Med. (2017) 5:857–68. doi: 10.1016/S2213-2600(17)30349-1
169. Sumida H, Asano Y, Tamaki Z, Aozasa N, Taniguchi T, Toyama T, et al. Prediction of therapeutic response before and during i.v. cyclophosphamide pulse therapy for interstitial lung disease in systemic sclerosis: a longitudinal observational study. J Dermatol. (2018) 45:1425–33. doi: 10.1111/1346-8138.14669
170. Diaz-Torne C, Ortiz MDA, Moya P, Hernandez MV, Reina D, Castellvi I, et al. The combination of IL-6 and its soluble receptor is associated with the response of rheumatoid arthritis patients to tocilizumab. Semin Arthritis Rheum. (2018) 47:757–64. doi: 10.1016/j.semarthrit.2017.10.022
171. Shirakashi M, Nakashima R, Tsuji H, Tanizawa K, Handa T, Hosono Y, et al. Efficacy of plasma exchange in anti-MDA5-positive dermatomyositis with interstitial lung disease under combined immunosuppressive treatment. Rheumatology. (2020) 59:3284–92. doi: 10.1093/rheumatology/keaa123
172. Assassi S, Li N, Volkmann ER, Mayes MD, Runger D, Ying J, et al. Predictive significance of serum interferon inducible protein score for response to treatment in systemic sclerosis related interstitial lung disease. Arthritis Rheumatol. (2020). doi: 10.1002/art.41627. [Epub ahead of print].
173. Ikeda K, Chiba H, Nishikiori H, Azuma A, Kondoh Y, Ogura T, et al. Serum surfactant protein D as a predictive biomarker for the efficacy of pirfenidone in patients with idiopathic pulmonary fibrosis: a post-hoc analysis of the phase 3 trial in Japan. Respir Res. (2020) 21:316. doi: 10.1186/s12931-020-01582-y
174. Oldham JM, Ma SF, Martinez FJ, Anstrom KJ, Raghu G, Schwartz DA, et al. TOLLIP, MUC5B, and the response to N-Acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2015) 192:1475–82. doi: 10.1164/rccm.201505-1010OC
175. Newton CA, Zhang D, Oldham JM, Kozlitina J, Ma SF, Martinez FJ, et al. Telomere length and use of immunosuppressive medications in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. (2019) 200:336–47. doi: 10.1164/rccm.201809-1646OC
176. Adegunsoye A, Morisset J, Newton CA, Oldham JM, Vittinghoff E, Linderholm AL, et al. Leukocyte telomere length and mycophenolate therapy in chronic hypersensitivity pneumonitis. Eur Respir J. (2020) 57:2002872. doi: 10.1183/13993003.02872-2020
177. Dressen A, Abbas AR, Cabanski C, Reeder J, Ramalingam TR, Neighbors M, et al. Analysis of protein-altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study. Lancet Respir Med. (2018) 6:603–14. doi: 10.1016/S2213-2600(18)30135-8
178. Wells AU, Kokosi MA. POINT: should BAL be routinely performed in the diagnostic evaluation of idiopathic pulmonary fibrosis? Yes. Chest. (2017) 152:917–9. doi: 10.1016/j.chest.2017.08.1173
179. Mooney JJ, Collard HR. COUNTERPOINT: should BAL be routinely performed in the diagnostic evaluation of idiopathic pulmonary fibrosis? No. Chest. (2017) 152:919–22. doi: 10.1016/j.chest.2017.08.1172
180. Adderley N, Humphreys CJ, Barnes H, Ley B, Premji ZA, Johannson KA. Bronchoalveolar lavage fluid lymphocytosis in chronic hypersensitivity pneumonitis: a systematic review and meta-analysis. Eur Respir J. (2020) 56:2000206. doi: 10.1183/13993003.00206-2020
181. Raghu G, Remy-Jardin M, Ryerson CJ, Myers JL, Kreuter M, Vasakova M, et al. Diagnosis of hypersensitivity pneumonitis in adults. an official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2020) 202:e36–69. doi: 10.1164/rccm.202005-2032ST
182. Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. Diagnosis of idiopathic pulmonary fibrosis. an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. (2018) 198:e44–68. doi: 10.1164/rccm.201807-1255ST
183. Suhara K, Miyazaki Y, Okamoto T, Ishizuka M, Tsuchiya K, Inase N. Fragmented gelsolins are increased in rheumatoid arthritis-associated interstitial lung disease with usual interstitial pneumonia pattern. Allergol Int. (2016) 65:88–95. doi: 10.1016/j.alit.2015.08.002
184. Hara A, Sakamoto N, Ishimatsu Y, Kakugawa T, Nakashima S, Hara S, et al. S100A9 in BALF is a candidate biomarker of idiopathic pulmonary fibrosis. Respir Med. (2012) 106:571–80. doi: 10.1016/j.rmed.2011.12.010
185. Suga M, Iyonaga K, Okamoto T, Gushima Y, Miyakawa H, Akaike T, et al. Characteristic elevation of matrix metalloproteinase activity in idiopathic interstitial pneumonias. Am J Respir Crit Care Med. (2000) 162:1949–56. doi: 10.1164/ajrccm.162.5.9906096
186. Nishikiori H, Chiba H, Ariki S, Kuronuma K, Otsuka M, Shiratori M, et al. Distinct compartmentalization of SP-A and SP-D in the vasculature and lungs of patients with idiopathic pulmonary fibrosis. BMC Pulm Med. (2014) 14:196. doi: 10.1186/1471-2466-14-196
187. Moor CC, Oppenheimer JC, Nakshbandi G, Aerts J, Brinkman P, Maitland-van der Zee AH, et al. Exhaled breath analysis by use of eNose technology: a novel diagnostic tool for interstitial lung disease. Eur Respir J. (2021) 57:2002042. doi: 10.1183/13993003.congress-2020.4395
188. Fujisawa T, Hozumi H, Kono M, Enomoto N, Hashimoto D, Nakamura Y, et al. Prognostic factors for myositis-associated interstitial lung disease. PLoS ONE. (2014) 9:e98824. doi: 10.1371/journal.pone.0098824
189. Li Y, Gao X, Li Y, Jia X, Zhang X, Xu Y, et al. Predictors and mortality of rapidly progressive interstitial lung disease in patients with idiopathic inflammatory myopathy: a series of 474 patients. Front Med. (2020) 7:363. doi: 10.3389/fmed.2020.00363
190. Goh NS, Veeraraghavan S, Desai SR, Cramer D, Hansell DM, Denton CP, et al. Bronchoalveolar lavage cellular profiles in patients with systemic sclerosis-associated interstitial lung disease are not predictive of disease progression. Arthritis Rheum. (2007) 56:2005–12. doi: 10.1002/art.22696
191. Boomars KA, Wagenaar SS, Mulder PG, van Velzen-Blad H, van den Bosch JM. Relationship between cells obtained by bronchoalveolar lavage and survival in idiopathic pulmonary fibrosis. Thorax. (1995) 50:1087–92. doi: 10.1136/thx.50.10.1087
192. Schmidt K, Martinez-Gamboa L, Meier S, Witt C, Meisel C, Hanitsch LG, et al. Bronchoalveoloar lavage fluid cytokines and chemokines as markers and predictors for the outcome of interstitial lung disease in systemic sclerosis patients. Arthritis Res Ther. (2009) 11:R111. doi: 10.1186/ar2766
193. Gochuico BR, Avila NA, Chow CK, Novero LJ, Wu HP, Ren P, et al. Progressive preclinical interstitial lung disease in rheumatoid arthritis. Arch Intern Med. (2008) 168:159–66. doi: 10.1001/archinternmed.2007.59
194. Norman KC, O'Dwyer DN, Salisbury ML, DiLillo KM, Lama VN, Xia M, et al. Identification of a unique temporal signature in blood and BAL associated with IPF progression. Sci Rep. (2020) 10:12049. doi: 10.1038/s41598-020-67956-w
195. Furusawa H, Cardwell JH, Okamoto T, Walts AD, Konigsberg IR, Kurche JS, et al. Chronic hypersensitivity pneumonitis, an interstitial lung disease with distinct molecular signatures. Am J Respir Crit Care Med. (2020) 202:1430–44. doi: 10.1164/rccm.202001-0134OC
196. Pankratz DG, Choi Y, Imtiaz U, Fedorowicz GM, Anderson JD, Colby TV, et al. Usual interstitial pneumonia can be detected in transbronchial biopsies using machine learning. Ann Am Thorac Soc. (2017) 14:1646–54. doi: 10.1513/AnnalsATS.201612-947OC
197. Raghu G, Flaherty KR, Lederer DJ, Lynch DA, Colby TV, Myers JL, et al. Use of a molecular classifier to identify usual interstitial pneumonia in conventional transbronchial lung biopsy samples: a prospective validation study. Lancet Respir Med. (2019) 7:487–96. doi: 10.1016/S2213-2600(19)30059-1
198. Richeldi L, Scholand MB, Lynch DA, Colby TV, Myers JL, Groshong SD, et al. Utility of a molecular classifier as a complement to HRCT to identify usual interstitial pneumonia. Am J Respir Crit Care Med. (2020) 203:211–20. doi: 10.1164/rccm.202003-0877OC
199. Kim EJ, Collard HR, King TE Jr. Rheumatoid arthritis-associated interstitial lung disease: the relevance of histopathologic and radiographic pattern. Chest. (2009) 136:1397–405. doi: 10.1378/chest.09-0444
200. Khanna D, Tashkin DP, Denton CP, Renzoni EA, Desai SR, Varga J. Etiology, risk factors, and biomarkers in systemic sclerosis with interstitial lung disease. Am J Respir Crit Care Med. (2020) 201:650–60. doi: 10.1164/rccm.201903-0563CI
201. Churg A, Sin DD, Everett D, Brown K, Cool C. Pathologic patterns and survival in chronic hypersensitivity pneumonitis. Am J Surg Pathol. (2009) 33:1765–70. doi: 10.1097/PAS.0b013e3181bb2538
202. Attanoos RL, Alchami FS, Pooley FD, Gibbs AR. Usual interstitial pneumonia in asbestos-exposed cohorts - concurrent idiopathic pulmonary fibrosis or atypical asbestosis? Histopathology. (2016) 69:492–8. doi: 10.1111/his.12951
203. Raghu G, Remy-Jardin M, Myers J, Richeldi L, Wilson KC. The 2018. diagnosis of idiopathic pulmonary fibrosis guidelines: surgical lung biopsy for radiological pattern of probable usual interstitial pneumonia is not mandatory. Am J Respir Crit Care Med. (2019) 200:1089–92. doi: 10.1164/rccm.201907-1324ED
204. Lynch DA, Sverzellati N, Travis WD, Brown KK, Colby TV, Galvin JR, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. (2018) 6:138–53. doi: 10.1016/S2213-2600(17)30433-2
205. Snetselaar R, van Batenburg AA, van Oosterhout MFM, Kazemier KM, Roothaan SM, Peeters T, et al. Short telomere length in IPF lung associates with fibrotic lesions and predicts survival. PLoS ONE. (2017) 12:e0189467. doi: 10.1371/journal.pone.0189467
Keywords: interstitial lung disease, biomarker, progressive fibrosing ILD, idiopathic pulmonary fibrosis, hypersensitivity pneumonitis, connective tissue disease-associated interstitial lung disease
Citation: Bowman WS, Echt GA and Oldham JM (2021) Biomarkers in Progressive Fibrosing Interstitial Lung Disease: Optimizing Diagnosis, Prognosis, and Treatment Response. Front. Med. 8:680997. doi: 10.3389/fmed.2021.680997
Received: 15 March 2021; Accepted: 06 April 2021;
Published: 10 May 2021.
Edited by:
Sydney Montesi, Massachusetts General Hospital and Harvard Medical School, United StatesReviewed by:
Barry Shea, Brown University, United StatesTraci Adams, University of Texas Southwestern Medical Center, United States
Copyright © 2021 Bowman, Echt and Oldham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Willis S. Bowman, wbowman@ucdavis.edu