 Marcelo T. Bozza
Marcelo T. Bozza Viktória Jeney
Viktória Jeney- 1Laboratório de Inflamação e Imunidade, Departamento de Imunologia, Universidade Federal Do Rio de Janeiro, Rio de Janeiro, Brazil
- 2MTA-DE Lendület Vascular Pathophysiology Research Group, Research Centre for Molecular Medicine, Faculty of Medicine, University of Debrecen, Debrecen, Hungary
Damage associated molecular patterns (DAMPs) are endogenous molecules originate from damaged cells and tissues with the ability to trigger and/or modify innate immune responses. Upon hemolysis hemoglobin (Hb) is released from red blood cells (RBCs) to the circulation and give a rise to the production of different Hb redox states and heme which can act as DAMPs. Heme is the best characterized Hb-derived DAMP that targets different immune and non-immune cells. Heme is a chemoattractant, activates the complement system, modulates host defense mechanisms through the activation of innate immune receptors and the heme oxygenase-1/ferritin system, and induces innate immune memory. The contribution of oxidized Hb forms is much less studied, but some evidence show that these species might play distinct roles in intravascular hemolysis-associated pathologies independently of heme release. This review aims to summarize our current knowledge about the formation and pro-inflammatory actions of heme and other Hb-derived DAMPs.
Introduction
Red blood cells (RBCs)—the most abundant cell type of the human body—deliver oxygen to the tissues during their lifespan of about 120 days. Under homeostasis, destruction of aged RBCs is usually an unrecognized event that takes place in macrophages of splenic and hepatic sinusoids (1). After RBC phagocytosis macrophages efficiently handle the high hemoglobin (Hb) load, break down heme, and redistribute iron for further use, or store it in a catalytically inactive form in ferritin (FT) (1). Some RBCs lyse intravascularly even under physiological conditions releasing Hb and heme to the circulation. To prevent the deleterious effects of extracellular Hb and heme they are scavenged by the acute phase plasma proteins haptoglobin (Hp) and hemopexin (Hx), respectively, and removed from the circulation rapidly (2). On the other hand, oversaturation of this defense system leads to the accumulation of different redox forms of Hb and heme in the circulation.
Heme is a well-known pro-oxidant that feature relies—at least in part—on the ability of heme iron to catalize the generation of hydroxyl-radicals in the Fenton reaction, due to its capability of acting as both an electron donor and an acceptor (3). Another important source of radical species that could mediate heme-induced toxicity is the conversion of organic hydroperoxides (ROOH) into highly reactive alkoxyl (RO•) and peroxyl (ROO•) radicals (4, 5). These radicals trigger lipid peroxidation forming alkyl radicals that in the presence of O2 will generate more peroxyl radicals, thus amplifying free radical reactions (6). Finally, heme can also promote reactive oxygen species (ROS) generation through enzymatic reactions mediated by NADPH oxidases (7–11) and by the mitochondria (11).
Besides being a pro-oxidant, three lines of evidence indicated that heme is an inflammatory molecule with unique properties: (i) sterile intra or extra vascular hemolysis cause inflammation, that is controlled by Hx and heme oxygenase-1 (HO-1) (12); (ii) injection of heme in experimental animals triggers local and systemic inflammation (11–14); (iii) heme activates innate immune cells in vitro acting as a chemoatractant, inducing cytokine production, ROS generation, and cell death (7, 8, 11, 15, 16). The observations that heme causes macrophage activation dependently of the innate immune receptors TLR4 and NLRP3 were important to a paradigm shift, defining heme as a prototipical damage-associated molecular pattern (DAMP) (11, 16–18). The requirement of TLR4 or NLRP3 to the pathological consequences of experimental sterile hemolysis suggest that heme-induced activation of these pathways contributes to the pathology (11, 12, 19, 20). Importantly, the tissue damage triggered by the actions of labile heme also critically contributes to the pathogenesis of severe infections such as malaria (21–24) and sepsis (14, 25). Growing evidence shows that the complement system can be activated by heme which mechanism play a role in the pathomechanism of certain hemolytic diseases (20, 26–28). On the other hand heme can activate defense mechanisms to establish tolerance and to foster survival of the host in diverse pathological conditions via the induction of the HO-1/FT system (17, 23, 29, 30). Recent investigation showed that heme can induce innate immune memory as well (31).
Growing evidence suggest that besides labile heme other Hb-related DAMPs e.g., metHb, ferrylHb as well as covalently crosslinked Hb multimers can be considered as alarmins (32–34). These species might play distinct, heme-independent roles in intravascular hemolysis-associated pathologies. The multiple mechanisms by which Hb-derived DAMPs modulate cell activation and inflammation, contributing to pathology, are object of intense research. In this review we aim to give an overview of the most recent development of this dynamically evolving field.
Hb Inside of the RBCs
Hb, the major oxygen-transport protein consist of 2 different subunits, α and β, that compose a α2β2 tetrahedron. Each of the four subunits contains a heme prosthetic group with a central Fe2+ (ferrous) ion. Heme iron is critically involved in O2 binding. Each ml of human blood contains ~0.3 g of Hb, most of it is compartmentalized within RBCs.
Circulating RBCs are continuously exposed to high levels of ROS of both endogenous and exogenous origin [reviewed in (35)]. When Hb binds O2, Hb auto-oxidation frequently occurs in which the central heme Fe2+ is oxidized into Fe3+ (ferric, metHb) with the concomitant reduction of O2 into superoxide anion () (Figure 1). This reaction is a major source of endogenous ROS inside the RBCs. Cytochrome-b5 reductase, an NADH-dependent enzyme present in RBCs convert metHb to Hb, therefore metHb content in intact RBC generally stays below 1%.
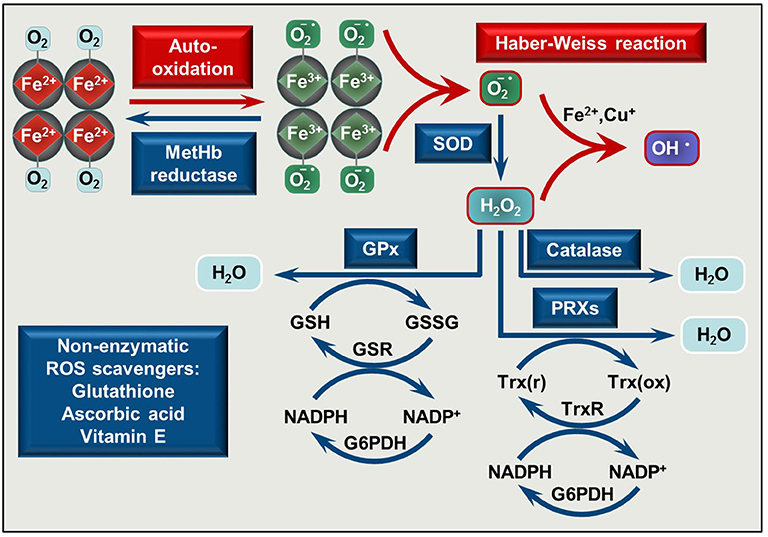
Figure 1. Pro-oxidant and antioxidant mechanisms is RBCs. O2 binding to Hb initiates Hb auto-oxidation in which process metHb (Fe3+) and superoxide anion () are formed. MetHb is reduced by metHb reductase, while is converted to H2O2 by superoxide dismutase (SOD). In the presence of transition metals such as Fe2+ or Cu+ a reaction between and H2O2 occurs yielding hydroxyl radical (OH•) (Haber Weiss reaction). Catalase, glutathione peroxidases (GPx), and peroxiredoxins (PRXs) decompose H2O2. The antioxidant system is completed with non-enzymatic low molecular weight scavengers, such as glutathione, ascorbic acid, and vitamin E. SOD, superoxide dismutase; GPx, glutathione peroxidase; PRXs, peroxiredoxins; GSH, reduced glutathione; GSSG, glutathione disulfide; GSR, glutathione-disulfide reductase; NADP+, nicotinamide adenine dinucleotide phosphate; NADPH, reduced NADP; G6PDH, glucose-6-phosphate dehydrogenase; Trx(r), reduced thioredoxin; Trx(ox), oxidized thioredoxin; TrxR, thioredoxin reductase.
A highly effective antioxidant defense system protects RBCs from the continuously produced ROS. This system consists of enzymes, such as Cu/Zn superoxide dismutase that converts superoxide anion to hydrogen-peroxide (H2O2), catalase, glutathione peroxidase, and peroxiredoxins which decompose H2O2 to H2O [reviewed in (35–37)] and non-enzymatic low molecular weight scavengers, such as glutathione, ascorbic acid, and vitamin E (Figure 1). When ROS production exceeds the capability of ROS neutralization, RBC membrane damage occurs which impairs oxygen delivery.
During their lifespan in the circulation RBCs lose about 20% of their initial Hb content via vesiculation (38). This process is considered an efficient mechanism to remove damaged membrane patches, senescent cell antigens and intracellular inclusion bodies (Heinz bodies) from the otherwise healthy RBCs, therefore they can stay longer in the circulation (39). Approximately after 120 days in the circulation RBCs are completely worn out, and they are cleaned from the circulation by hemophagocytic macrophages, mainly in the spleen, via a non-inflammatory process which allows efficient and safe recycling of the RBC components, particularly the heme iron (40–42).
Hb Outside of the RBCs
Diverse inherited or acquired conditions can trigger uncontrolled destruction of RBCs in the vasculature or in the extravascular space. Upon RBC lysis a large amounts of Hb is released into the circulation, or into the surrounding tissues.
Elimination of Cell Free Hb and Limitations of the Clearance System
Following RBC lysis extracellular Hb is promptly removed from the circulation. Hp, an acute phase plasma protein is in the first line of defense [reviewed in (43)]. Hp binds extracellular Hb avidly, protects Hb from oxidation (44–48), and facilitates its clearance from the circulation through endocytosis via the CD163 macrophage scavenger receptor (49). Although abundant in the plasma (0.41–1.65 mg/ml), the amount of Hp allows the clearance of ~3 g of Hb, <1% of the Hb amount in the circulation. Therefore, massive hemolysis with more than 1% of RBC lysis, Hp is depleted from the plasma and cell-free Hb is eliminated from the circulation via alternative mechanisms. These include (i) a low-affinity pathway through CD163 by macrophages (50) and (ii) renal excretion which is accompanied by profound oxidative stress and organ damage (51, 52).
Failure of Hb Clearance: Nitric Oxide Depletion, Hb Oxidation, and Heme Release
Once the capacity of the Hp/CD163 system is overwhelmed, cell free Hb accumulates in the plasma. Hb exhibits a high affinity for nitric oxide (NO), the important endogenously produced gas that plays a major role in the regulation of vascular tone [reviewed in (53, 54)]. Scavenging of NO by Hb triggers vasoconstriction that contributes to clinical complications in diverse forms of hereditary or acquired hemolytic anemias (55). Furthermore, non-compartmentalized Hb cannot benefit from the highly efficient antioxidant defense system present in intact RBCs, and Hb tends to oxidize. One-electron oxidation of Hb occurs when Hb reacts with NO resulting metHb. Also auto-oxidation of oxyHb triggers metHb generation with the concomitant production of superoxide anions (Figure 2A). Two-electron oxidation of Hb occurs when Hb reacts with peroxides, such as H2O2 or lipid hydroperoxides leading to the formation of ferryl (Fe4+ = O 2−) Hb (Figure 2B). When metHb reacts with peroxides ferrylHb radical is formed [Hb•+(Fe4+= O 2−)] in which the unpaired electron is located at either the globin chain or at the porphyrin ring (56–59). The ferryl oxidation state of iron is very unstable, therefore these high-valence Hb forms are short-lived intermediates that decay quickly (60).
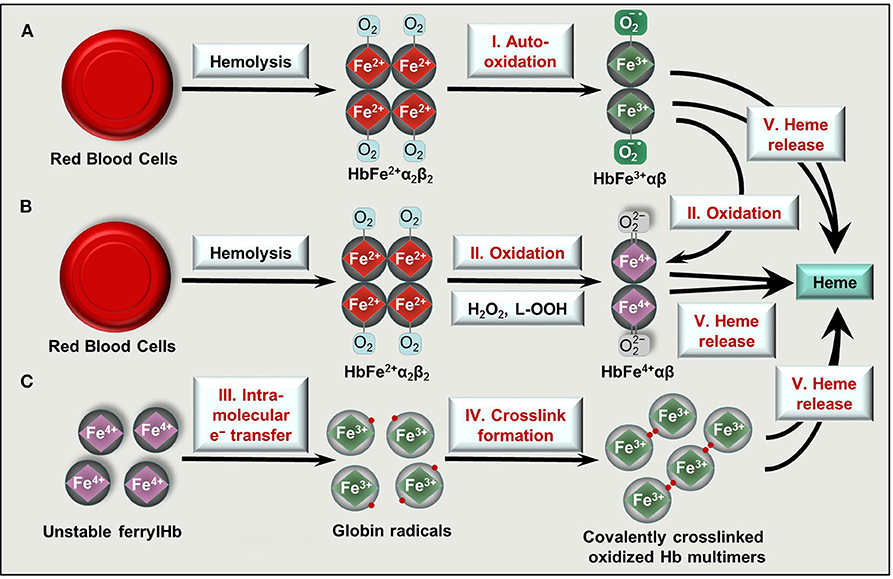
Figure 2. Formation of oxidized Hb forms and labile heme upon hemolysis. Hb tetramers () is released from RBCs following intra- or extravascular hemolysis. (A) Hb outside of RBCs dimerize and can undergo spontaneous auto-oxidation (reaction I) to metHb (HbFe3+αβ). (B) Two-electron oxidation (reactions II) of Hb and metHb by H2O2 or lipid hydroperoxides (L-OOH) lead to the formation of ferrylHb (HbFe4+αβ) or ferrylHb radicals, respectively. (C) FerrylHb get stabilized via intramolecular electron transfer (reaction III) between iron and the globin chain forming globin radicals. Globin radicals get stabilized via covalent crosslinking (reaction IV) producing covalently crosslinked Hb multimers. Oxidized Hb forms (metHb, ferrylHb, covalently crosslinked Hb) release their heme prosthetic group (reactions V).
Ferryl iron can oxidize the neighboring oxidation-prone amino acid residues of the globin chains (i.e., αTyr-24, αTyr-42, αHis-20, βTyr-35, βTyr-130, and βCys-93) with the concomitant reduction of Fe4+ into Fe3+ (37, 61, 62). This intramolecular electron transfer between the ferryl iron and the amino acids yields metHb globin radicals in which the unpaired electrons are located on the oxidized amino acid residues (37, 61, 62). Reactions between globin radicals or between globin and porphyrin-centered radicals lead to the formation of globin-globin and porphyrin-globin adducts, respectively (Figure 2C). These structurally altered Hb forms are less efficiently removed from the circulation because both high-affinity (Hb-Hp/CD163) and low-affinity (Hb/CD163) endocytosis pathways are compromised (50, 63).
The prosthetic heme group is tightly bound in Hb, while this bound is weakened in oxidized Hb forms. Both metHb and ferrylHb releases heme moiety (Figure 2) which is captured by the acute phase plasma protein Hx (64). Hx-heme complexes are taken up mainly by macrophages and hepatocytes through the scavenger receptor LDL receptor-related protein 1/CD91 (65, 66). Similarly to Hp, Hx is also depleted from the plasma upon massive intravascular hemolysis, leading to the appearance of labile heme, that is, a redox active form of heme which is loosely bound to molecules, other than hemoproteins including albumin, α1-microglobulin and lipoproteins such as LDL and HDL.
Recycling of heme iron is a critical component of systemic iron metabolism. Iron is released from the heme molecule via the action of heme oxygenases (HOs), mainly HO-1, the inducible isoenzyme that catabolizes free heme into equimolar amounts of Fe2+, carbon monoxide (CO), and biliverdin (67). HO-1 induction and heme degradation products exhibit various cytoprotective mechanisms (29). Heme-mediated HO-1 induction and iron release is associated with the upregulation of ferritin, the major intracellular iron storage protein, assuring that iron is stored in a catalytically inactive still bioavailable form inside the cells (68).
Activation of the Innate Immune System by Labile Heme and Oxidized Hb Forms
Hemolytic and hemorrhagic episodes are often accompanied by inflammation even in the absence of pathogens (17, 69). Accumulating evidence suggest that upon hemolysis RBCs release large amounts of DAMPs including RBC microvesicles, heme, ATP, heat shock protein 70, interleukin-33 that induce pro-inflammatory responses in different cells (70, 71). Here we will focus on the contribution of Hb-derived DAMPs to the hemolysis-induced sterile inflammatory responses (Figure 3).
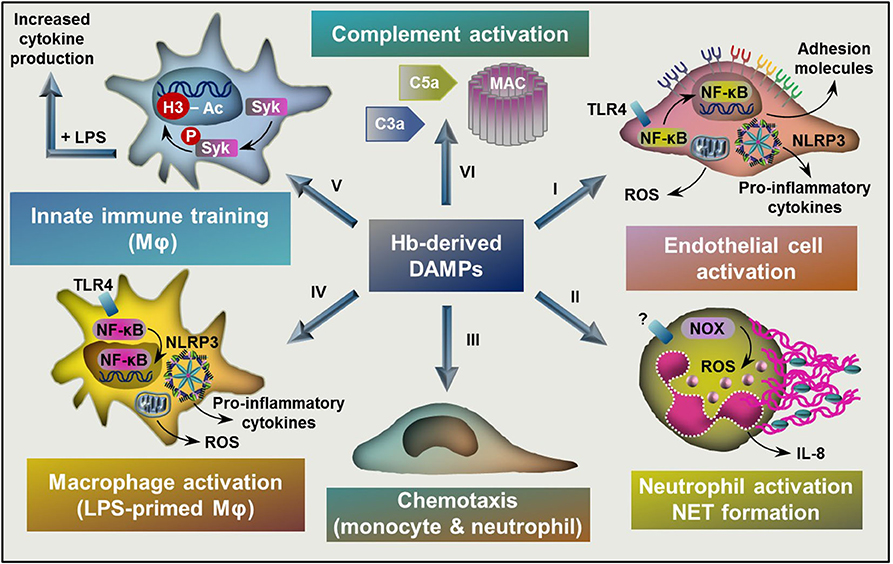
Figure 3. Targets of Hb-derived DAMPs. (I) Labile heme and ferrylHb induces endothelial cell activation characterized by NF-κB activation, elevated ROS production, and increased expression of adhesion molecules and pro-inflammatory cytokines. (II) Heme activates neutrophils characterized by elevated ROS production through the activation of NOX, increased production of IL-8 and NET formation. (III) Heme and ferrylHb induces monocyte and neutrophil chemotaxis. (IV) Labile heme and ferrylHb induces ROS production, NLRP3 activation, and pro-inflammatory cytokine production in LPS-primed macrophages. (V) Heme induces innate immune training through triggering epigenetic changes, such as acetylation of H3 at lysine-27 in monocytes and macrophages in a Syk-dependent manner. (VI) Heme induces complement activation leading to the formation of C3a and C5a activation fragments and the assembly of MAC. NF-κB, nuclear factor kappa B; ROS, reactive oxygen species; NOX, NADPH oxidase; NET, neutrophil extracellular trap; TLR4, toll-like receptor 4, NLRP3, NLR family pyrin domain containing 3; LPS, lipopolysaccharide; Syk, Spleen tyrosine kinase; H3, histone 3, MAC, membrane attack complex.
Endothelial Cells (ECs) as First Line Targets of Hb-Derived DAMPs
A monolayer of ECs cover the entire vasculature and the lymphatic system providing a semi-permeable barrier between blood and tissue, and lymph and tissue, respectively. Under physiological conditions, ECs are involved in many processes including the regulation of metabolic homeostasis, vascular hemodynamics, vascular permeability, coagulation, and cell trafficking [reviewed in (72)]. Besides of these numerous functions, ECs are one of the first cell types to detect pathogen-associated molecular patterns (PAMPs) and DAMPs in the bloodstream, therefore ECs have important immunological functions in the early innate immune system activation as danger signal sensors [reviewed in (72)]. ECs are equipped with a series of pathogen-associated pattern recognition receptors (PRRs) including toll-like receptors (TLRs) and nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), as well as diverse chemokine receptors [reviewed in (73)]. Growing evidence shows that ECs respond to various Hb-derived DAMPs.
Heme-Mediated TLR4-Dependent EC Activation (Adhesion Molecules and Barrier Function)
ECs respond to a variety of inflammatory stimuli e.g., IL-1, tumor necrosis factor α (TNF-α), lipopolysaccharid (LPS) by upregulating the expression of cellular adhesion molecules including intracellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and E selectin (74, 75). These adhesion molecules anchor leukocytes to the endothelial surface and facilitate their transmigration into the inflamed tissue. Interestingly heme, similarly to that of IL-1, TNF-α, or LPS upregulates the expressions of adhesion molecules (76) in a TLR4-dependent manner (19, 20).
This heme-mediated TLR4-dependent mechanism has been connected to vaso-occlusive crisis in sickle cell disease (Figure 3) (19). Recent evidence shows that TLR4-dependent upregulation of endothelial P-selectin triggers an unconventional route of complement activation by non-covalent binding of C3 activation fragments on the surface of ECs, which mechanism contributes to liver injury in hemolytic diseases such as sickle cell disease (20).
Besides increased expression of cell surface adhesion molecules, increased endothelial permeability contributes to inflammatory cell extravasation upon hemolysis. Many attempts were made to identify the molecular mechanism of this phenomenon which revealed that ferrylHb and free heme trigger the loss of endothelial integrity (33, 77–80). Heme-induced loss of endothelial barrier function is dependent on the activation of the p38/heat shock protein 27 pathway (79) and associated with TLR4-dependent production of ROS and necroptosis (77).
FerrylHb-Mediated TLR4-Independent EC Activation (Adhesion Molecules and Barrier Function)
Besides heme, ferrylHb but not Hb or metHb induces up-regulation of adhesion molecules ICAM-1, VCAM-1, and E-selectin, and increase endothelial cell monolayer permeability in human ECs (Figure 3) (33). Interestingly, ferrylHb-mediated responses are dependent on the activation of nuclear factor kappaB (NF-κB), requires actin polymerization, involves the activation of the c-Jun N-terminal kinase and the p38 mitogen-activated protein kinase signal transduction pathways but not dependent on TLR4 activation (33). The facts that (i) ferrylHb and heme trigger endothelial activation with the use different signaling mechanisms and that (ii) metHb – that can release heme more avidly than ferrylHb – does not induce EC activation suggest that ferrylHb-mediated EC activation cannot be simply considered as a consequence of heme release from ferrylHb. The putative receptors associated with ferrylHb-induced EC activation are currently unknown.
Induction of Cytokine Production by Hb-Derived DAMPs in ECs
Growing evidence suggest that endothelial cells are not only sentinels of the activation of the innate immune system but actively participate in cytokine production upon hemolysis. Recently heme and ferrylHb were identified as activators of the NLRP3 inflammasome leading to processing and secretion of active IL-1β in LPS-primed macrophages (11, 34). Previously it has been shown that ECs respond to classical DAMPs such as extracellular ATP and high mobility group box 1 protein (HMGB1) by the activation of NLRP3 inflammasome and the subsequent production of IL-1β (81, 82). Based on these information we investigated whether heme and the different Hb redox forms induce NLRP3 inflammasome activation in ECs (32). We showed that heme but not the different Hb redox forms induced NLRP3 inflammasome activation and IL-1β production ECs (Figure 3). Heme-induced inflammasome activation in ECs requires LPS priming, structural integrity of the heme molecule, and ROS production (32). Recent data suggest that globin-derived peptides formed during Hb oxidation are also capable to induce NLRP3 inflammasome activation and IL-1β production in ECs (83). Besides IL-1β production, Hb-derived DAMPs, namely metHb but not Hb has been implicated in IL-6 and IL-8 production in ECs (84). In the same experimental setting heme did not induce IL-6 and IL-8 production in ECs, so we can assume that metHb-induced EC response was independent of heme release.
Hb-Derived Molecules as Chemoattractants
Heme is chemoatractant in vivo, which notion is supported by the finding that peritoneal injection of heme causes the recruitment of neutrophils and intravenous administration of heme causes leukocyte infiltration in various organs (7, 11, 15, 85). Heme-induced neutrophil recruitment is independent of TLR4 activation (16), but depends on the endogenous production of leukotriene B4 by macrophages (86) and the activation of the NLRP3 inflammasome (11).
Recent studies showed that besides heme, ferrylHb, but not Hb and metHb triggers peritoneal infiltration of monocytes and neutrophils (33, 34). The chemotactic effect of ferrylHb is less likely to be dependent on heme release exclusively, which is supported by two facts; first, ferrylHb is a more powerful inducer of leukocyte infiltration than heme and second, metHb that has the ability to release heme at the same or even higher rate as ferrylHb (87) fails to trigger leukocyte recruitment (34).
Actions on Neutrophils
Neutrophil granulocytes play a fundamental role in innate and adaptive immunity. Upon infection or inflammation, neutrophils are the first leukocytes migrating from the blood into the affected tissues. Neutrophils are equipped with sensors of PAMPs and DAMPs, they kill and phagocytose pathogens and clear cellular debris (88). In the recent years, it has become evident that neutrophils not only sense PAMPs but can recognize and respond to endogenous DAMPs as well. As it was mentioned before, heme and ferrylHb are potent triggers of neutrophil infiltration (7, 15, 33, 85). Moreover, heme has been shown to activate neutrophils characterized by elevated ROS production and increased expression of the pro-inflammatory cytokine IL-8 (Figure 3) (7, 15). Heme is a potent chemoattractant of neutrophils in vitro in a mechanism characteristic of a G protein-coupled receptor activation (7, 15). Heme-induced neutrophil chemotaxis and ROS production are independent of the coordinated iron present in heme, while requires the vinyl groups in the porphyrin ring (15, 89).
Upon activation neutrophils release extracellular traps—meshes composed of chromatin and neutrophil granular proteins—which plays a critical role in immobilization of invading pathogens (90). Recently heme has been identified as a potent inducer of neutrophil extracellular trap (NET) formation in TNF-α-primed neutrophils in vitro and in vivo (91). Accumulating evidence show that heme-mediated NET formation plays a pathogenic role in vaso-occlusion crises in sickle cell disease, in transfusion-related acute lung injury, in systemic inflammation in paroxysmal nocturnal hemoglobinuria as well as in malaria (91–94). Interestingly, heme-induced NET formation requires the coordinated heme iron, is dependent on NADPH oxidase and ROS formation but occurs independently of TLR4 (95). These results suggest that at least two different signaling pathways are activated by heme on neutrophils. One that triggers chemotaxis and is independent of the heme iron while requires the vinyl groups, and one that triggers the NET release, requires the iron but not the vinyl groups of the porphyrin ring. The putative receptors associated with these activities are currently unknown.
Activation of Macrophages by Hb-Derived DAMPs
Macrophages are effector cells of the innate immune system, which respond to a variety of PAMPs and DAMPs. Macrophages are present in all vertebrate tissues and have highly heterogeneous phenotypes depending on the environmental cues encountered.
TLR4 Activation by Heme in Macrophages
Heme profoundly affect macrophage physiology through multiple pathways (17, 18). The requirement of TLR4 to the induction of TNF production by heme on macrophages was the first demonstration of a receptor-mediated effect of heme (16). The coordinated iron and the vinyl groups are essential for heme to induce TLR4-dependent TNF production. The effect of heme on macrophages through TLR4 is exquisitely different from the effect of LPS, the canonical agonist of TLR4. While LPS triggers the activation of the Myeloid differentiation primary response 88 (MyD88) and the TIR-domain-containing adapter-inducing interferon-β (TRIF) pathways (96) on macrophages and DCs, heme activates only the MYD-88 pathway and is unable to induce the expression of type I interferon or co-stimulatory molecules (16).
Heme-Induced Macrophage Necroptosis
High amounts of free heme due to hemolysis is involved with the loss of macrophages, specially in the absence of HO-1 (97). Heme induces macrophage necroptotic cell death in a mechanism that requires TNF, ROS, and the kinases RIPK1 and RIPK3 (98). Although required to heme-induced TNF production, TLR4 is not essential for the necroptosis induced by heme in the presence of exogenous TNF. An important study demonstrated that heme triggers tissue macrophage differentiation by inducing the transcriptional factor Spic in monocytes through a mechanism dependent on the degradation of the transcriptional repressor Bach1 (99). During pathological hemolysis, the axis formed by heme, Bach1, and Spic is critically involved in the homeostatic response to the macrophage loss. Another compensatory response to hemolysis and heme is the formation of aggresome-like induced structures (ALIS) on macrophages, p62/SQTM1 aggregates containing ubiquitinated proteins (100, 101). The heme-induced ALIS formation on macrophages requires mitochondrial ROS, NRF2 and HO-1, while is independent of TLR4. Moreover, iron from heme is necessary, while both Fe2+ and Fe3+ are sufficient to trigger ALIS formation (100). The physiopathological role of heme-iduced ALIS formation is currently unknown.
NLRP3 Activation by Heme in Macrophages
Heme has a synergistic effect with microbial molecules on macrophages, increasing the production of inflammatory cytokines in a mechanism dependent of ROS and spleen tyrosine kinase (Syk) (13). Moreover, heme induces NLRP3 inflammasome activation leading to processing and secretion of active IL-1β in LPS-primed macrophages (11). Heme-mediated NLRP3 inflammasome activation is found to be dependent on the coordinated heme iron, and also involves activation of Syk, elevated ROS production by NOX2 and the mitochondria and K(+) efflux, contributing to intravascular hemolysis-induced lethality (11). An interesting study demonstrated that heme reduces the host resistance to bacterial infection (102). Treatment of macrophages with heme, but not with Hb, free iron, or the heme analogs protoporphyrin IX and tin-protoporphyrin IX, causes a dose-dependent inhibition of E. coli phagocytosis by macrophages (102). This inhibitory effect of heme on macrophage phagocytosis and chemotaxis occurs through the activation of the GTP-binding Rho family protein Cdc42 by DOCK8, a guanine nucleotide exchange factor, disrupting actin cytoskeletal dynamics (102). Together, these results indicate possible signaling pathways for therapeutic intervention during hemolytic infectious conditions.
NLRP3 Activation by ferrylHb in Macrophages
Besides heme, the involvement of different Hb redox forms was investigated in hemolysis-associated NLRP3 inflammasome activation in macrophages. That study revealed that ferrylHb but not Hb or metHb induce active IL-1β production in LPS-primed macrophages in an NLRP3-dependent manner (34). Based on the fact that metHb cannot induce IL-1β production in LPS-primed macrophages it is unlikely that heme release plays a critical role in the ferrylHb-triggered response. FerrylHb-induced NLRP3 activation is associated with elevated ROS production but the detailed molecular mechanism needs to be further explored (34).
Hemorrhage-Associated Macrophage Subsets
Besides the two extreme canonical macrophage phenotypes, the pro-inflammatory M1 and the anti-inflammatory M2, many other specific and distinct macrophage subsets exist (103). Both extracellular Hb and heme are implicated in macrophage polarization triggering the formation of hemorrhage-associated M(Hb) and M(heme) subsets, respectively (103–106). These hemorrhage-associated macrophage subsets were first identified in advanced human atherosclerotic lesions with intraplaque hemorrhage (103–106). M(Hb) macrophages represent a subpopulation of CD68+ macrophages and their characteristic markers are the macrophage mannose receptor 1 and the CD163 receptor through which macrophages recognize and endocytose Hp-Hb complexes (103–105). Additionally, due to their role in Hb clearance, M(Hb) macrophages exhibit increased HO-1 and ferroportin expressions thereby facilitating heme catabolism and cellular efflux of excess iron (105). Reduced labile iron content in M(Hb) macrophages is associated with less ROS production which is linked to increased activity of the transcription factor liver X receptor-α and the induction of cholesterol efflux (105). Because of this, M(Hb) macrophages are protected from lipid accumulation and produce anti-inflammatory factors, such as IL-10 (105). M(heme) macrophage polarization is driven by extracellular heme, and similarly to that of M(Hb) this subset is protected from oxidative stress and lipid accumulation (107).
Activation of Microglia by Hb-Derived DAMPs
Microglia are the primary innate immune effector cells of the central nervous system (CNS) with a similar function to macrophages. Intracerebral and subarachnoid hemorrhages (ICH and SAH, respectively) are associated with activation of microglia and growing evidence suggest that inflammation is the key contributor of secondary brain injury induced by ICH or SAH (108, 109). Microglia have an important function in hematoma resolution by phagocytosing RBCs which process is mediated by the class B scavenger receptor CD36 (110, 111). CD36 expression is regulated by peroxisome proliferator-activated receptor γ (PPARγ), and activation of PPARγ has been shown to promote hematoma resolution and decrease neuronal damage following ICH (111). Incomplete removal of RBCs leads to hemolysis and the production of oxidized Hb forms and free heme (112, 113). Microglia plays a critical role in removing these toxic Hb derivatives from the central nervous system through CD163-Hp-Hb and CD91-Hx-heme scavenging mechanisms and heme degradation by HO enzymes (114–117). These mechanisms can attenuate bleeding-associated neuronal damage, though we have to note that upon significant intrathecal hemolysis the Hb-heme elimination system is largely overwhelmed leading to the accumulation of oxidized Hb forms and free heme in the CNS (112–114).
These Hb-derived DAMPs trigger neuroinflammation following ICH and SAH. In line of this notion it has been shown that heme induces TLR4-mediated inflammatory injury via the activation of MyD88/TRIF pathways in microglia following ICH (118). Importantly, mice deficient of Tlr4 or anti-TLR4 treatment reduce heme-induced neurologic deficit, brain edema, and inflammation. Not only heme, but also metHb has been identified as a TLR4 agonist that triggers the secretion of TNF-α by the microglia (119). A number of studies support the notion that TLR4 contributes to the brain injury due to ICH (120–123). Additionally, heme induces the release of IL-1α but not IL-1β in primary mixed glia (124). Targeting these inflammatory pathways with anti-TLR4 antibody or with IL-1 receptor antagonist attenuate intrathecal hemorrhage-associated inflammatory injury (118, 124). A recent study showed that besides heme and metHb, large amounts of covalently crosslinked Hb multimers (dimers and tetramers) accumulate in the cerebrospinal fluid of preterm infants following IVH (112). Further work needed to address whether these Hb multimers are implicated in the inflammatory microglia activation following IVH.
Induction of Innate Immune Memory by Heme
Trained immunity or innate immune memory is the ability of the innate immune system to adapt its function after previous encounters with pathogens or their products (125). This mechanism not only provides protection against reinfection but also contributes cross-protection between infections with different pathogens (125, 126). The major cell types in which trained immunity occurs are myeloid cells, natural killer cells, and innate lymphoid cells. Trained immunity is activated by PAMPs such as LPS or β-glucan via PRR signaling, resulting changes in transcription programs through epigenetic regulation that can persist for up to several weeks (125, 126). Epigenetic reprogramming – driven by histone acetylation, for example at lysine-9 (H3K9ac) and lysine-27 (H3K27ac) of H3 histones that almost exclusively determines transcriptional capability – is a critical determinant of trained immunity (127).
Recently it was reported that heme is a potent inducer of trained immunity in monocytes and macrophages both in vitro and in vivo (31). Heme pretreatment increased pro-inflammatory cytokine (TNF-α, IL-6, IL-8) release from macrophages upon secondary challenge by LPS (Figure 3) (31). Such effect of heme was independent on the pro-oxidant nature of heme, which notion is supported by the fact that (i) trained immunity is induced by protoporphyrin IX, lacking iron and (ii) it is not prevented by the glutathione precursor N-acetyl cysteine (31). Heme pretreatment triggered epigenetic changes, such as acetylation of H3k27. Comparing heme and β-glucan-induced training in monocytes revealed overlapping as well as distinct epigenetic and transcriptional responses between the two triggers (31). Common pathways, regulated by both heme and β-glucan included lysosome maturation and metabolism. Genes only induced by heme are mainly involved in inflammatory pathways, and as expected heme/iron related metabolism (31). Another remarkable difference between heme- and β-glucan-induced training is that heme-mediated training relies on the activation of Syk and c-Jun n-terminal kinase, but independent on the activation of the Mammalian Target of Rapamycin which is largely involved in β-glucan training (31). This finding reinforces the critical role of Syk signaling on heme-induced macrophage activation (11, 13). At present, the mechanism by which heme triggers Syk phosphorylation is unknown (17). Interestingly, heme seems to be a Janus-faced training molecule in vivo resulting that the outcome of heme pretreatment largely depends on the experimental conditions (31).
Complement Activation and the Thromboinflammatory Loop
Originally the complement system has been considered as a simple mechanism to induce bacterial lysis. Recently it became evident that complement has diverse functions in both physiologic and pathologic conditions (128). The complement system senses PAMPs and DAMPs and translate the danger information into an adequate cellular innate or adaptive immune response (129). The complement system is a cascade of more than 40 proteins, which can be initiated by different ways. There are three known distinct ways for complement activation: the classical, the lectin-mediated, and the alternative pathway (AP) (130). Activation of each of the three pathways leads to a common terminal pathway in which the inactive C3 protein is cleaved into the functional fragments C3a and C3b, and the membrane attack complex (MAC) is formed (130). These products of complement activation mediate a diverse inflammatory response that includes opsonization and phagocytosis, bacterial killing, immune cell recruitment, endothelial and epithelial cell activation, platelet activation and interaction with the adaptive immune system (128).
It has long been known that hemolytic disorders, such as sickle-cell disease, beta-thalassemia major, thrombotic thrombocytopenic purpura, and paroxysmal nocturnal hemoglobinuria are associated with complement over-activation (131–136). Recent evidence suggest that heme has a direct role in hemolysis-associated complement activation (Figure 3). In line of this notion heme has been shown to activate the complement AP and trigger the deposition of C3 activation fragments on the surface of RBCs (28). A detailed work showed that C3a, C5a, and sC5b9 activation fragments are formed during heme-mediated activation of the complement AP in normal human serum (Figure 3) (26). Additionally, heme-exposed ECs also activate the AP resulting in cell-bound C3 and MAC, which mechanism contributes to endothelial damage and thrombosis in atypical hemolytic uremic syndrome (26). Drug-induced intravascular hemolysis or injection of heme trigger C3 deposition in the kidneys and subsequent renal damage which can be attenuated by the heme scavenger Hx. Also, deficiency of C3 attenuates hemolysis-induced kidney injury in mice, suggesting that heme-mediated complement activation and C3 deposition play a fundamental role in renal damage upon intravascular hemolysis (27).
Clinical and epidemiological studies revealed that RBC abnormalities such as abnormal hematocrit, sickle cell disease, thalassemia, hemolytic anemias, and malaria are associated with increased incidence of both arterial and venous thrombosis (137). Recently it has been shown that heme activates the tissue factor (TF)-dependent extrinsic coagulation pathway both in vitro and in vivo which can be attenuated by an anti-TF antibody (138, 139). Importantly, inhibition of TF-induced coagulation activation reduces microvascular stasis and lung vaso-occlusion in sickle mice (140). By activating both inflammatory and hemostatic pathways extracellular heme can trigger a thromboinflammatory vicious cycle that can contribute to the pathogenesis of hemolytic diseases. Recently the heme-induced thrombogenecity was studied in an ex vivo human whole blood model. Heme-induced thromboinflammation was attenuated by the inhibition of the complement component C5 and the TLR coreceptor CD14 (141). Inhibition of the thromboinflammatory loop can be a meaningful therapeutic target in hemolytic diseases (141).
Modulation of Host Defense Mechanism by Labile Heme
High concentrations of labile heme are observed in infectious conditions, such as malaria and sepsis, both in humans and experimental animals (14, 17, 21, 29). As in sterile hemolytic conditions, the axis Hp/Hx and HO-1/FT heavy chain (FTH) also provides critical host protection against free heme on infectious disease with increased hemolysis (14, 21–25, 29). Two complementary mechanisms comprise the host response to infection. Resistance, a primarily attribute of the immune system, is associated with the reduction or elimination of infectious agents, while tolerance is the capacity of limiting the pathological consequences of an infection (142, 143). Heme can affect the host responses in multiple ways, modulating both the disease tolerance and the resistance to infection. In a series of important studies, it has been demonstrated that heme contributes to the pathogenesis of malaria by increasing tissue damage, while HO-1 and FTH contributes to disease tolerance irrespective of changes on pathogen loads (21–24). A recent study indicates that sickle cell trait with low grade hemolysis is beneficial in malaria infection due to an increased in disease tolerance associated with higher HO-1 expression (23). CO, generated by the catabolism of heme by HO-1, binds to heme inhibiting its release from the Hb during malaria, thus preventing pathology (21). Moreover, the antioxidant effects of HO-1 inhibits the hepatocyte apoptosis induced by the synergistic effects of heme and TNF, preventing hepatic failure, and death in a mouse model of malaria (144).
In a mouse model of endotoxemia heme enhances the plasma concentrations of TNF and IL-6, drastically increasing the lethality induced by LPS (13). This increased lethality in mice is observed even when the challenge with LPS occurs after 6 days of treatment with heme and correlates with increased numbers of tissue macrophages (31). Heme reduces blood glucose levels dependently of TLR4, contributing to the severity of sepsis, while FTH reverts this effect contributing to glucose and tissue homeostasis (25). In mouse models of severe bacterial infection, heme increases multi organ failure, and lethality irrespective to a change on pathogen load (14). An interesting study has shown that heme reduces resistance to Gram-negative infection in mice predisposing to pathogen dissemination through the suppression of phagocytic function and independently of bacterial growth due to nutritional advantage (102). These results suggest that upon acute bacterial infection heme can be deleterious due to an increase on tissue damage and bacterial loads.
Heme can also modify immunoglobulin-mediated immune responses. This activity relies on the ability of heme to bind to immunoglobulins of different isotypes (IgG, IgA and IgM) leading to the formation of heme-immunoglobulin complexes that exhibit increased reactivities toward various self and bacterial antigens (145). Besides that, it has been shown that heme-IgG complex can interact with previously unrecognized bacterial antigens and intact bacteria through binding to an enlarged panel of structurally unrelated epitopes (146). Heme-induced expansion of the antibody repertoire may represent an inducible innate-type host defense mechanism against infections (146).
Conclusions
Upon hemolysis a large amount of Hb is released from RBCs that is oxidized in the extracellular milieu. Cell free Hb, its oxidation products and heme that is released from oxidized Hb forms are potential DAMPs. Among these numerous Hb oxidation products heme is the most widely studied molecule, and its contribution as a DAMP in hemolysis-associated pathologies has been confirmed. Because of structural alterations oxidized Hb forms (metHb and ferrylHb) bind heme less avidly than Hb, therefore pro-inflammatory actions of oxidized Hb forms was thought to be attributed to their ability to release the heme prosthetic group. This idea is challenged by recent studies suggesting that oxidized Hb forms, in particular ferrylHb exhibit pro-inflammatory actions independently of heme release. A lot of work needs to be done to further explore the colorful picture of Hb-derived DAMPs, their targeted cells and the mechanisms of their actions. Comprehensive understanding of hemolysis/hemorrhage-associated inflammation could contribute to the development of novel therapeutics intended to interrupt these pathological events.
Author Contributions
MB and VJ planned and wrote the manuscript. VJ draw the figures. All authors contributed to the article and approved the submitted version.
Funding
This research was funded by the Hungarian National Research, Development and Innovation Office (NKFIH), Grant No. K131535 and by the Hungarian Academy of Sciences, MTA-DE Lendület Vascular Pathophysiology Research Group, Grant No. 96050. MB received support from the Brazilian agencies FAPERJ, CNPq, and CAPES.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ALIS, aggresome-like induced structures; AP: alternative pathway; ATP, adenosine triphosphate; CNS, central nervous system; CO, carbon monoxide; DAMPs, damage-associated molecular patterns; EC, endothelial cell; ferrylHb, ferryl hemoglobin; FT, ferritin; FTH, ferritin heavy chain; GPXs, glutathione peroxidases; Hb, hemoglobin; HO, heme oxygenase; H2O2, hydrogen-peroxide; Hp, haptoglobin; Hsp, heat shock protein; Hx, hemopexin; H3K9ac, histone 3 acetylation at lysine-9; H3K27ac, histone 3 acetylation at lysine-27; ICAM-1, intracellular adhesion molecule-1; ICH, intracerebral hemorrhage; IgG, immunoglobulin G; IL, interleukin; LPS, lipopolysaccharide; MAC, membrane attack complex; metHb, met(ferric) hemoglobin; MyD88, myeloid differentiation primary response gene 88; NADPH, nicotinamide adenine dinucleotide phosphate; NET, neutrophil extracellular trap; NLR, NOD-like receptor; NLRP3, NLR family pyrin domain containing 3; NO, nitric oxide; NOD, nucleotide-binding oligomerization domain; NOX, NADPH oxidase; PAMPs, pathogen-associated molecular patterns; PMNs, polymorphonuclear cells; PPARγ, Peroxisome proliferator-activated receptor γ; PRR, pattern recognition receptor; PRXs, peroxiredoxins; RBC, red blood cell; ROS, reactive oxygen species; SAH, subarachnoid hemorrhage; SOD, superoxide dismutase; Syk, spleen tyrosine kinase; TLR, toll-like receptor; TNF-α, tumor necrosis factor-alpha; TRIF, TIR-domain-containing adapter-inducing interferon-β; VCAM-1, vascular cell adhesion molecule-1.
References
1. Ganz T. Macrophages and systemic iron homeostasis. J Innate Immun. (2012) 4:446–53. doi: 10.1159/000336423
2. Schaer DJ, Vinchi F, Ingoglia G, Tolosano E, Buehler PW. Haptoglobin, hemopexin, and related defense pathways-basic science, clinical perspectives, and drug development. Front Physiol. (2014) 5:415. doi: 10.3389/fphys.2014.00415
3. Sadrzadeh SM, Graf E, Panter SS, Hallaway PE, Eaton JW. Hemoglobin. A biologic fenton reagent. J Biol Chem. (1984) 259:14354–6.
4. Tappel AL. Unsaturated lipide oxidation catalyzed by hematin compounds. J Biol Chem. (1955) 217:721–33.
5. VanderZee J, Barr DP, Mason RP. ESR spin trapping investigation of radical formation from the reaction between hematin and tert-butyl hydroperoxide. Free Radical Bio Med. (1996) 20:199–206. doi: 10.1016/0891-5849(95)02031-4
6. Ryter SW, Tyrrell RM. The Heme synthesis and degradation pathways: role in oxidant sensitivity - heme oxygenase has both pro- and antioxidant properties. Free Radical Bio Med. (2000) 28:289–309. doi: 10.1016/S0891-5849(99)00223-3
7. Graca-Souza AV, Arruda MA, de Freitas MS, Barja-Fidalgo C, Oliveira PL. Neutrophil activation by heme: implications for inflammatory processes. Blood. (2002) 99:4160–5. doi: 10.1182/blood.V99.11.4160
8. Arruda MA, Barcellos-de-Souza P, Sampaio AL, Rossi AG, Graca-Souza AV, Barja-Fidalgo C. NADPH oxidase-derived ROS: key modulators of heme-induced mitochondrial stability in human neutrophils. Exp Cell Res. (2006) 312:3939–48. doi: 10.1016/j.yexcr.2006.08.022
9. Moraes JA, Barcellos-de-Souza P, Rodrigues G, Nascimento-Silva V, Silva SV, Assreuy J, et al. Heme modulates smooth muscle cell proliferation and migration via NADPH oxidase: a counter-regulatory role for heme oxygenase system. Atherosclerosis. (2012) 224:394–400. doi: 10.1016/j.atherosclerosis.2012.07.043
10. Barcellos-de-Souza P, Moraes JA, de-Freitas-Junior JC, Morgado-Diaz JA, Barja-Fidalgo C, Arruda MA. Heme modulates intestinal epithelial cell activation: involvement of NADPHox-derived ROS signaling. Am J Physiol Cell Physiol. (2013) 304:C170–9. doi: 10.1152/ajpcell.00078.2012
11. Dutra FF, Alves LS, Rodrigues D, Fernandez PL, de Oliveira RB, Golenbock DT, et al. Hemolysis-induced lethality involves inflammasome activation by heme. Proc Natl Acad Sci USA. (2014) 111:E4110–8. doi: 10.1073/pnas.1405023111
12. Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, Archer DR, et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest. (2013) 123:4809–20. doi: 10.1172/JCI64578
13. Fernandez PL, Dutra FF, Alves L, Figueiredo RT, Mourao-Sa D, Fortes GB, et al. Heme amplifies the innate immune response to microbial molecules through spleen tyrosine kinase (Syk)-dependent reactive oxygen species generation. J Biol Chem. (2010) 285:32844–51. doi: 10.1074/jbc.M110.146076
14. Larsen R, Gozzelino R, Jeney V, Tokaji L, Bozza FA, Japiassu AM, et al. A central role for free heme in the pathogenesis of severe sepsis. Sci Transl Med. (2010) 2:51ra71. doi: 10.1126/scitranslmed.3001118
15. Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, Graca-Souza A, et al. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem. (2007) 282:24430–6. doi: 10.1074/jbc.M703570200
16. Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, et al. Characterization of heme as activator of toll-like receptor 4. J Biol Chem. (2007) 282:20221–9. doi: 10.1074/jbc.M610737200
17. Dutra FF, Bozza MT. Heme on innate immunity and inflammation. Front Pharmacol. (2014) 5:115. doi: 10.3389/fphar.2014.00115
18. Soares MP, Bozza MT. Red alert: labile heme is an alarmin. Curr Opin Immunol. (2016) 38:94–100. doi: 10.1016/j.coi.2015.11.006
19. Belcher JD, Chen C, Nguyen J, Milbauer L, Abdulla F, Alayash AI, et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood. (2014) 123:377–90. doi: 10.1182/blood-2013-04-495887
20. Merle NS, Paule R, Leon J, Daugan M, Robe-Rybkine T, Poillerat V, et al. P-selectin drives complement attack on endothelium during intravascular hemolysis in TLR-4/heme-dependent manner. Proc Natl Acad Sci USA. (2019) 116:6280–5. doi: 10.1073/pnas.1814797116
21. Pamplona A, Ferreira A, Balla J, Jeney V, Balla G, Epiphanio S, et al. Heme oxygenase-1 and carbon monoxide suppress the pathogenesis of experimental cerebral malaria. Nat Med. (2007) 13:703–10. doi: 10.1038/nm1586
22. Ferreira A, Balla J, Jeney V, Balla G, Soares MP. A central role for free heme in the pathogenesis of severe malaria: the missing link? J Mol Med. (2008) 86:1097–111. doi: 10.1007/s00109-008-0368-5
23. Ferreira A, Marguti I, Bechmann I, Jeney V, Chora A, Palha NR, et al. Sickle hemoglobin confers tolerance to Plasmodium infection. Cell. (2011) 145:398–409. doi: 10.1016/j.cell.2011.03.049
24. Gozzelino R, Andrade BB, Larsen R, Luz NF, Vanoaica L, Seixas E, Coutinho A, et al. Metabolic adaptation to tissue iron overload confers tolerance to malaria. Cell Host Microbe. (2012) 12:693–704. doi: 10.1016/j.chom.2012.10.011
25. Weis S, Carlos AR, Moita MR, Singh S, Blankenhaus B, Cardoso S, et al. Metabolic adaptation establishes disease tolerance to sepsis. Cell. (2017) 169:1263–75 e14. doi: 10.1016/j.cell.2017.05.031
26. Frimat M, Tabarin F, Dimitrov JD, Poitou C, Halbwachs-Mecarelli L, Fremeaux-Bacchi Vet al. Complement activation by heme as a secondary hit for atypical hemolytic uremic syndrome. Blood. (2013) 122:282–92. doi: 10.1182/blood-2013-03-489245
27. Merle NS, Grunenwald A, Rajaratnam H, Gnemmi V, Frimat M, Figueres ML, et al. Intravascular hemolysis activates complement via cell-free heme and heme-loaded microvesicles. JCI Insight. (2018) 3:e96910. doi: 10.1172/jci.insight.96910
28. Pawluczkowycz AW, Lindorfer MA, Waitumbi JN, Taylor RP. Hematin promotes complement alternative pathway-mediated deposition of C3 activation fragments on human erythrocytes: potential implications for the pathogenesis of anemia in malaria. J Immunol. (2007) 179:5543–52. doi: 10.4049/jimmunol.179.8.5543
29. Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol. (2010) 50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600
30. Ramos S, Carlos AR, Sundaram B, Jeney V, Ribeiro A, Gozzelino R, et al. Renal control of disease tolerance to malaria. Proc Natl Acad Sci USA. (2019) 116:5681–6. doi: 10.1073/pnas.1822024116
31. Jentho E, Novakovic B, Ruiz-Moreno C, Kourtzelis I, Martins R, Chavakis T, et al. Heme induces innate immune memory. bioRxiv. [Preprint]. (2019). doi: 10.1101/2019.12.12.874578
32. Erdei J, Toth A, Balogh E, Nyakundi BB, Banyai E, Ryffel B, et al. Induction of NLRP3 inflammasome activation by heme in human endothelial cells. Oxid Med Cell Longev. (2018) 2018:4310816. doi: 10.1155/2018/4310816
33. Silva G, Jeney V, Chora A, Larsen R, Balla J, Soares MP. Oxidized hemoglobin is an endogenous proinflammatory agonist that targets vascular endothelial cells. J Biol Chem. (2009) 284:29582–95. doi: 10.1074/jbc.M109.045344
34. Nyakundi BB, Toth A, Balogh E, Nagy B, Erdei J, Ryffel B, et al. Oxidized hemoglobin forms contribute to NLRP3 inflammasome-driven IL-1beta production upon intravascular hemolysis. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:464–75. doi: 10.1016/j.bbadis.2018.10.030
35. Mohanty JG, Nagababu E, Rifkind JM. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front Physiol. (2014) 5:84. doi: 10.3389/fphys.2014.00084
36. Siems WG, Sommerburg O, Grune T. Erythrocyte free radical and energy metabolism. Clin Nephrol. (2000) 53:S9–17.
37. Jeney V, Eaton JW, Balla G, Balla J. Natural history of the bruise: formation, elimination, and biological effects of oxidized hemoglobin. Oxid Med Cell Longev. (2013) 2013:703571. doi: 10.1155/2013/703571
38. Willekens FL, Roerdinkholder-Stoelwinder B, Groenen-Dopp YA, Bos HJ, Bosman GJ, van den Bos AG, et al. Hemoglobin loss from erythrocytes in vivo results from spleen-facilitated vesiculation. Blood. (2003) 101:747–51. doi: 10.1182/blood-2002-02-0500
39. Willekens FL, Werre JM, Groenen-Dopp YA, Roerdinkholder-Stoelwinder B, de Pauw B, Bosman GJ. Erythrocyte vesiculation: a self-protective mechanism? Br J Haematol. (2008) 141:549–56. doi: 10.1111/j.1365-2141.2008.07055.x
40. Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. (2017) 168:344–61. doi: 10.1016/j.cell.2016.12.034
41. Bratosin D, Mazurier J, Tissier JP, Estaquier J, Huart JJ, Ameisen JC, et al. Cellular and molecular mechanisms of senescent erythrocyte phagocytosis by macrophages. A review. Biochimie. (1998) 80:173–95. doi: 10.1016/S0300-9084(98)80024-2
42. de Back DZ, Kostova EB, van Kraaij M, van den Berg TK, van Bruggen R. Of macrophages and red blood cells; a complex love story. Fronti Physiol. (2014) 5:9. doi: 10.3389/fphys.2014.00009
43. Alayash AI. Haptoglobin: old protein with new functions. Clin Chim Acta. (2011) 412:493–8. doi: 10.1016/j.cca.2010.12.011
44. Miller YI, Altamentova SM, Shaklai N. Oxidation of low-density lipoprotein by hemoglobin stems from a heme-initiated globin radical: antioxidant role of haptoglobin. Biochemistry. (1997) 36:12189–98. doi: 10.1021/bi970258a
45. Buehler PW, Abraham B, Vallelian F, Linnemayr C, Pereira CP, Cipollo JF, et al. Haptoglobin preserves the CD163 hemoglobin scavenger pathway by shielding hemoglobin from peroxidative modification. Blood. (2009) 113:2578–86. doi: 10.1182/blood-2008-08-174466
46. Pimenova T, Pereira CP, Gehrig P, Buehler PW, Schaer DJ, Zenobi R. Quantitative mass spectrometry defines an oxidative hotspot in hemoglobin that is specifically protected by haptoglobin. J Proteome Res. (2010) 9:4061–70. doi: 10.1021/pr100252e
47. Banerjee S, Jia Y, Siburt CJ, Abraham B, Wood F, Bonaventura C, et al. Haptoglobin alters oxygenation and oxidation of hemoglobin and decreases propagation of peroxide-induced oxidative reactions. Free Radic Biol Med. (2012) 53:1317–26. doi: 10.1016/j.freeradbiomed.2012.07.023
48. Andersen CB, Torvund-Jensen M, Nielsen MJ, de Oliveira CL, Hersleth HP, Andersen NH, et al. Structure of the haptoglobin-haemoglobin complex. Nature. (2012) 489:456–9. doi: 10.1038/nature11369
49. Kristiansen M, Graversen JH, Jacobsen C, Sonne O, Hoffman HJ, Law SK, et al. Identification of the haemoglobin scavenger receptor. Nature. (2001) 409:198–201. doi: 10.1038/35051594
50. Schaer DJ, Schaer CA, Buehler PW, Boykins RA, Schoedon G, Alayash AI, et al. CD163 is the macrophage scavenger receptor for native and chemically modified hemoglobins in the absence of haptoglobin. Blood. (2006) 107:373–80. doi: 10.1182/blood-2005-03-1014
51. Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. (2013) 121:1276–84. doi: 10.1182/blood-2012-11-451229
52. Murray RK, Connell GE, Pert JH. The role of haptoglobin in the clearance and distribution of extracorpuscular hemoglobin. Blood. (1961) 17:45–53. doi: 10.1182/blood.V17.1.45.45
53. Rother RP, Bell L, Hillmen P, Gladwin MT. The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin: a novel mechanism of human disease. JAMA. (2005) 293:1653–62. doi: 10.1001/jama.293.13.1653
54. Helms CC, Gladwin MT, Kim-Shapiro DB. Erythrocytes and vascular function: oxygen and nitric oxide. Front Physiol. (2018) 9:125. doi: 10.3389/fphys.2018.00125
55. Kato GJ, Steinberg MH, Gladwin MT. Intravascular hemolysis and the pathophysiology of sickle cell disease. J Clin Invest. (2017) 127:750–60. doi: 10.1172/JCI89741
56. Harel S, Kanner J. The generation of ferryl or hydroxyl radicals during interaction of haemproteins with hydrogen peroxide. Free Radic Res Commun. (1988) 5:21–33. doi: 10.3109/10715768809068555
57. Patel RP, Svistunenko DA, Darley-Usmar VM, Symons MC, Wilson MT. Redox cycling of human methaemoglobin by H2O2 yields persistent ferryl iron and protein based radicals. Free Radic Res. (1996) 25:117–23. doi: 10.3109/10715769609149916
58. Jia Y, Buehler PW, Boykins RA, Venable RM, Alayash AI. Structural basis of peroxide-mediated changes in human hemoglobin: a novel oxidative pathway. J Biol Chem. (2007) 282:4894–907. doi: 10.1074/jbc.M609955200
59. Alayash AI, Patel RP, Cashon RE. Redox reactions of hemoglobin and myoglobin: biological and toxicological implications. Antioxid Redox Signal. (2001) 3:313–27. doi: 10.1089/152308601300185250
60. Reeder BJ, Cutruzzola F, Bigotti MG, Hider RC, Wilson MT. Tyrosine as a redox-active center in electron transfer to ferryl heme in globins. Free Radic Biol Med. (2008) 44:274–83. doi: 10.1016/j.freeradbiomed.2007.06.030
61. Deterding LJ, Ramirez DC, Dubin JR, Mason RP, Tomer KB. Identification of free radicals on hemoglobin from its self-peroxidation using mass spectrometry and immuno-spin trapping: observation of a histidinyl radical. J Biol Chem. (2004) 279:11600–7. doi: 10.1074/jbc.M310704200
62. Ramirez DC, Chen YR, Mason RP. Immunochemical detection of hemoglobin-derived radicals formed by reaction with hydrogen peroxide: involvement of a protein-tyrosyl radical. Free Radic Biol Med. (2003) 34:830–9. doi: 10.1016/S0891-5849(02)01437-5
63. Vallelian F, Pimenova T, Pereira CP, Abraham B, Mikolajczyk MG, Schoedon G, et al. The reaction of hydrogen peroxide with hemoglobin induces extensive alpha-globin crosslinking and impairs the interaction of hemoglobin with endogenous scavenger pathways. Free Radic Biol Med. (2008) 45:1150–8. doi: 10.1016/j.freeradbiomed.2008.07.013
64. Hrkal Z, Vodrazka Z, Kalousek I. Transfer of heme from ferrihemoglobin and ferrihemoglobin isolated chains to hemopexin. Eur J Biochem. (1974) 43:73–8. doi: 10.1111/j.1432-1033.1974.tb03386.x
65. Hvidberg V, Maniecki MB, Jacobsen C, Hojrup P, Moller HJ, Moestrup SK. Identification of the receptor scavenging hemopexin-heme complexes. Blood. (2005) 106:2572–9. doi: 10.1182/blood-2005-03-1185
66. Herz J, Strickland DK. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest. (2001) 108:779–84. doi: 10.1172/JCI200113992
67. Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci USA. (1968) 61:748–55. doi: 10.1073/pnas.61.2.748
68. Eisenstein RS, Garcia-Mayol D, Pettingell W, Munro HN. Regulation of ferritin and heme oxygenase synthesis in rat fibroblasts by different forms of iron. Proc Natl Acad Sci USA. (1991) 88:688–92. doi: 10.1073/pnas.88.3.688
69. Arruda MA, Graca-Souza AV, Barja-Fidalgo C. Heme and innate immunity: new insights for an old molecule. Mem Inst Oswaldo Cruz. (2005) 100:799–803. doi: 10.1590/S0074-02762005000700022
70. Mendonca R, Silveira AA, Conran N. Red cell DAMPs and inflammation. Inflamm Res. (2016) 65:665–78. doi: 10.1007/s00011-016-0955-9
71. Jeney V. Pro-inflammatory actions of red blood cell-derived DAMPs. Exp Suppl. (2018) 108:211–33. doi: 10.1007/978-3-319-89390-7_9
72. Mai J, Virtue A, Shen J, Wang H, Yang XF. An evolving new paradigm: endothelial cells–conditional innate immune cells. J Hematol Oncol. (2013) 6:61. doi: 10.1186/1756-8722-6-61
73. Opitz B, Eitel J, Meixenberger K, Suttorp N. Role of toll-like receptors, NOD-like receptors and RIG-I-like receptors in endothelial cells and systemic infections. Thromb Haemost. (2009) 102:1103–9. doi: 10.1160/TH09-05-0323
74. Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA Jr. Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. J Clin Invest. (1985) 76:2003–11. doi: 10.1172/JCI112200
75. Pohlman TH, Stanness KA, Beatty PG, Ochs HD, Harlan JM. An endothelial cell surface factor(s) induced in vitro by lipopolysaccharide, interleukin 1, and tumor necrosis factor-alpha increases neutrophil adherence by a CDw18-dependent mechanism. J Immunol. (1986) 136:4548–53.
76. Wagener FA, Feldman E, de Witte T, Abraham NG. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proc Soc Exp Biol Med. (1997) 216:456–63. doi: 10.3181/00379727-216-44197
77. Singla S, Sysol JR, Dille B, Jones N, Chen J, Machado RF. Hemin causes lung microvascular endothelial barrier dysfunction by necroptotic cell death. Am J Respir Cell Mol Biol. (2017) 57:307–14. doi: 10.1165/rcmb.2016-0287OC
78. Kuck JL, Bastarache JA, Shaver CM, Fessel JP, Dikalov SI, May JM, et al. Ascorbic acid attenuates endothelial permeability triggered by cell-free hemoglobin. Biochem Biophys Res Commun. (2018) 495:433–7. doi: 10.1016/j.bbrc.2017.11.058
79. Rafikova O, Williams ER, McBride ML, Zemskova M, Srivastava A, Nair V, et al. Hemolysis-induced lung vascular leakage contributes to the development of pulmonary hypertension. Am J Respir Cell Mol Biol. (2018) 59:334–45. doi: 10.1165/rcmb.2017-0308OC
80. Wagener BM, Hu PJ, Oh JY, Evans CA, Richter JR, Honavar J, et al. Role of heme in lung bacterial infection after trauma hemorrhage and stored red blood cell transfusion: a preclinical experimental study. PLoS Med. (2018) 15:e1002522. doi: 10.1371/journal.pmed.1002522
81. Li YX, Wang P, Yang XF, Wang WR, Zhang JY, He YH, et al. SIRT1 inhibits inflammatory response partly through regulation of NLRP3 inflammasome in vascular endothelial cells. Mol Immunol. (2016) 77:148–56. doi: 10.1016/j.molimm.2016.07.018
82. Chen Y, Pitzer AL, Li X, Li PL, Wang L, Zhang Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J Cell Mol Med. (2015) 19:2715–27. doi: 10.1111/jcmm.12657
83. Posta N, Csosz E, Oros M, Petho D, Potor L, Kallo G, et al. Hemoglobin oxidation generates globin-derived peptides in atherosclerotic lesions and intraventricular hemorrhage of the brain, provoking endothelial dysfunction. Lab Invest. (2020) 1–17. doi: 10.1038/s41374-020-0403-x
84. Liu XY, Spolarics Z. Methemoglobin is a potent activator of endothelial cells by stimulating IL-6 and IL-8 production and E-selectin membrane expression. Am J Physiol Cell Physiol. (2003) 285:C1036–46. doi: 10.1152/ajpcell.00164.2003
85. Wagener FADTG, Eggert A, Boerman OC, Oyen WJG, Verhofstad A, Abraham NG, et al. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. (2001) 98:1802–11. doi: 10.1182/blood.V98.6.1802
86. Monteiro AP, Pinheiro CS, Luna-Gomes T, Alves LR, Maya-Monteiro CM, Porto BN, et al. Leukotriene B4 mediates neutrophil migration induced by heme. J Immunol. (2011) 186:6562–7. doi: 10.4049/jimmunol.1002400
87. Kassa T, Jana S, Meng F, Alayash AI. Differential heme release from various hemoglobin redox states and the upregulation of cellular heme oxygenase-1. Febs Open Bio. (2016) 6:876–84. doi: 10.1002/2211-5463.12103
88. Rosales C, Demaurex N, Lowell CA, Uribe-Querol E. Neutrophils: their role in innate and adaptive immunity. J Immunol Res. (2016) 2016:1469780. doi: 10.1155/2016/1469780
89. Kono M, Saigo K, Takagi Y, Kawauchi S, Wada A, Hashimoto M, et al. Morphological and flow-cytometric analysis of haemin-induced human neutrophil activation: implications for transfusion-related acute lung injury. Blood Transfus. (2013) 11:53–60. doi: 10.2450/2012.0141-11
90. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
91. Chen G, Zhang D, Fuchs TA, Manwani D, Wagner DD, Frenette PS. Heme-induced neutrophil extracellular traps contribute to the pathogenesis of sickle cell disease. Blood. (2014) 123:3818–27. doi: 10.1182/blood-2013-10-529982
92. Kono M, Saigo K, Takagi Y, Takahashi T, Kawauchi S, Wada A, et al. Heme-related molecules induce rapid production of neutrophil extracellular traps. Transfusion. (2014) 54:2811–9. doi: 10.1111/trf.12700
93. van Bijnen STA, Wouters D, van Mierlo GJ, Muus P, Zeerleder S. Neutrophil activation and nucleosomes as markers of systemic inflammation in paroxysmal nocturnal hemoglobinuria: effects of eculizumab. J Thromb Haemost. (2015) 13:2004–11. doi: 10.1111/jth.13125
94. Knackstedt SL, Georgiadou A, Apel F, Abu-Abed U, Moxon CA, Cunnington AJ, et al. Neutrophil extracellular traps drive inflammatory pathogenesis in malaria. Sci Immunol. (2019) 4:eaaw0336. doi: 10.1126/sciimmunol.aaw0336
95. Ohbuchi A, Kono M, Kitagawa K, Takenokuchi M, Imoto S, Saigo K. Quantitative analysis of hemin-induced neutrophil extracellular trap formation and effects of hydrogen peroxide on this phenomenon. Biochem Biophys Rep. (2017) 11:147–53. doi: 10.1016/j.bbrep.2017.07.009
96. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. (2010) 140:805–20. doi: 10.1016/j.cell.2010.01.022
97. Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood. (2010) 116:6054–62. doi: 10.1182/blood-2010-03-272138
98. Fortes GB, Alves LS, de Oliveira R, Dutra FF, Rodrigues D, Fernandez PL, et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood. (2012) 119:2368–75. doi: 10.1182/blood-2011-08-375303
99. Haldar M, Kohyama M, So AYL, Wumesh KC, Wu XD, Briseno CG, et al. Heme-mediated SPI-C induction promotes monocyte differentiation into iron-recycling macrophages. Cell. (2014) 156:1223–34. doi: 10.1016/j.cell.2014.01.069
100. Vasconcellos LRC, Dutra FF, Siqueira MS, Paula-Neto HA, Dahan J, Kiarely E, et al. Protein aggregation as a cellular response to oxidative stress induced by heme and iron. Proc Natl Acad Sci USA. (2016) 113:E7474–82. doi: 10.1073/pnas.1608928113
101. Travassos LH, Vasconcellos LRC, Bozza MT, Carneiro LAM. Heme and iron induce protein aggregation. Autophagy. (2017) 13:625–6. doi: 10.1080/15548627.2016.1271515
102. Martins R, Maier J, Gorki AD, Huber KV, Sharif O, Starkl P, et al. Heme drives hemolysis-induced susceptibility to infection via disruption of phagocyte functions. Nat Immunol. (2016) 17:1361–72. doi: 10.1038/ni.3590
103. Chinetti-Gbaguidi G, Colin S, Staels B. Macrophage subsets in atherosclerosis. Nat Rev Cardiol. (2015) 12:10–7. doi: 10.1038/nrcardio.2014.173
104. Vinchi F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, Jeney V. Atherogenesis and iron: from epidemiology to cellular level. Front Pharmacol. (2014) 5:94. doi: 10.3389/fphar.2014.00094
105. Finn A, Nakano M, Polavarapu R, Karmali V, Saeed O, Zhao XQ, et al. Hemoglobin directs macrophage differentiation and prevents foam cell formation in human atherosclerotic plaques. Circulation. (2011) 124:166–177. doi: 10.1016/j.jacc.2011.10.852
106. Boyle JJ, Harrington HA, Piper E, Elderfield K, Stark J, Landis RC, et al. Coronary intraplaque hemorrhage evokes a novel atheroprotective macrophage phenotype. Am J Pathol. (2009) 174:1097–108. doi: 10.2353/ajpath.2009.080431
107. Boyle JJ, Johns M, Kampfer T, Nguyen AT, Game L, Schaer DJ, et al. Activating transcription factor 1 directs Mhem atheroprotective macrophages through coordinated iron handling and foam cell protection. Circ Res. (2012) 110:20–33. doi: 10.1161/CIRCRESAHA.111.247577
108. Zhou Y, Wang Y, Wang J, Anne Stetler R, Yang QW. Inflammation in intracerebral hemorrhage: from mechanisms to clinical translation. Prog Neurobiol. (2014) 115:25–44. doi: 10.1016/j.pneurobio.2013.11.003
109. Schneider UC, Xu R, Vajkoczy P. Inflammatory events following subarachnoid hemorrhage (SAH). Curr Neuropharmacol. (2018) 16:1385–95. doi: 10.2174/1570159X16666180412110919
110. Connor J, Pak CC, Schroit AJ. Exposure of phosphatidylserine in the outer leaflet of human red-blood-cells - relationship to cell-density, cell age, and clearance by mononuclear-cells. J Biol Chem. (1994) 269:2399–404.
111. Zhao XR, Sun GH, Zhang J, Strong R, Song WT, Gonzales N, et al. Hematoma resolution as a target for intracerebral hemorrhage treatment: role for peroxisome proliferator-activated receptor gamma in microglia/macrophages. Ann Neurol. (2007) 61:352–62. doi: 10.1002/ana.21097
112. Erdei J, Toth A, Nagy A, Nyakundi BB, Fejes Z, Nagy B, et al. The role of hemoglobin oxidation products in triggering inflammatory response upon intraventricular hemorrhage in premature infants. Front Immunol. (2020) 11:228. doi: 10.3389/fimmu.2020.00228
113. Nyakundi BB, Erdei J, Toth A, Balogh E, Nagy A, Nagy B Jr, et al. Formation and detection of highly oxidized hemoglobin forms in biological fluids during hemolytic conditions. Oxid Med Cell Longev. (2020) 2020:8929020. doi: 10.1155/2020/8929020
114. Bulters D, Gaastra B, Zolnourian A, Alexander S, Ren DX, Blackburn SL, et al. Haemoglobin scavenging in intracranial bleeding: biology and clinical implications. Nat Rev Neurol. (2018) 14:416–32. doi: 10.1038/s41582-018-0020-0
115. Galea J, Cruickshank G, Teeling JL, Boche D, Garland P, Perry VH, et al. The intrathecal CD163-haptoglobin-hemoglobin scavenging system in subarachnoid hemorrhage. J Neurochem. (2012) 121:785–92. doi: 10.1111/j.1471-4159.2012.07716.x
116. Schallner N, Pandit R, LeBlanc R, Thomas AJ, Ogilvy CS, Zuckerbraun BS, et al. Microglia regulate blood clearance in subarachnoid hemorrhage by heme oxygenase-1. J. Clin. Investigat. (2015) 125:2609–25. doi: 10.1172/JCI78443
117. Ma B, Day JP, Phillips H, Slootsky B, Tolosano E, Dore S. Deletion of the hemopexin or heme oxygenase-2 gene aggravates brain injury following stroma-free hemoglobin-induced intracerebral hemorrhage. J Neuroinflammation. (2016) 13:26. doi: 10.1186/s12974-016-0490-1
118. Lin S, Yin Q, Zhong Q, Lv FL, Zhou Y, Li JQ, et al. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J Neuroinflammation. (2012) 9:46. doi: 10.1186/1742-2094-9-46
119. Kwon MS, Woo SK, Kurland DB, Yoon SH, Palmer AF, Banerjee U, et al. Methemoglobin is an endogenous toll-like receptor 4 ligand-relevance to subarachnoid hemorrhage. Int J Mol Sci. (2015) 16:5028–46. doi: 10.3390/ijms16035028
120. Teng W, Wang L, Xue W, Guan C. Activation of TLR4-mediated NfκB signaling in hemorrhagic brain in rats. Mediat Inflamm. (2009) 2009:473276. doi: 10.1155/2009/473276
121. Sansing LH, Harris TH, Welsh FA, Kasner SE, Hunter CA, Kariko K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann Neurol. (2011) 70:646–56. doi: 10.1002/ana.22528
122. Fang H, Wang PF, Zhou Y, Wang YC, Yang QW. Toll-like receptor 4 signaling in intracerebral hemorrhage-induced inflammation and injury. J Neuroinflammation. (2013) 10:27. doi: 10.1186/1742-2094-10-27
123. Wang YC, Wang PF, Fang H, Chen J, Xiong XY, Yang QW. Toll-like receptor 4 antagonist attenuates intracerebral hemorrhage-induced brain injury. Stroke. (2013) 44:2545–52. doi: 10.1161/STROKEAHA.113.001038
124. Greenhalgh AD, Brough D, Robinson EM, Girard S, Rothwell NJ, Allan SM. Interleukin-1 receptor antagonist is beneficial after subarachnoid haemorrhage in rat by blocking haem-driven inflammatory pathology. Dis Model Mech. (2012) 5:823–33. doi: 10.1242/dmm.008557
125. Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, et al. Trained immunity: a program of innate immune memory in health and disease. Science. (2016) 352:aaf1098. doi: 10.1126/science.aaf1098
126. Quintin J, Cheng SC, van der Meer JW, Netea MG. Innate immune memory: towards a better understanding of host defense mechanisms. Curr Opin Immunol. (2014) 29:1–7. doi: 10.1016/j.coi.2014.02.006
127. van der Heijden C, Noz MP, Joosten LAB, Netea MG, Riksen NP, Keating ST. Epigenetics and trained immunity. Antioxid Redox Signal. (2018) 29:1023–40. doi: 10.1089/ars.2017.7310
128. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol. (2015) 6:257. doi: 10.3389/fimmu.2015.00257
129. Kohl J. Self, non-self, and danger: a complementary view. Adv Exp Med Biol. (2006) 586:71–94. doi: 10.1007/0-387-34134-X_6
130. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol. (2015) 6:262. doi: 10.3389/fimmu.2015.00262
131. Mold C, Tamerius JD, Phillips G Jr. Complement activation during painful crisis in sickle cell anemia. Clin Immunol Immunopathol. (1995) 76:314–20. doi: 10.1006/clin.1995.1131
132. deCiutiis AC, Peterson CM, Polley MJ, Metakis LJ. Alternative pathway activation in sickle cell disease and beta-thalassemia major. J Natl Med Assoc. (1978) 70:503–6.
133. Ruiz-Torres MP, Casiraghi F, Galbusera M, Macconi D, Gastoldi S, Todeschini M, et al. Complement activation: the missing link between ADAMTS-13 deficiency and microvascular thrombosis of thrombotic microangiopathies. Thromb Haemostasis. (2005) 93:443–52. doi: 10.1160/TH04-07-0450
134. Hochsmann B, Murakami Y, Osato M, Knaus A, Kawamoto M, Inoue N, et al. Complement and inflammasome overactivation mediates paroxysmal nocturnal hemoglobinuria with autoinflammation. J Clin Invest. (2019) 129:5123–36. doi: 10.1101/635573
135. Logue GL, Rosse WF, Adams JP. Mechanisms of immune lysis of red blood cells in vitro. I. Paroxysmal nocturnal hemoglobinuria cells. J Clin Invest. (1973) 52:1129–37. doi: 10.1172/JCI107279
136. Roumenina LT, Chadebech P, Bodivit G, Vieira-Martins P, Grunenwald A, Boudhabhay I, et al. Complement activation in sickle cell disease: Dependence on cell density, hemolysis and modulation by hydroxyurea therapy. Am J Hematol. (2020) 95:456–64. doi: 10.1002/ajh.25742
137. Byrnes JR, Wolberg AS. Red blood cells in thrombosis. Blood. (2017) 130:1795–9. doi: 10.1182/blood-2017-03-745349
138. Sparkenbaugh EM, Chantrathammachart P, Wang S, Jonas W, Kirchhofer D, Gailani D, et al. Excess of heme induces tissue factor-dependent activation of coagulation in mice. Haematologica. (2015) 100:308–14. doi: 10.3324/haematol.2014.114728
139. de Souza GR, Hounkpe BW, Fiusa MML, Colella MP, Annichino-Bizzacchi JM, Traina F, et al. Tissue factor-dependent coagulation activation by heme: a thromboelastometry study. PLoS ONE. (2017) 12:e0176505. doi: 10.1371/journal.pone.0176505
140. Sparkenbaugh EM, Chen C, Brzoska T, Nguyen J, Wang S, Vercellotti GM, et al. Thrombin activation of PAR-1 contributes to microvascular stasis in mouse models of sickle cell disease. Blood. (2020) 135:1783–7. doi: 10.1182/blood.2019003543
141. Thomas AM, Gerogianni A, McAdam MB, Floisand Y, Lau C, Espevik T, et al. Complement component C5 and TLR molecule CD14 mediate heme-induced thromboinflammation in human blood. J Immunol. (2019) 203:1571–8. doi: 10.4049/jimmunol.1900047
142. Schneider DS, Ayres JS. Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat Rev Immunol. (2008) 8:889–95. doi: 10.1038/nri2432
143. Medzhitov R, Schneider DS, Soares MP. Disease tolerance as a defense strategy. Science. (2012) 335:936–41. doi: 10.1126/science.1214935
144. Seixas E, Gozzelino R, Chora A, Ferreira A, Silva G, Larsen R, et al. Heme oxygenase-1 affords protection against noncerebral forms of severe malaria. Proc Natl Acad Sci USA. (2009) 106:15837–42. doi: 10.1073/pnas.0903419106
145. Dimitrov JD, Vassilev TL, Andre S, Kaveri SV, Lacroix-Desmazes S. Functional variability of antibodies upon oxidative processes. Autoimmun Rev. (2008) 7:574–8. doi: 10.1016/j.autrev.2008.04.009
Keywords: hemoglobin, heme, TLR4 (toll-like receptor 4), NLRP3, DAMP, hemolysis (red blood cells)
Citation: Bozza MT and Jeney V (2020) Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front. Immunol. 11:1323. doi: 10.3389/fimmu.2020.01323
Received: 01 April 2020; Accepted: 26 May 2020;
Published: 30 June 2020.
Edited by:
Nicola Conran, Campinas State University, BrazilReviewed by:
Lubka T. Roumenina, INSERM U1138 Centre de Recherche des Cordeliers, FranceSwee Lay Thein, National Institutes of Health (NIH), United States
Copyright © 2020 Bozza and Jeney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viktória Jeney, jeney.viktoria@med.unideb.hu