 Esther von Stebut1*†
Esther von Stebut1*† Wolf-Henning Boehncke2,3†
Wolf-Henning Boehncke2,3† Kamran Ghoreschi4†
Kamran Ghoreschi4† Tommaso Gori5,6†
Tommaso Gori5,6† Ziya Kaya7†
Ziya Kaya7† Diamant Thaci8†
Diamant Thaci8† Andreas Schäffler9†
Andreas Schäffler9†- 1Department of Dermatology, University of Cologne, Cologne, Germany
- 2Division of Dermatology and Venereology, Geneva University Hospitals, Geneva, Switzerland
- 3Department of Pathology and Immunology, Faculty of Medicine, University de Geneva, Geneva, Switzerland
- 4Department of Dermatology, Venereology and Allergology, Charité—Universitätsmedizin Berlin, Berlin, Germany
- 5Center of Cardiology—Cardiology I, University Medical Center Mainz, Mainz, Germany
- 6German Center for Cardiovascular Research, University Center Mainz, Mainz, Germany
- 7Department of Internal Medicine III, University of Heidelberg, Heidelberg, Germany
- 8Institute and Comprehensive Center of Inflammation Medicine, University of Lübeck, Lübeck, Germany
- 9Department of Internal Medicine III, Giessen University Hospital, Giessen, Germany
Interleukin 17A (IL-17A) is one of the currently known six members of the IL-17 cytokine family and is implicated in immune responses to infectious pathogens and in the pathogenesis of inflammatory autoimmune diseases like psoriasis. Psoriatic skin is characterized by high expression of IL-17A and IL-17F, which act on immune and non-immune cell types and strongly contribute to tissue inflammation. In psoriatic lesions, IL-17A, IL-17E, and IL-17F are involved in neutrophil accumulation, followed by the formation of epidermal micro abscesses. IL-17A together with other Th17 cytokines also upregulates the production of several chemokines that are implicated in psoriasis pathogenesis. IL17A-targeting antibodies show an impressive clinical efficacy in patients with psoriasis. Studies have reported an improvement of at least 75% as measured by the psoriasis area and severity index (PASI) in >80% of patients treated with anti-IL-17A therapy. Psoriasis skin manifestations, cardiovascular as well as metabolic disease in psoriasis appear to share pathogenic mechanisms evolving around IL-17A and its proinflammatory role. Thus, anti-IL-17A therapy not only improves skin manifestations of psoriasis, but also cardiovascular inflammation as well as metabolic factors and different domains of psoriatic arthritis (PsA) including peripheral arthritis, enthesitis, dactylitis, and axial involvement. This review summarizes the biological role of IL-17A, before reviewing currently available data on its role in the physiology and pathophysiology of the skin, as well as the cardiovascular and the metabolic system. In conclusion, clinical recommendations for patients with moderate to severe psoriasis based on the current available data are given.
Introduction
Increased cardiovascular mortality is a common feature amongst many chronic inflammatory disorders. Interestingly, several lines of evidence suggest that effective systemic anti-inflammatory therapy may reduce this risk (1–6): A recent large randomized study showed that canakinumab, an antibody blocking interleukin (IL)-1β, is protective against myocardial infarction in a population at risk whereas its immunosuppressive effect also exposed patients to an increased risk of infections (7). Also, it is a well-known clinical observation that inflammatory diseases are accompanied by metabolic implications such as hyperglycemia, insulin resistance, and increased fatty acids (8). On the other hand, several metabolic diseases have inflammatory implications. Most important, there is a chronic and low-grade state of inflammation (“metaflammation”) in obesity and type 2 diabetes, both systemically and locally in visceral adipose tissue (“adipoflammation”) and other tissues (9–11). Antagonistic therapeutic approaches to proinflammatory molecules such as IL-1β, IL-1 receptor (IL-1R), tumor necrosis factor (TNF), and C-C chemokine ligand (CCL)2 were shown to improve glycemic control and metabolic parameters in patients with type 2 diabetes (12).
Psoriasis—one of the most common skin diseases with a prevalence of ~2%—is amongst the few non-communicable diseases the World Health Organization identified as a major global health problem (13). This is due to its chronic course, stigmatizing character due to the readily visible red scaly plaques, and its association with numerous other major diseases, such as hypertension, myocardial infarction, or diabetes (14). Over the last decade, targeted therapies have substantially improved our capacity to reduce signs and symptoms of psoriasis patients to an extent that—with the advent of biologics blocking IL-17A—“clear skin” became a feasible treatment goal (15). As strategies to block interleukin 1 have not been effective in treating psoriasis (16), the question arises whether IL-17A or other cytokines like TNFα, IL-12/23 (p40), or IL-23 might be a therapeutic target that would allow achieving therapeutic effects beyond the skin and joints, namely regarding the cardiometabolic situation of the patients.
In this review, we will briefly summarize what is known about the biological role of IL-17A, before reviewing currently available data on its role in the physiology and pathophysiology of the skin, the cardiovascular, and the metabolic system. Finally, we will share our point of view regarding the research agenda in the field and possible therapeutic implications.
Biology of IL-17A and IL-17F
The IL-17 cytokine family is formed by six members. As numerous other cytokines, IL-17 cytokines are part of the adaptive and innate immunity (17). IL-17A and IL-17F are significantly implicated in immune responses to infectious pathogens and in the pathogenesis of inflammatory autoimmune diseases like psoriasis. Furthermore, evidence indicates an additional proinflammatory activity of IL-17C and IL-17E within the pathogenesis of psoriasis (18–21).
IL-17A and IL-17F share the highest structural homology inside the IL-17 cytokine family and by this fulfill very similar biological functions (22). IL-17A/IL17F homo- and heterodimers bind to heterodimeric receptors (IL-17R) (23). The IL-17RA unit is part of the receptors for IL-17A, IL-17F, and IL-17E. Downstream of the IL-17R, signaling proteins are activated that result in the activation of transcription factors like nuclear factor kappa B (NFκB), activator protein 1 and CCAAT-enhancer-binding protein. One protein that transmits the signals from the IL-17R at an early stage is the NFκB activator 1 (ACT1) (24). Of note, the gene encoding ACT1 TNF receptor-associated factor 3 interacting protein 2 (TRAF3IP2) is one of the genes with common variants that are highly associated with the susceptibility to psoriasis and psoriatic arthritis (24).
Several factors have been reported to induce IL-17A and IL-17F expression in lymphoid cells. This has been intensively studied in CD4+ T helper (Th) cells. The critical cytokines that promote the differentiation of IL-17-producing Th (Th17) cells from naive Th cells are typically combinations of TGF-β1 with STAT3-activating cytokines like IL-6 or IL-21 (25). Cytokines like IL-1β and IL-23 can potentiate IL-17A and IL-17F expression. Moreover, the combination of IL-6, IL-1β, and IL-23 can induce IL-17A and IL-17F production in Th cells (25). The latter cytokine, IL-23, is responsible for the pathogenicity of autoreactive Th17 cells and in addition promotes IL-17 production in different lymphoid cell types including CD3+CD4−CD8− populations as well as innate lymphoid cells (26, 27).
IL-17A in Dermatology
Organ-Specific Pathophysiology
IL-17A-producing T cells can be protective for the organism but depending on their target cells can also cause tissue damage. Such diverse effects of IL-17A become evident in skin immunity. Effective immune responses against cutaneous pathogens require IL-17A expression. Examples are skin infections with bacteria like Mycobacteria tuberculosis or with fungi like Candida albicans (28, 29). IL-17A contributes to pathogen defense mechanisms by promoting neutrophil recruitment or production of antimicrobial peptides. However, high levels of IL-17A released during autoinflammatory or autoimmune conditions can also result in pathological skin changes. For instance, IL-17A has been reported to be present in Hidradenitis suppurativa, a devastating neutrophil-rich inflammation with formation of nodules and abscesses (30). The best studied inflammatory skin disease with a predominant role of IL-17A is psoriasis.
As confirmed in many studies, psoriatic skin is characterized by high expression of IL-17A and IL-17F (31). More recently, other IL-17 family members, namely IL-17C and IL-17E, have been reported to be expressed in psoriatic skin (18). While IL-17A and IL-17F are mainly produced by immune cells (e.g., Th cells, γδ T cells), the cellular sources of IL-17E also include epithelial cells like keratinocytes. Other sources comprise neutrophils and mast cells (32).
IL-17A and IL-17F act on immune and non-immune cell types and strongly contribute to tissue inflammation. For instance, they are involved in neutrophil recruitment. In the skin of psoriatic lesions, neutrophil accumulation is followed by the formation of epidermal micro abscesses (33, 34). IL-17 together with other Th17 cytokines also upregulates the production of several chemokines that are implicated in psoriasis pathogenesis, including CCL20, C-X-C motif chemokine ligand (CXCL)8, or CXCL1 (33, 34). Likewise, the production of matrix metalloproteinases (MMP) like MMP9 and antimicrobial peptides like β-defensin 2, LL37, and S100 proteins are controlled by IL-17A and IL-17F.
Experimental and Clinical Evidence for a Role of IL-17A in Psoriatic Inflammation
First antibodies that target IL-17A or its receptor IL-17RA are approved for the treatment of psoriasis. Secukinumab and ixekizumab both neutralize IL-17A, while brodalumab binds to the IL-17RA. The clinical efficacy of these IL17-targeting antibodies in psoriasis is demonstrated in several randomized studies and registries, as a clinical improvement of at least 75% as measured by the PASI is observed in >80% of patients treated (35–37). Similar PASI responses are achieved in clinical trials testing antibodies neutralizing IL-23 (p19), which is a potent inducer of IL-17A production by immune cells (38–40). In the recent five years there has been growing clinical evidence that IL-17A inhibitors are effective and safe in treating moderate to severe plaque type psoriasis.
A fully human monoclonal antibody that selectively neutralizes IL-17A, a key cornerstone cytokine involved in the development of psoriasis, is an approved treatment for patients with moderate to severe plaque psoriasis. Almost 10 years ago, the first clinical trial data with secukinumab were published, and at an early stage it was obvious that the targeted treatment with IL-17A antagonists has a higher capability of improving psoriasis compared to established previous therapy options. The ultimate treatment goal of PASI 75 (75% decrease of symptoms compared to baseline) and improvement of quality of life as assessed by dermatology life quality index (DLQI) of <5 was easily achieved by almost all patients treated with secukinumab (35). Furthermore, the IL-17A inhibitor secukinumab 300 mg s.c. treatment in head-to-head double-blind randomized trials was superior to established biological treatments like etanercept (TNF inhibitor) and ustekinumab (anti-IL12/23), raising the bar, where more than 80% of the patients achieved a PASI 90 and DLQI of 0/1 (35, 41, 42).
Beside short-term data, secukinumab has shown long-lasting efficacy and safety. Recently, high sustained efficacy and safety of 300 mg secukinumab treatment through 5 years in moderate to severe psoriasis was published (43). Direct comparison with first-line conventional treatment in Germany with fumaric acid esters showed clinically meaningful and statistically highly significant differences in patients naïve to any previous systemic treatment (44). Early treatment with secukinumab even as a first-line systemic treatment became possible, not only according to label but also in daily practice. The recent German guideline recommends secukinumab in patients with moderate to severe psoriasis, who in the judgment of the dermatologist have less chance to respond to other first-line conventional systemic therapies (45); other agents blocking the IL-17 pathway had not been approved for use at the time of guideline publication. Secukinumab has shown efficacy and safety in the complete spectrum of psoriasis manifestations, including nails, scalp, palms and soles, joints and symptoms like itch, scaling, and skin redness (46, 47).
Ixekizumab is a humanized monoclonal antibody with a selective inhibition of IL-17A and approved for the treatment of moderate to severe psoriasis. After initial s.c. treatment with 160 mg, followed by 80 mg every other week in the first 12 weeks, ixekizumab showed similar high PASI 90 responses as other IL-17 inhibitors in direct comparison to etanercept and ustekinumab (48, 49). Also, long-term 4-year continuous treatment with ixekizumab has recently been presented (50). Ixekizumab has also received first line approval from the EMA, but due to its late approval date has not yet been included in the most recent guidelines.
Clinical studies with anti-IL-17 (ixekizumab and secukinumab) are ongoing in adolescents and children, and results are not yet presented.
Brodalumab, a monoclonal antibody with high binding affinity to IL-17 receptor A (IL-17RA), is a treatment modality approved by the FDA for second line and by the EMA for the first line treatment of moderate to severe psoriasis. Treatment with 210 mg every 2 weeks shows very rapid onset of response with high clearing capacity where ~40–50% of treated patients had a PASI 100 (51). Up to now, there is no approval for PsA.
Importantly, targeting IL-17A is also effective in psoriatic arthritis, demonstrating that the psoriatic inflammation at distant sites also depends on the action of IL-17A (52). Also, different domains of PsA showed response to secukinumab including peripheral arthritis, enthesitis, dactylitis, and axial involvement (53, 54). In line, ixekizumab also improved different domains of psoriasis and PsA (55).
All IL-17 inhibitors have a similar safety profile, which is comparable with other approved biologics. Unlike TNF-alpha inhibitors, an increased risk of tuberculosis occurrence or reactivation was not observed. Unique for this biologic group is an increased risk for muco-cutaneous candida infection, mostly mild or moderate and easily manageable (56). Patients with active chronic inflammatory bowel disease (IBD) should not receive IL-17 inhibitors because of potential aggravation of IBD. Nevertheless, latest analyses of 21 clinical trials showed incidence rates of IBD comparable to untreated patients with psoriasis or psoriatic arthritis (57). Newest real-life data are confirming safety and tolerability of the IL-17 inhibitors also in daily practice, mirroring the evidence from clinical trials (58).
Currently, the effects of IL-17A neutralization on psoriasis-associated comorbidities are being investigated. Psoriasis patients with metabolic syndrome seem to have higher serum levels of IL-17, leptin, and c-reactive protein (CRP), while adiponectin levels seem to be lower (59–61). One could assume that the systemic suppression of IL-17 levels may be of benefit for patients with comorbidities like metabolic syndrome and cardiovascular disease.
Potential Therapeutic Implications
A common feature of IL-17 inhibitors is their fast mode of onset. Therefore, they might be particularly well-suited to treat patients with wide-spread and/or active/non-stable disease. Looking at the two currently available antibodies directly targeting IL-17A—secukinumab and ixekizumab—these still seem to be sufficiently different from each other to validate an attempt to use one in case the other has failed. There is an increasing number of reports of patients who did respond to ixekizumab after having failed to respond to secukinumab (62).
The third molecule currently available with a related mechanism of action, brodalumab, targets the IL-17RA chain. This results in inhibition of effects mediated by IL-17A as well as additional members of the IL-17 family. This mode of action might have two clinically relevant consequences:
1. As other IL-17s also play a pro-inflammatory role in psoriasis (17, 18), the efficacy of brodalumab might be particularly high. Indeed, indirect comparisons point into this direction (63).
2. Blocking the IL-17 receptor might result in a feedback-loop with substantial or even higher amounts of the mediator present. Therefore, drug withdrawal might result in a fast relapse of the disease. This has indeed been observed (64). Consequently, brodalumab may need to be given continuously, or switched directly to another active treatment.
Cardiovascular Inflammation in Psoriasis
Experimental and Clinical Evidence
An association of increased cardiovascular disease with psoriasis has been known for a long time, and the incidence of thromboembolic events in patients with untreated psoriasis has long been known to be higher than in age-matched controls (65). In addition, severe psoriasis is associated with a 50% increased risk of cardiovascular mortality (66), and life expectancy in patients with severe psoriasis is reduced (females: −4.4 years, males: −3.5 years) (67). Complicating the identification of a cause-effect relationship, it is also known that psoriasis patients are more likely to have an increased body mass index (68), are more likely to have metabolic syndrome (69) and type 2 diabetes (70) as well as depression (71, 72). Thus, one cannot exclude that elevated cumulative cardiovascular risk factors contribute to their increased rate of cardiovascular disease, even though evidence from large studies supports the existence of an independent relationship between psoriasis and cardiovascular disease (73, 74).
Recent evidence indicated that IL-17A may represent one of the main links between cardiovascular disease manifestations and psoriatic inflammation. Mouse models for psoriasis demonstrate that psoriasis is a T cell-mediated disease in which—among other factors—IL-17A is released from skin-infiltrating T cells to induce pathology via neutrophil recruitment (75–77). A T cell-mediated production of IL-17A has also been shown for human psoriasis (78).To mimic this situation and to study downstream events, mice overexpressing IL-17A only in keratinocytes have been generated (79). On their back skin, these mice developed skin lesions with extensive scaling and epidermal barrier loss. Histologically, the skin showed typical features of human psoriasis with hyperkeratosis, acanthosis, and infiltration with T cells and neutrophils. CD4+ T cells infiltrating the skin carrying a Th17 signature were abundant. Interestingly, the life span of these K14-IL-17ind mice was significantly shorter, and premature death started to occur at around 4 weeks of life, with most of the mice having died after 6 months (80). A thorough investigation established that hypertension, resulting in an increased heart/body ratio with cardiomyocyte hypertrophy, and a pathologic endothelial function were typical features of this murine model compatible with an increased cardiovascular risk. Signs for other cardiovascular risk factors such as increased body weights, hyperlipidemia, and/or hyperglycemia were not found.
To establish a direct link between the psoriatic phenotype and cardiovascular death, treatment with anti-TNF-α or anti-IL-6 was performed (80). Interestingly, these anti-inflammatory treatments not only efficiently reduced skin inflammation, they also abrogated pathologic endothelial stiffness, suggesting that the systemic inflammation present in K14-IL-17Aind mice was directly responsible for cardiovascular death. These results also suggested that IL-6 and TNF-α act downstream of IL-17A to promote cardiovascular comorbidity (80). In line with this, in recent reviews, a strong link between vascular inflammation, neutrophil recruitment, and IL-17 in mouse models has been highlighted (81, 82).
The CARIMA (Evaluation of Cardiovascular Risk Markers in Psoriasis Patients Treated with Secukinumab) study was a 52-week, randomized, double-blind, placebo-controlled, exploratory trial in patients with moderate to severe plaque psoriasis (83). Patients with prior cardiovascular disease were excluded. The primary outcome was endothelial function measured by flow-mediated dilation (FMD). In line with prior observations (84, 85), baseline FMD in the 151 psoriasis patients included was significantly lower in psoriasis patients than healthy volunteers, suggesting that subclinical alterations of endothelial function are present even in the absence of measurable clinical manifestations. At week 12, baseline adjusted mean FMD was higher in patients receiving secukinumab vs. those receiving placebo, but this difference did not reach significance. At week 52, the FMD was significantly higher than at baseline in patients receiving secukinumab for 52 weeks (+2.1%). Other relevant CV markers were unchanged. Thus, CARIMA indicates that secukinumab might have a beneficial effect on CV risk by improving the endothelial function of patients with plaque psoriasis. In line, adalimumab induced improvement of FMD in 14 psoriasis patients (84). Other studies with small patient cohorts showed mixed or no effect on FMD after anti-TNF treatment (85, 86). All in all, the increase in FMD at week 52 in anti-IL-17A-treated patients in the CARIMA study may be clinically relevant, since a 1% increase in absolute FMD is associated with a 13% decrease in the relative CV risk (87).
Other studies assessed the carotid plaque burden in psoriasis patients during biologic treatment (88, 89). Interestingly, psoriasis patients receiving anti-TNF treatment exhibited decreased vascular inflammation and a gender-dependent effect on carotid plaque progression after 1 year (88). Further evidence of the effect of anti-IL-17A treatment on cardiovascular disease in psoriasis was provided by Elnabawi et al. (89). In this prospective observational study, 121 patients without prior systemic therapy received treatment with various biologicals for 1 year; 169 patients with topical and/or phototherapy served as controls. Before and after treatment, total coronary plaque burden and subcomponents in coronary computed tomography angiography were determined. Interestingly, biologic therapy was associated with a 5% reduction of non-calcified plaques, a reduction of the necrotic cores, and no effect on fibrous burden. Compared to anti-TNF or anti-IL-12/23 treatment, anti-IL-17A was most efficient in reducing non-calcified plaque burden (12% compared to 5 and 2%, respectively).
Potential Therapeutic Implications
The effect of biologic treatment on subclinical cardiovascular disease in psoriasis patients is unclear so far. Some studies have convincingly shown improvement after therapy and others showed no changes; the majority of studies have assessed the effect of anti-TNF treatment in small patient collectives. However, the most recent studies (83, 89) with highly sensitive parameters for cardiovascular surrogate parameters, and comparably large patient cohorts as well as control groups have indicated that anti-IL-17A treatment (and potentially other biologics) may represent a promising agent to alter not only skin disease, but also cardiovascular comorbidity. Larger, randomized trials addressing this effect are needed.
IL-17A in the Cardiovascular System
The improvement of cardiovascular disease might be a direct consequence of a specific or relatively unspecific anti-inflammatory therapy in general. A first prospective, randomized, placebo-controlled, phase-III study could demonstrate in high risk patients after myocardial infarction that specific neutralization of IL-1 by canakinumab (CANTOS trial) can significantly reduce cardiovascular mortality (7). In contrast, a relatively unspecific anti-inflammatory therapy by methotrexate (CIRT trial) failed to achieve significant effects on cardiovascular mortality (90). Based on these considerations, highly specific and molecular therapies targeted against single cytokines or their receptors such as the IL-17A system appear to be promising regarding cardiometabolic comorbidities in psoriasis.
The Role of Inflammatory Cytokines in the Pathogenesis of Atherosclerosis
Atherosclerosis and thromboembolic diseases are maladaptive chronic inflammatory diseases whose pathogenesis and progression are determined by an interaction between immuno-inflammatory mechanisms, hemodynamic disturbances, activation of platelets and humoral coagulation factors, and lipid accumulation. The resulting ischemic diseases (e.g., myocardial infarction, cardiomyopathies or stroke) represent a prognosis-relevant comorbidity frequently associated with most inflammatory conditions, including those, like psoriasis, in which IL-17A appears to play an important role (91–93).
Interestingly, the pathophysiology of atherosclerosis and psoriasis share common aspects (94, 95). These similarities include an inflammatory background associated with local formation of modified autoantigens that are targeted by both the innate and adaptive immune system and in which Th1 cells have an important initiator function. The extravasation and migration into the vessel wall of these inflammatory cells is initially stimulated by adhesion molecules whose production is controlled by the secretion of pro-inflammatory cytokines and chemokines in response to alterations in the laminarity of blood flow. Several cytokines such as IL-2, interferon gamma (IFN-γ), and, most importantly, TNF-α, have important modulatory functions. Of note, the same mediators are also known to be linked to endothelial dysfunction and vascular calcification in other inflammatory diseases such as autoimmune diseases, inflammatory bowel syndromes, and rheumatoid arthritis. Cytokines and mediators including TNF-α, IL-1β, IL-6, and IFN-γ, along with MMP, are also responsible, upon stimulation, of the destabilization of atherosclerotic plaques, leading to ischemic events (96).
Experimental and Clinical Evidence for Proatherogenic and Atheroprotective Effects of IL-17A
The concept that IL-17 might have a pro-atherogenic role is supported by several lines of evidence (Figures 1A–C). The initial activation of Th17 cells may result from a variety of mechanisms, including inflammatory stimuli triggered by lipid accumulation, external inflammatory stimuli, or hemodynamic conditions. Additionally, exogenous stimuli, like sodium salt load, might also stimulate the induction of Th17 cells as shown recently in murine and human assays (97). Importantly, the IL-17A receptor is ubiquitously expressed in the vessel wall; among the pleiotropic activities of this receptor the induction of the production of TNF-α, IL-1β, CCL2, and adhesion molecules like intercellular adhesion molecule 1 (ICAM-1) provide important links to the pathogenesis of atherosclerosis (98). In a hypercholesterolemic rat model, IL-17A inhibition was associated with a 50% reduction of the atheroma area and with a reduction in the maximum percent stenosis, and it lead to lower expression of the chemokine CCL5 and lower intraplaque levels of IL-6, TNF-α, adhesion molecules such as vascular cell adhesion molecule 1 (VCAM-1), and the prothrombic molecule TF; in addition, it was associated with features suggestive of reduced plaque vulnerability such as an increase in intravascular smooth muscle cells and in the collagen content of the fibrous cap, a reduced expression of metalloproteinases, and a reduced apoptosis in the lesion. Further, cytokine/chemokine expression was reduced, suggesting a modulatory role of IL-17A in vascular inflammation (99). These data were confirmed in similar murine models in which exogenous application of recombinant IL-17A stimulated pathological changes associated with increased plaque instability, and, vice versa, IL-17A inhibition produced a regression of atherosclerosis (100–103).
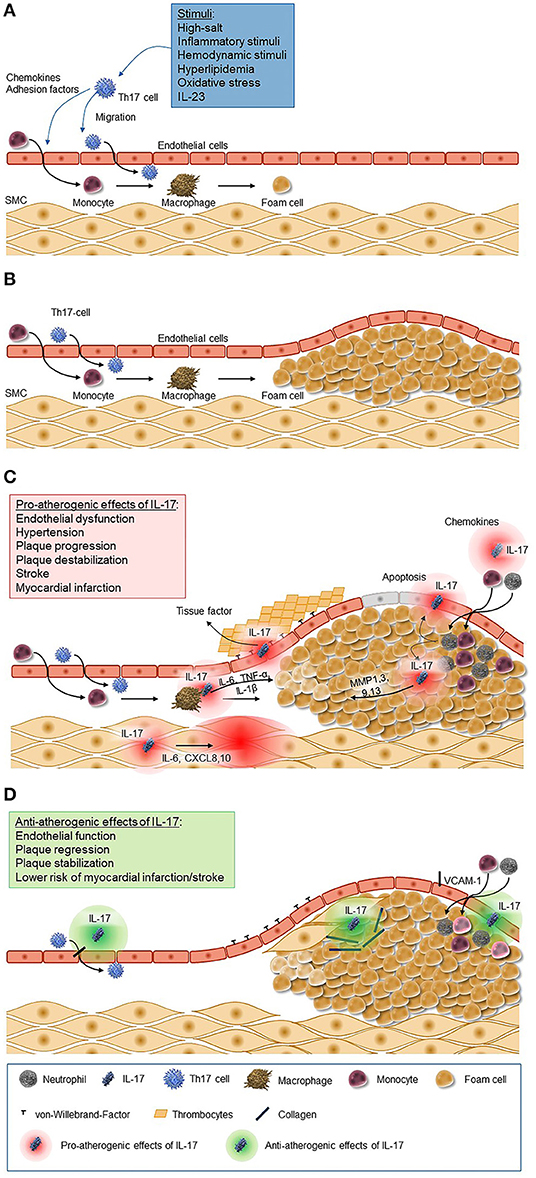
Figure 1. Pro-atherogenic and anti-atherogenic effects of IL-17. (A) Chemokines and adhesion factors are secreted by Th17 cells and cells of the innate immune system in response to a variety of stimuli (e.g., inflammatory stimuli, hemodynamic stimuli, and hyperlipidemia). These chemokines trigger migration and differentiation of monocytes and Th17 cells through the epithelium, which leads to the activation of macrophages and macrophage-mediated foam cell formation. (B) Foam cells accumulate to plaques as a key process of atherosclerosis. (C) IL-17 induces the release of chemokines leading to the recruitment of neutrophils and monocytes to the atherosclerotic lesion and their transformation to macrophages and foam cells and accumulation in fatty streaks. Further it stimulates macrophages to produce inflammatory cytokines such as IL-6, TNF-α, and IL-1β, matrix metalloproteinase from fibroblasts, endothelial cells, and epithelial cells, and it induces apoptosis of vascular endothelial cells. The increased secretion of endothelium-mediated von-Willebrand-factor promotes platelets adhesion and aggregation. (D) IL-17, however, also has antiatherogenic effects as it stimulates the proliferation of smooth muscle cells and the production of collagen and inhibits the expression of adhesion molecules. CXCL, C-X-C motif chemokine ligand; IL, interleukin; MMP, matrix metallopeptidase; SMC, smooth muscle cells; VCAM, vascular cell adhesion molecule.
Several researchers have shown that IL-17A is also required for the development of ischemic and dilated cardiomyopathy (DCM), since Th17 cells have been observed to infiltrate the heart during experimental autoimmune myocarditis (104, 105). IL-17RA−/− mice were protected from inducing DCM, although they developed myocarditis comparable to WT controls (104). IL-17RA−/− mice had diminished infiltration of neutrophils, which also have been shown to play an important role in the recruitment of monocytes (106, 107). In this model, cardiac fibroblasts were stimulated by IL-17A to produce key cytokines and chemokines that are critical downstream effectors in the recruitment and differentiation of myeloid cells. The authors concluded that IL-17A, while less important for the early stages of myocardtis, is required for the progression to DCM (105). These results were similar to the findings of Liu et al. who showed that IL-17 contributes to cardiac fibrosis and suggested that myocardial fibrosis induced by IL-17 is dependent on the PKCß/ERK1/2/NF-κB signaling pathways (108). In addition, Yuan and colleagues found increased numbers of Th17 cells in the blood of patients with DCM and an elevated level of Th17 cytokines in their serum (109).
Other research demonstrated that IL-17A mediates inflammation and the inappropriate increased vascular superoxide () production in response to high-fat diet-induced atherosclerosis, one of the crucial determinants of endothelial dysfunction in the setting of vascular disease (102). Ex vivo, IL-17A stimulation of human plaque fragments induced a pro-inflammatory, pro-thrombotic, plaque destabilizing and cell-attracting phenotype (110); in vivo in humans, higher IL-17A plasma levels were found in patients with coronary artery and carotid disease (111), an observation which might however be interpreted both as causation or as activation of compensatory mechanisms. Importantly, in these studies, the expression of IL-17A was maximal in human carotid artery plaques derived from symptomatic patients with stroke or transient ischemic attack, a piece of evidence that fits well with the concept that IL-17A is associated with plaque instability (112). Even further, a study in coronary thrombus aspirates from patients who had suffered an acute myocardial infarction demonstrated that up to 10–30% of the occluding thrombus mass is represented by neutrophil extracellular traps; in this study, IL-17A and IL-17F appeared to be important constituents of fresh, but not chronic, thrombi, indicating a possible role also for IL-17-dependent inflammation in acute thrombosis (113).
From a functional standpoint, IL-17A was also shown to promote endothelial dysfunction and angiotensin II-induced hypertension in a recent publication: in this murine model, angiotensin II infusion increased IL-17 production from T cells, and IL-17 knockout mice did not develop sustained hypertension, endothelial dysfunction, or evidence of vascular oxidative stress after chronic infusion of this potent vasoconstrictor (114). In another mouse model of IL-17A and enhanced green fluorescent protein (EGFP) co-overexpression in keratinocytes simulating clinical psoriasis, evidence of vascular dysfunction (including increased cardiovascular mortality) and arterial hypertension, along with large aortic wall cellular infiltrates, was observed. IL-17 may also contribute to atherogenesis by inducing the maturation and differentiation of macrophages; the subsequent activation of these precursors of foam cells by oxidized low-density lipoprotein (oxLDL) is considered to be the first step in the formation of atherosclerotic plaques (115).
Despite this convincing evidence, however, some uncertainty remains, as other publications have shown significantly smaller atherosclerotic lesions in mice with increased IL-17 expression (an observation that was reversed with IL-17 inhibition) and an association between IL-17 expression and plaque stability in human carotid artery plaques (116). Although some of the differences observed across different studies can be at least partially explained by differences in the study design, the method used to inhibit IL-17A, the animal model, and the site of the atherosclerotic lesion, the existence of controversial results needs to be acknowledged. Finally, evidence that IL-17A might also have atheroprotective effects also exists (Figure 1D): low serum levels of IL-17A have been associated with a higher risk of cardiovascular recurrences in coronary artery disease patients, an observation which might suggest that IL-17A might exert some form of preconditioning-like protection, and a murine IL-17A knockout model conferred resistance to high-fat diet-induced weight gain (102). Similar results were also obtained by Simon et al. using data from 981 patients with acute myocardial infarction, demonstrating that low serum levels of IL-17 and high soluble VCAM-1 levels are associated with a higher risk of major cardiovascular events of death and recurrent myocardial infarction. They concluded that these results raise possible concern about the use of inhibitors of the IL-17 pathway in clinical settings associated with a high cardiovascular risk (117).
Finally, a large meta-analysis including 38 randomized controlled trials showed no significant difference in risk of major adverse cardiovascular events (myocardial infarction, cerebrovascular accident, or cardiovascular death) in patients with psoriasis (n = 18,024) treated with biologic therapies including anti-IL-12/23, TNF-α inhibitors, and anti-IL-17A agents (118).
Potential Therapeutic Implications of Anti-inflammatory Therapy on Cardiovascular Function
From a therapeutic perspective, preliminary data show that pharmacological inhibition of cytokines is associated with a significant improvement in endothelial function: for instance, a single subcutaneous injection of the TNF-α inhibitor etanercept is associated with an improvement in flow-mediated dilation in postmenopausal women (119), and a preliminary study reported a significant improvement in resting endothelial function after therapy of psoriasis with anti-TNF-α and anti-IL-12p40, suggesting an activation of resting endothelial function in response to anti-inflammatory therapy (Karbach, unpublished). Whether this also occurs with a specific inhibition of IL-17A was tested in the CARIMA study described above, which showed a significant improvement after 54, but not 12, weeks of therapy with secukinumab (83). In sum, psoriasis and vascular disease appear to (at least partially) share common pathogenetic mechanisms, and an anti-inflammatory therapy (with a focus on the IL-17A signaling cascade) appears to not only reduce the skin manifestations, but also to improve (cardio)vascular function.
IL-17A in the Metabolic System
Besides LDL cholesterol (lipocentric view), the main metabolic factors causing cardiovascular complications (discussed in chapter 5) comprise hyperglycemia (glucocentric view) and hyperinsulinemia (insulin resistance, either as overt type 2 diabetes or as pre-diabetes). These two conditions become strongly activated during inflammation (and also during infection) via the so called “energy appeal reaction.” The molecular basis of this energy appeal reaction has been evolutionarily conserved and is characterized by an immuno-metabolic crosstalk leading to insulin resistance. Since the adipose tissue as a central organ in insulin resistance as well as adipokines, chemokines and cytokines such as IL-17A are involved in these mechanisms, the following chapter will summarize the underlying molecular mechanisms.
Effect of Inflammatory Processes on Insulin Resistance
During 600 million years of evolution, Drosophila's fat body developed into distinct organs such as adipose tissue, liver, and cells of the hematopoietic and the immune system (120). Since all of these organs were initially combined in this fat body, there must have been a molecular crosstalk between the metabolism on one hand and the immune system on the other hand. This kind of interaction has been conserved and is still present in humans (121). The molecular cross-talk between both systems represents a central homeostatic mechanism that is regulated by common pro-inflammatory molecules with a dichotomous function such as TNF, IL-6, IL-1, and many others (10, 122, 123). As demonstrated and reviewed previously (121), there is an additional need of up to 500 kJ for activation of the monocytic cell pool during inflammation. Energy has to be re-allocated to the cells of the immune system (energy appeal reaction) and this re-distribution is mediated by induction of systemic and local insulin resistance of tissues such as liver, muscle, and fat (124). Taken together, physiological insulin resistance during infection or inflammation re-distributes glucose and fatty acids to cells of the immune system. This situation perfectly resembles type 2 diabetes. Reduced insulin sensitivity as well as reduced insulin secretion are culprits in type 2 diabetes. Levels of glucose and fatty acids are chronically elevated. Glucotoxicity and lipotoxicity are the main mediators of diabetic complications (125).
Most importantly, there is a high grade of dynamic plasticity (126) of visceral adipose tissue. In the obese state, a shift from anti-inflammatory M2 macrophages (responsible to IL-4, IL-13; secreting IL-10) to proinflammatory M1 macrophages (responsible to lipopolysaccharide, IFN-γ; secreting IL-6 and TNF) occurs. Generally, local CD4+ T cells are being replaced by CD8+ T cells, and increasing numbers of granulocytes and even eosinophils can be registered (127–130). Moreover, specific CD4+ T cell subsets such as IL-17-secreting Th17 cells and IL-22-producing Th22 are infiltrating the adipose tissue and represent local mediators of inflammation and insulin resistance (127–130). There is also a shift from anti-inflammatory adipokines such as adiponectin to proinflammatory adipokines such as leptin or resistin (12, 126). Taken together, adipoflammation mediates local and systemic insulin resistance as well as metabolic inflammation.
There are several common immuno-metabolic signaling pathways in adipocytes, macrophages, and pancreatic beta-cells such as the Toll-like receptor-(TLR-)2, TLR-4, NFκB pathway, and the inflammasome/caspase-1/IL-1β pathway (12). Macrophage-derived IL-6 for example upregulates lipolysis in adipocytes leading to an increased flux of fatty acids and glycerol into the liver (12). These mechanisms upregulate hepatic glucose production, a main finding in diabetes. It is important to remember that insulin receptor signaling does not work well when cytokines and lipids are around. Lipids and cytokines are known to activate certain types of kinases that cause an inhibitory phosphorylation of insulin receptor substrate-(IRS-)1 and IRS-2 (120). Phosphorylation takes place at the wrong sites leading to less effective insulin signaling. Taken together, the most important finding of metabolic research during the last decades is given by the fact that nutrients are able to act through pathogen-sensing pathways. Similarly, stress pathways such as hypoxia, reactive oxygen species (ROS), endoplasmic reticulum activation, lipids, fatty acids, and cytokines are integrated with insulin signaling via inhibitory mechanisms by c-Jun N-terminal kinase (JNK) activation (120). JNK represents the central kinase connecting stress and inflammation with insulin signaling. As a conclusion, inflammation, and stress regularly induce insulin resistance by common mechanisms.
Leptin and IL-17 represent a suitable molecular example for the reciprocal regulation between immune system and energy metabolism. Mice with a T cell-specific deficiency of hepatocyte progenitor kinase-like/germinal center kinase-like kinase (HGK, also termed MAP4K4) develop systemic inflammation together with insulin resistance, a condition that could be ameliorated by neutralization of IL-6 or IL-17 (130). In this model, it was proven that HGK downregulates IL-6 production in T cells via phosphorylation and subsequent degradation of TNF-receptor-associated factor-2 (TRAF2) (130). Moreover, HGK-deficient and IL-6-overproducing T cells undergo differentiation into IL-6/IL-17-co-expressing cells that accumulate in adipose tissue (130). Accumulation of these cells in adipose tissue enhances the release of leptin from adipocytes, which in turn cooperates with IL-6 to induce Th17 differentiation. Local and systemic insulin resistance are driven by this coregulation.
IL-17 Links Inflammation With Insulin Resistance and Adipocyte Dysfunction
From an epidemiological point of view (69, 131–133), obesity and psoriasis are connected via the HLA-Cw6 locus (134), and obesity is ~35-fold more common in psoriasis. Vice versa, in patients with severe psoriasis, obesity and metabolic syndrome are more common (1.79-fold and 2.26-fold, respectively). Prevalence of the metabolic syndrome increases with increasing severity of psoriasis (135). With respect to diabetes (133, 136, 137), higher rates of psoriasis (~1.76-fold), especially of severe psoriasis (~2.1-fold), have been reported, and elevated levels of TNF seem mainly to be responsible via JNK activation and subsequent IRS-1 phosphorylation (138).
Based on the considerations mentioned above, it seems reasonable that the proinflammatory cytokine IL-17 interferes with insulin signaling and insulin sensitivity (Figure 2) and contributes to adipoflammation and metaflammation in obesity and diabetes (139). Since IL-17 activates the IκB kinase (IKK)/NFκB pathway, inhibitory phosphorylation of IRS-1 directly by IKK and indirectly by JNK activation in response to other proinflammatory cytokines is able to attenuate insulin sensitivity (140) on a molecular basis. Moreover, classical proinflammatory pathways in adipocytes such as TNF- and LPS-induced IL-6 and CCL20 production are augmented by IL-17 (141).
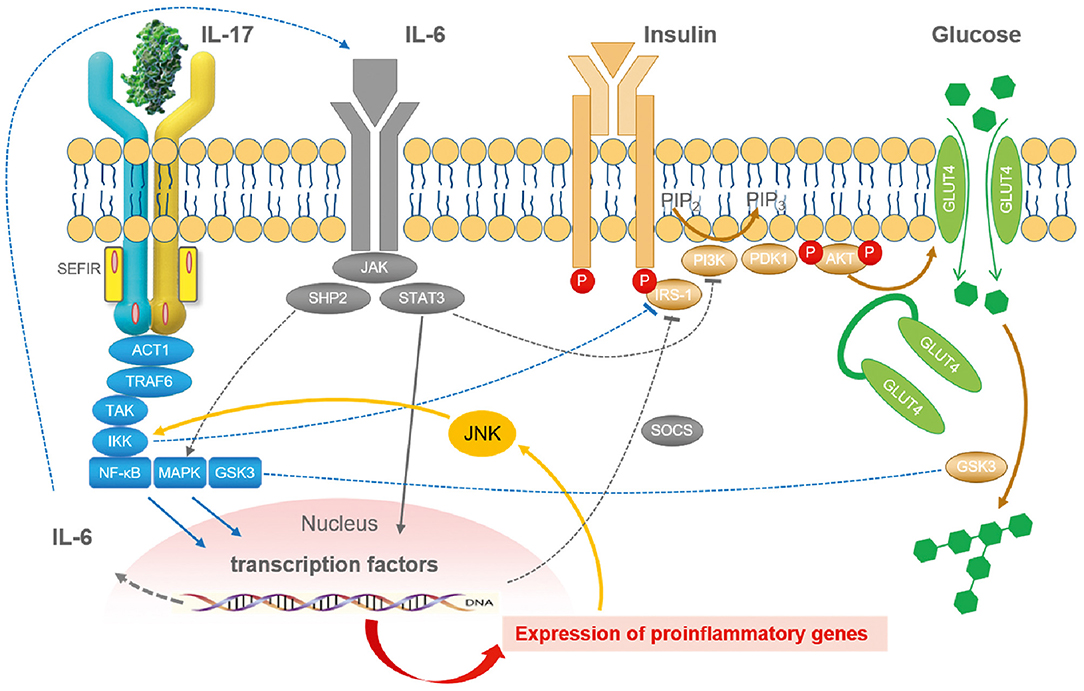
Figure 2. The integrated role of IL-17 in the cytokine- and inflammation-related regulatory network of insulin signaling. In general, inflammation reduces insulin sensitivity and insulin receptor signaling via IRS-1 by a myriad of different mediators. Cellular glucose disposal needs glucose transport via specific glucose transporters such as GLUT4 in adipose tissue and in striated muscle. Activation of the NFκB and the JNK pathway via different lipids and cytokines such as IL-6 and IL-17 (but also TNF, IL-1, and others) decrease insulin signaling. Lipids and cytokines are known to activate certain types of kinases that cause an inhibitory phosphorylation of IRS-1. Phosphorylation takes place at the wrong sites leading to less effective insulin signaling. Stress pathways such as hypoxia, ROS, ER activation, lipids, fatty acids, and cytokines are all integrated with insulin signaling via inhibitory mechanisms by JNK activation. Taken together, inflammation and stress always induce insulin resistance by common mechanisms. JNK represents the central kinase connecting stress and inflammation with insulin signaling. ACT1, nuclear factor-kappa-B activator; AKT, protein kinase AKT; ER, endoplasmic reticulum; Glut4, glucose transporter-4; GSK3, glycogen synthase kinase-3; IKK, I-kappa-B-kinase; IRS, insulin receptor substrate; JNK, Janus kinase; MAPK, mitogen-activated protein kinase; PDK1, pyruvate dehydrogenase kinase-1; NFκB, nuclear factor-kappa-B; PI3K, phosphoinositol-3-kinase; ROS, reactive oxygen species; SEFIR, similar expression to FGFR1; SHP2, tyrosine phosphatase SHP2; SOCS, suppressor of cytokine signaling; STAT3, signal transducer and activator of transcription-3; TRAF6, TNF receptor-associated factor-6 [Figure modified according to Straub (121) and Reich (137)].
Interestingly, IL-17A/IL-17A receptor signaling can promote adipogenic transdifferentiation of myoblast cells via activation of the main adipogenic transcription factor PPARγ (142). In contrast, in human mesenchymal stem cells and in adipocytes, IL-17 inhibits adipocyte differentiation (143, 144) and the expression of adipokines and adipogenic transcription factors. In differentiated adipocytes, IL-17 upregulates lipolysis and IL-6/IL-8 secretion (143), whereas it inhibits the expression of adipogenic transcription factors and the cellular uptake of glucose (127). Most importantly, IL-17-deficient mice are characterized by enhanced insulin sensitivity and increased glucose uptake (127). In human co-culture experiments, macrophage-derived IL-1β was shown to enhance IL-17 production (145). Since macrophages do express IL-17 receptors, there might exist a positive and paracrine feedback loop that enhances local visceral adipose tissue inflammation. Importantly, in KK-Ay diabetic mice, antibody-mediated neutralization of elevated IL-17 concentrations improved insulin resistance and increased glucose uptake by skeletal muscle, but not by adipose tissue (146). Additionally, the expression of adipogenic differentiation markers and adiponectin was upregulated.
Most interestingly, IL-17, adipose tissue inflammation, and psoriasis might be directly connected on a molecular basis. Adipocytes are able to synthesize and to secrete significant quantities of VEGF (vascular endothelial growth factor) that can be further enhanced in the context of hyperinsulinemia. Since VEGF induces keratinocyte inflammation by an interaction with IL-17, hyperinsulinemia in the context of visceral obesity and/or type 2 (pre)-diabetes might further increase severity or susceptibility of psoriasis (147, 148).
Potential Therapeutic Implications of Anti-IL-17 in Metabolic Diseases
From an evolutionary point of view, immune system, and metabolism are connected and cross-regulated by common proinflammatory molecules. Based on epidemiological data, the inflammatory state in severe psoriasis is positively associated with obesity and type 2 diabetes. IL-17, one of the main and causative proinflammatory cytokines in psoriasis, mechanistically links inflammation with insulin resistance and adipocyte dysfunction (127, 143, 144). In contrast, neutralization of IL-17 improves insulin sensitivity and glucose uptake in animal studies (130, 146). Considering these data, it is intriguing to speculate whether long-term neutralization of IL-17 in humans improves co-existent type 2 diabetes or even reduces the risk of diabetes manifestation. This hypothesis has to be investigated by randomized, placebo-controlled, clinical long-term studies in patients with poorly controlled type 2 diabetes and in pre-diabetic patients, respectively. With respect to available in vitro data and to data from experimental animal studies, neutralization of IL-17 should rather improve a coexistent type 2 diabetes than worsen it (127, 146). In addition, biologicals targeting the IL-17 or IL-12/23 pathway seem to be weight-neutral and do not worsen preexistent type 2 diabetes or increase the risk of diabetes manifestation (148–155).
Future Clinical Perspectives
Psoriasis skin manifestations, cardiovascular as well as metabolic disease in psoriasis appear to share pathogenic mechanisms evolving around IL-17 and its proinflammatory role (Figure 3). First evidence suggests that anti-IL-17A therapy not only improves skin manifestations of psoriasis, but also cardiovascular inflammation as well as (potentially) metabolic factors. If other anti-inflammatory drugs (e.g., biologics, immunosuppressants) have similar effects is currently not known.
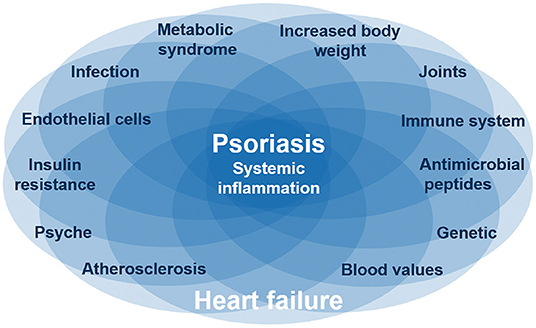
Figure 3. Common pathways for psoriasis, cardiovascular disease, and metabolic syndrome. Several diseases like psoriasis and heart failure are based on systematic inflammation, which is linked by IL-17, one of the main and causative proinflammatory cytokines in psoriasis. These inflammatory diseases are associated with certain interacting factors like obesity and depression. The complex interaction between inflammatory diseases and different factors is reflected by individual organ manifestations of the same inflammatory disease. Nevertheless, it is still unclear which factors are originating from the inflammation or are causing the inflammation.
What do we recommend to our patients with moderate to severe psoriasis based on these findings? First, thorough screening for subclinical cardiovascular diseases should be performed, including assessment of additional cardiovascular risk factors to help minimize the potential risk of developing cardiovascular events. Since severe psoriasis with a high inflammatory burden is associated with increased cardiovascular problems, effective control of skin inflammation is critical. In addition, signs for existing cardiovascular or metabolic disease need special attention and treatment (if required). Second, based on the findings discussed in the present review, the existence of systemic comorbidity (psoriasis arthritis, cardiovascular disease, metabolic disease) should trigger usage of anti-inflammatory systemic treatment. Among the various options, it appears that the best evidence so far is present for an effect of anti-IL-17. Controlled studies addressing this, however, are missing.
In summary, a picture emerges that underlines the complex interplay of several or even multiple cytokines or isoforms thereof in the context of psoriasis and its comorbidities. Clearly, synergies exist resulting in increased inflammation when two cytokines jointly affect a given target cell, e.g., with regard to IL-8 production of synoviocytes or skin fibroblasts stimulated with TNF-α plus IL-17A or -F (156). This opens up an opportunity for a more “complete” normalization of an inflamed target tissue through blockade of more than one cytokine, as again exemplified by the more complete normalization of the gene expression profile of fibroblasts stimulated with the supernatant of Th17 cells through simultaneous blockade of IL-17A and -F in comparison to blocking either one (156).
Further evaluation of the therapeutic potential of strategies aimed to blocking more than one IL-17 isoform would substantially benefit from a deepened understanding of the synergies amongst these isoforms as well as their quite different and sometimes even opposite functions in a tissue-specific manner (17).
Author Contributions
All authors contributed equally to conception and writing of the paper. All authors contributed to manuscript revision, read, and approved the submitted version.
Conflict of Interest
ES received honoraria as a speaker or advisor from Novartis, Miltenyi, and Celgene. She received a research grant to study therapeutic modulation of regulatory T cells and myeloid cells in psoriasis patients from Novartis. W-HB received honoraria as a speaker or advisor from Abbvie, Almirall, BMC, Celgene, Janssen, Leo, Lilly, Novartis, UCB. He received a research grant to study effects of to facitinib on monocytes and macrophages from Pfizer. KG has been a consultant, lecturer, or investigator for AbbVie, Almirall, Biogen, Boehringer Ingelheim, Brisol-Myers Squibb, Celgene, Eli Lilly, Janssen-Cilag, MSD Sharp & Dohme, Novartis, Pfizer, Roche, UCB Pharma. TG has received honoraria as a speaker or advisor from several pharmaceutical and medical device companies including Abbott Vascular, St Jude medicals, Neovasc, Bayer, Novartis. ZK has received an honorarium as an advisor from Novartis. DT has been a consultant and advisor and/or received speaking fees and/or grants and/or served as an investigator in clinical trials for the following companies: AbbVie, Almirall, Amgen, Bioskin, Boehringer Ingelheim, BMS, Celgene, Dignity, Eli Lilly, Galapagos, GSK, LEO Pharma, Janssen-Cilag, MSD, Morphosis, Novartis, Pfizer, Regeneron, Roche, Sandoz, Sanofi, and UCB. AS has received an honorarium as an advisor from Novartis.
Acknowledgments
The authors thank Wolfgang Exel Ph.D. of Novartis Pharma GmbH, Germany for providing medical writing/editorial support, which was funded by Novartis Pharma GmbH, Germany, in accordance with Good Publication Practice guidelines.
References
1. Prodanovich S, Ma F, Taylor JR, Pezon C, Fasihi T, Kirsner RS. Methotrexate reduces incidence of vascular diseases in veterans with psoriasis or rheumatoid arthritis. J Am Acad Dermatol. (2005) 52:262–7. doi: 10.1016/j.jaad.2004.06.017
2. Ludwig RJ, Herzog C, Rostock A, Ochsendorf FR, Zollner TM, Thaci D, et al. Psoriasis: a possible risk factor for development of coronary artery calcification. Br J Dermatol. (2007) 156:271–6. doi: 10.1111/j.1365-2133.2006.07562.x
3. Kristensen SL, Ahlehoff O, Lindhardsen J, Erichsen R, Lamberts M, Khalid U, et al. Inflammatory bowel disease is associated with an increased risk of hospitalization for heart failure: a Danish Nationwide Cohort study. Circ Heart Fail. (2014) 7:717–22. doi: 10.1161/CIRCHEARTFAILURE.114.001152
4. Listing J, Kekow J, Manger B, Burmester GR, Pattloch D, Zink A, et al. Mortality in rheumatoid arthritis: the impact of disease activity, treatment with glucocorticoids, TNFalpha inhibitors and rituximab. Ann Rheum Dis. (2015) 74:415–21. doi: 10.1136/annrheumdis-2013-204021
5. Egeberg A, Skov L, Joshi AA, Mallbris L, Gislason GH, Wu JJ, et al. The relationship between duration of psoriasis, vascular inflammation, and cardiovascular events. J Am Acad Dermatol. (2017) 77:650–6.e653. doi: 10.1016/j.jaad.2017.06.028
6. Boehncke W-H. Systemic inflammation and cardiovascular comorbidity in psoriasis patients: causes and consequences. Front Immunol. (2018) 9:579. doi: 10.3389/fimmu.2018.00579
7. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
8. Boehncke S, Thaci D, Beschmann H, Ludwig RJ, Ackermann H, Badenhoop K, et al. Psoriasis patients show signs of insulin resistance. Br J Dermatol. (2007) 157:1249–51. doi: 10.1111/j.1365-2133.2007.08190.x
9. Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci USA. (1994) 91:4854–8. doi: 10.1073/pnas.91.11.4854
10. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance. J Clin Invest. (1995) 95:2409–15. doi: 10.1172/JCI117936
11. Schäffler A, Scholmerich J. Innate immunity and adipose tissue biology. Trends Immunol. (2010) 31:228–35. doi: 10.1016/j.it.2010.03.001
12. Donath MY. Targeting inflammation in the treatment of type 2 diabetes: time to start. Nat Rev Drug Discov. (2014) 13:465–76. doi: 10.1038/nrd4275
13. World Health Organization. Global Report on Psoriasis. (2016). Available online at: https://ifpa-pso.com/our-actions/advocacy/who-global-report/ (accessed December 27, 2019).
15. Puig L. PASI90 response: the new standard in therapeutic efficacy for psoriasis. J Eur Acad Dermatol Venereol. (2015) 29:645–8. doi: 10.1111/jdv.12817
16. Jung N, Hellmann M, Hoheisel R, Lehmann C, Haase I, Perniok A, et al. An open-label pilot study of the efficacy and safety of anakinra in patients with psoriatic arthritis refractory to or intolerant of methotrexate (MTX). Clin Rheumatol. (2010) 29:1169–73. doi: 10.1007/s10067-010-1504-5
17. Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol. (2018) 9:1682. doi: 10.3389/fimmu.2018.01682
18. Senra L, Stalder R, Alvarez Martinez D, Chizzolini C, Boehncke WH, Brembilla NC. Keratinocyte-derived IL-17E contributes to inflammation in psoriasis. J Invest Dermatol. (2016) 136:1970–80. doi: 10.1016/j.jid.2016.06.009
19. Vandeghinste N, Klattig J, Jagerschmidt C, Lavazais S, Marsais F, Haas JD, et al. Neutralization of IL-17C reduces skin inflammation in mouse models of psoriasis and atopic dermatitis. J Invest Dermatol. (2018) 138:1555–63. doi: 10.1016/j.jid.2018.01.036
20. Garzorz-Stark N, Eyerich K. Psoriasis pathogenesis: keratinocytes are back in the spotlight. J Invest Dermatol. (2019) 139:995–6. doi: 10.1016/j.jid.2019.01.026
21. Senra L, Mylonas A, Kavanagh RD, Fallon PG, Conrad C, Borowczyk-Michalowska J, et al. IL-17E (IL-25) enhances innate immune responses during skin inflammation. J Invest Dermatol. (2019) 139:1732–42.e17. doi: 10.1016/j.jid.2019.01.021
22. Zhang X, Angkasekwinai P, Dong C, Tang H. Structure and function of interleukin-17 family cytokines. Protein Cell. (2011) 2:26–40. doi: 10.1007/s13238-011-1006-5
23. Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. (2009) 9:556–67. doi: 10.1038/nri2586
24. Huffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E, et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet. (2010) 42:996–9. doi: 10.1038/ng.688
25. Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature. (2010) 467:967–71. doi: 10.1038/nature09447
26. Geremia A, Arancibia-Carcamo CV, Fleming MP, Rust N, Singh B, Mortensen NJ, et al. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J Exp Med. (2011) 208:1127–33. doi: 10.1084/jem.20101712
27. Sherlock JP, Joyce-Shaikh B, Turner SP, Chao CC, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-gammat+ CD3+CD4-CD8- entheseal resident T cells. Nat Med. (2012) 18:1069–76. doi: 10.1038/nm.2817
28. de Repentigny L, Goupil M, Jolicoeur P. Oropharyngeal candidiasis in HIV infection: analysis of impaired mucosal immune response to candida albicans in mice expressing the HIV-1 transgene. Pathogens. (2015) 4:406–21. doi: 10.3390/pathogens4020406
30. Moran B, Sweeney CM, Hughes R, Malara A, Kirthi S, Tobin AM, et al. Hidradenitis suppurativa is characterized by dysregulation of the Th17:Treg cell axis, which is corrected by anti-TNF therapy. J Invest Dermatol. (2017) 137:2389–95. doi: 10.1016/j.jid.2017.05.033
31. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol. (2007) 8:950–7. doi: 10.1038/ni1497
32. Brembilla NC, Stalder R, Senra L, Boehncke WH. IL-17A localizes in the exocytic compartment of mast cells in psoriatic skin. Br J Dermatol. (2017) 177:1458–60. doi: 10.1111/bjd.15358
33. Nograles KE, Zaba LC, Guttman-Yassky E, Fuentes-Duculan J, Suarez-Farinas M, Cardinale I, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol. (2008) 159:1092–102. doi: 10.1111/j.1365-2133.2008.08769.x
34. Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, et al. Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol. (2009) 129:2175–83. doi: 10.1038/jid.2009.65
35. Langley RG, Elewski BE, Lebwohl M, Reich K, Griffiths CE, Papp K, et al. Secukinumab in plaque psoriasis - results of two phase 3 trials. N Engl J Med. (2014) 371:326–38. doi: 10.1056/NEJMoa1314258
36. Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. (2015) 373:1318–28. doi: 10.1056/NEJMoa1503824
37. Gordon KB, Colombel JF, Hardin DS. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. (2016) 375:2102. doi: 10.1056/NEJMoa1512711
38. Blauvelt A, Papp KA, Griffiths CE, Randazzo B, Wasfi Y, Shen YK, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. (2017) 76:405–17. doi: 10.1016/j.jaad.2016.11.041
39. Papp KA, Blauvelt A, Bukhalo M, Gooderham M, Krueger JG, Lacour JP, et al. Risankizumab versus ustekinumab for moderate-to-severe plaque psoriasis. N Engl J Med. (2017) 376:1551–60. doi: 10.1056/NEJMoa1607017
40. Reich K, Papp KA, Blauvelt A, Tyring SK, Sinclair R, Thaci D, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. (2017) 390:276–88. doi: 10.1016/S0140-6736(17)31279-5
41. Thaçi D, Blauvelt A, Reich K, Tsai T-F, Vanaclocha F, Kingo K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. (2015) 73:400–9. doi: 10.1016/j.jaad.2015.05.013
42. Blauvelt A, Reich K, Tsai T-F, Tyring S, Vanaclocha F, Kingo K, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate-to-severe plaque psoriasis up to 1 year: results from the CLEAR study. J Am Acad Dermatol. (2017) 76:60–9 e69. doi: 10.1016/j.jaad.2016.08.008
43. Bissonnette R, Luger T, Thaci D, Toth D, Lacombe A, Xia S, et al. Secukinumab demonstrates high sustained efficacy and a favourable safety profile in patients with moderate-to-severe psoriasis through 5 years of treatment (SCULPTURE Extension Study). J Eur Acad Dermatol Venereol. (2018) 32:1507–14. doi: 10.1111/jdv.14878
44. Sticherling M, Mrowietz U, Augustin M, Thaçi D, Melzer N, Hentschke C, et al. Secukinumab is superior to fumaric acid esters in treating patients with moderate-to-severe plaque psoriasis who are naive to systemic treatments: results from the randomized controlled PRIME trial. Br J Dermatol. (2017) 177:1024–32. doi: 10.1111/bjd.15707
45. Nast A, Amelunxen L, Augustin M, Boehncke WH, Dressler C, Gaskins M, et al. S3 Guideline for the treatment of psoriasis vulgaris, update - short version part 1 - Systemic treatment. J Dtsch Dermatol Ges. (2018) 16:645–69. doi: 10.1111/ddg.13516
46. Gottlieb A, Sullivan J, van Doorn M, Kubanov A, You R, Parneix A, et al. Secukinumab shows significant efficacy in palmoplantar psoriasis: results from GESTURE, a randomized controlled trial. J Am Acad Dermatol. (2017) 76:70–80. doi: 10.1016/j.jaad.2016.07.058
47. Reich K, Sullivan J, Arenberger P, Mrowietz U, Jazayeri S, Augustin M, et al. Effect of secukinumab on the clinical activity and disease burden of nail psoriasis: 32-week results from the randomized placebo-controlled transfigure trial. Br J Dermatol. (2018) 181:954–66. doi: 10.1111/bjd.17351
48. Reich K, Pinter A, Lacour JP, Ferrandiz C, Micali G, French LE, et al. Comparison of ixekizumab with ustekinumab in moderate-to-severe psoriasis: 24-week results from IXORA-S, a phase III study. Br J Dermatol. (2017) 177:1014–23. doi: 10.1111/bjd.15666
49. Papp KA, Leonardi CL, Blauvelt A, Reich K, Korman NJ, Ohtsuki M, et al. Ixekizumab treatment for psoriasis: integrated efficacy analysis of three double-blinded, controlled studies (UNCOVER-1, UNCOVER-2, UNCOVER-3). Br J Dermatol. (2018) 178:674–81. doi: 10.1111/bjd.16050
50. Egeberg A, Bryld LE, Skov L. Drug survival of secukinumab and ixekizumab for moderate-to-severe plaque psoriasis. J Am Acad Dermatol. (2019) 30:150–1. doi: 10.1016/j.jaad.2019.05.082
51. Farahnik B, Beroukhim K, Abrouk M, Nakamura M, Zhu TH, Singh R, et al. Brodalumab for the treatment of psoriasis: a review of phase III trials. Dermatol Ther. (2016) 6:111–24. doi: 10.1007/s13555-016-0121-x
52. Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov. (2012) 11:763–76. doi: 10.1038/nrd3794
53. McInnes IB, Mease PJ, Kirkham B, Kavanaugh A, Ritchlin CT, Rahman P, et al. Secukinumab, a human anti-interleukin-17A monoclonal antibody, in patients with psoriatic arthritis (FUTURE 2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. (2015) 386:1137–46. doi: 10.1016/S0140-6736(15)61134-5
54. Mease PJ, McInnes IB, Kirkham B, Kavanaugh A, Rahman P, van der Heijde D, et al. Secukinumab inhibition of interleukin-17A in patients with psoriatic arthritis. N Engl J Med. (2015) 373:1329–39. doi: 10.1056/NEJMoa1412679
55. Nash P, Kirkham B, Okada M, Rahman P, Combe B, Burmester G-R, et al. Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet. (2017) 389:2317–27. doi: 10.1016/S0140-6736(17)31429-0
56. Deodhar A, Mease PJ, McInnes IB, Baraliakos X, Reich K, Blauvelt A, et al. Long-term safety of secukinumab in patients with moderate-to-severe plaque psoriasis, psoriatic arthritis, and ankylosing spondylitis: integrated pooled clinical trial and post-marketing surveillance data. Arthritis Res Ther. (2019) 21:111. doi: 10.1186/s13075-019-1882-2
57. Schreiber S, Colombel JF, Feagan BG, Reich K, Deodhar AA, McInnes IB, et al. Incidence rates of inflammatory bowel disease in patients with psoriasis, psoriatic arthritis and ankylosing spondylitis treated with secukinumab: a retrospective analysis of pooled data from 21 clinical trials. Ann Rheum Dis. (2019) 78:473–9. doi: 10.1136/annrheumdis-2018-214273
58. Torres T, Balato A, Conrad C, Conti A, Dapavo P, Ferreira P, et al. Secukinumab drug survival in patients with psoriasis: a multicenter, real-world, retrospective study. J Am Acad Dermatol. (2019) 81:273–5. doi: 10.1016/j.jaad.2019.02.031
59. Kaur S, Zilmer K, Kairane C, Kals M, Zilmer M. Clear differences in adiponectin level and glutathione redox status revealed in obese and normal-weight patients with psoriasis. Br J Dermatol. (2008) 159:1364–7. doi: 10.1111/j.1365-2133.2008.08759.x
60. Gerdes S, Osadtschy S, Buhles N, Baurecht H, Mrowietz U. Cardiovascular biomarkers in patients with psoriasis. Exp Dermatol. (2014) 23:322–5. doi: 10.1111/exd.12381
61. Coimbra S, Oliveira H, Neuparth MJ, Proenca JB, Figueiredo A, Rocha-Pereira P, et al. Systemic inflammation and proinflammatory interleukin-17 signalling persist at the end of therapy in patients with metabolic syndrome and psoriasis, reducing the length of remission. Br J Dermatol. (2016) 174:414–6. doi: 10.1111/bjd.14013
62. Bokor-Billmann T, Schakel K. No need to change the drug class: ixekizumab- following secukinumab-therapy in psoriasis. J Dermatolog Treat. (2019) 30:216–20. doi: 10.1080/09546634.2018.1506081
63. Loos AM, Liu S, Segel C, Ollendorf DA, Pearson SD, Linder JA. Comparative effectiveness of targeted immunomodulators for the treatment of moderate-to-severe plaque psoriasis: a systematic review and network meta-analysis. J Am Acad Dermatol. (2018) 79:135–44 e137. doi: 10.1016/j.jaad.2018.02.027
64. Masson Regnault M, Konstantinou MP, Khemis A, Poulin Y, Bourcier M, Amelot F, et al. Early relapse of psoriasis after stopping brodalumab: a retrospective cohort study in 77 patients. J Eur Acad Dermatol Venereol. (2017) 31:1491–6. doi: 10.1111/jdv.14387
65. McDonald CJ, Calabresi P. Thromboembolic disorders associated with psoriasis. JAMA Dermatology. (1973) 107:918. doi: 10.1001/archderm.1973.01620210078025
66. Mallbris L, Akre O, Granath F, Yin L, Lindelöf B, Ekbom A, et al. Increased risk for cardiovascular mortality in psoriasis inpatients but not in outpatients. Eur J Epidemiol. (2004) 19:225–30. doi: 10.1023/B:EJEP.0000020447.59150.f9
67. Gelfand JM, Troxel AB, Lewis JD, Kurd SK, Shin DB, Wang X, et al. The risk of mortality in patients with psoriasis: results from a population-based study. JAMA Dermatol. (2007) 143:1493–9. doi: 10.1001/archderm.143.12.1493
68. Naldi L, Chatenoud L, Linder D, Belloni Fortina A, Peserico A, Virgili AR, et al. Cigarette smoking, body mass index, and stressful life events as risk factors for psoriasis: results from an Italian case–control study. J Invest Dermatol. (2005) 125:61–7. doi: 10.1111/j.0022-202X.2005.23681.x
69. Gisondi P, Tessari G, Conti A, Piaserico S, Schianchi S, Peserico A, et al. Prevalence of metabolic syndrome in patients with psoriasis: a hospital-based case–control study. Br J Dermatol. (2007) 157:68–73. doi: 10.1111/j.1365-2133.2007.07986.x
70. Herron MD, Hinckley M, Hoffman MS, Papenfuss J, Hansen CB, Callis KP, et al. Impact of obesity and smoking on psoriasis presentation and management. JAMA Dermatol. (2005) 141:1527–34. doi: 10.1001/archderm.141.12.1527
71. Player MS, Peterson LE. Anxiety disorders, hypertension, and cardiovascular risk: a review. Int J Psychiatry Med. (2011) 41:365–77. doi: 10.2190/PM.41.4.f
72. Rieder E, Tausk F. Psoriasis, a model of dermatologic psychosomatic disease: psychiatric implications and treatments. Int J Dermatol. (2012) 51:12–26. doi: 10.1111/j.1365-4632.2011.05071.x
73. Mehta NN, Azfar RS, Shin DB, Neimann AL, Troxel AB, Gelfand JM. Patients with severe psoriasis are at increased risk of cardiovascular mortality: cohort study using the General Practice Research Database. Eur Heart J. (2010) 31:1000–6. doi: 10.1093/eurheartj/ehp567
74. Matusiewicz D, Koerber A, Schadendorf D, Wasem J, Neumann A. Childhood psoriasis — an analysis of German health insurance data. Pediatr Dermatol. (2014) 31:8–13. doi: 10.1111/pde.12205
75. Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, Nestle FO. Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med. (2004) 199:731–6. doi: 10.1084/jem.20031482
76. Weitz JE, Ritchlin CT. Mechanistic insights from animal models of psoriasis and psoriatic arthritis. Curr Rheumatol Rep. (2013) 15:377. doi: 10.1007/s11926-013-0377-4
77. Lowes MA, Suarez-Farinas M, Krueger JG. Immunology of psoriasis. Annu Rev Immunol. (2014) 32:227–55. doi: 10.1146/annurev-immunol-032713-120225
78. Hawkes JE, Chan TC, Krueger JG. Psoriasis pathogenesis and the development of novel targeted immune therapies. J Allergy Clin Immunol. (2017) 140:645–53. doi: 10.1016/j.jaci.2017.07.004
79. Croxford AL, Karbach S, Kurschus FC, Wortge S, Nikolaev A, Yogev N, et al. IL-6 regulates neutrophil microabscess formation in IL-17A-driven psoriasiform lesions. J Invest Dermatol. (2014) 134:728–35. doi: 10.1038/jid.2013.404
80. Karbach S, Croxford AL, Oelze M, Schuler R, Minwegen D, Wegner J, et al. Interleukin 17 drives vascular inflammation, endothelial dysfunction, and arterial hypertension in psoriasis-like skin disease. Arterioscler Thromb Vasc Biol. (2014) 34:2658–68. doi: 10.1161/ATVBAHA.114.304108
81. Kurschus FC, Moos S. IL-17 for therapy. J Dermatol Sci. (2017) 87:221–7. doi: 10.1016/j.jdermsci.2017.06.010
82. Sanda GE, Belur AD, Teague HL, Mehta NN. Emerging associations between neutrophils, atherosclerosis, and psoriasis. Curr Atheroscler Rep. (2017) 19:53. doi: 10.1007/s11883-017-0692-8
83. von Stebut E, Reich K, Thaci D, Koenig W, Pinter A, Korber A, et al. Impact of secukinumab on endothelial dysfunction and other cardiovascular disease parameters in psoriasis patients over 52 weeks. J Invest Dermatol. (2019) 139:1054–62. doi: 10.1016/j.jid.2018.10.042
84. Avgerinou G, Tousoulis D, Siasos G, Oikonomou E, Maniatis K, Papageorgiou N, et al. Anti-tumor necrosis factor alpha treatment with adalimumab improves significantly endothelial function and decreases inflammatory process in patients with chronic psoriasis. Int J Cardiol. (2011) 151:382–3. doi: 10.1016/j.ijcard.2011.06.112
85. Ramonda R, Puato M, Punzi L, Rattazzi M, Zanon M, Balbi G, et al. Atherosclerosis progression in psoriatic arthritis patients despite the treatment with tumor necrosis factor-alpha blockers: a two-year prospective observational study. Joint Bone Spine. (2014) 81:421–5. doi: 10.1016/j.jbspin.2014.02.005
86. Mazzoccoli G, Notarsanto I, de Pinto GD, Dagostino MP, De Cata A, D'Alessandro G, et al. Anti-tumor necrosis factor-alpha therapy and changes of flow-mediated vasodilatation in psoriatic and rheumatoid arthritis patients. Intern Emerg Med. (2010) 5:495–500. doi: 10.1007/s11739-010-0458-6
87. Inaba Y, Chen JA, Bergmann SR. Prediction of future cardiovascular outcomes by flow-mediated vasodilatation of brachial artery: a meta-analysis. Int J Cardiovasc Imaging. (2010) 26:631–40. doi: 10.1007/s10554-010-9616-1
88. Eder L, Joshi AA, Dey AK, Cook R, Siegel EL, Gladman DD, et al. Association of tumor necrosis factor inhibitor treatment with reduced indices of subclinical atherosclerosis in patients with psoriatic disease. Arthritis Rheumatol. (2018) 70:408–16. doi: 10.1002/art.40366
89. Elnabawi YA, Dey AK, Goyal A, Groenendyk JW, Chung JH, Belur AD, et al. Coronary artery plaque characteristics and treatment with biologic therapy in severe psoriasis: results from a prospective observational study. Cardiovasc Res. (2019) 115:721–8. doi: 10.1093/cvr/cvz009
90. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. (2019) 380:752–62. doi: 10.1056/NEJMoa1809798
91. Akhavanpoor M, Akhavanpoor H, Gleissner CA, Wangler S, Doesch AO, Katus HA, et al. The two faces of interleukin-17A in atherosclerosis. Curr Drug Targets. (2017) 18:863–73. doi: 10.2174/1389450117666161229142155
92. Nordlohne J, von Vietinghoff S. Interleukin 17A in atherosclerosis-regulation and pathophysiologic effector function. Cytokine. (2017) 122:154089. doi: 10.1016/j.cyto.2017.06.016
93. Robert M, Miossec P. Effects of Interleukin 17 on the cardiovascular system. Autoimmun Rev. (2017) 16:984–91. doi: 10.1016/j.autrev.2017.07.009
94. Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. (2005) 352:1685–95. doi: 10.1056/NEJMra043430
96. Rai V, Agrawal DK. The role of damage- and pathogen-associated molecular patterns in inflammation-mediated vulnerability of atherosclerotic plaques. Can J Physiol Pharmacol. (2017) 95:1245–53. doi: 10.1139/cjpp-2016-0664
97. Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. (2013) 496:518–22. doi: 10.1038/nature11868
98. Jovanovic DV, Di Battista JA, Martel-Pelletier J, Jolicoeur FC, He Y, Zhang M, et al. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-beta and TNF-alpha, by human macrophages. J Immunol. (1998) 160:3513–21.
99. Erbel C, Chen L, Bea F, Wangler S, Celik S, Lasitschka F, et al. Inhibition of IL-17A attenuates atherosclerotic lesion development in apoE-deficient mice. J Immunol. (2009) 183:8167–75. doi: 10.4049/jimmunol.0901126
100. Gao Q, Jiang Y, Ma T, Zhu F, Gao F, Zhang P, et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. J Immunol. (2010) 185:5820–7. doi: 10.4049/jimmunol.1000116
101. Smith E, Prasad KM, Butcher M, Dobrian A, Kolls JK, Ley K, et al. Blockade of interleukin-17A results in reduced atherosclerosis in apolipoprotein E-deficient mice. Circulation. (2010) 121:1746–55. doi: 10.1161/CIRCULATIONAHA.109.924886
102. Madhur MS, Funt SA, Li L, Vinh A, Chen W, Lob HE, et al. Role of interleukin 17 in inflammation, atherosclerosis, and vascular function in apolipoprotein e-deficient mice. Arterioscler Thromb Vasc Biol. (2011) 31:1565–72. doi: 10.1161/ATVBAHA.111.227629
103. Danzaki K, Matsui Y, Ikesue M, Ohta D, Ito K, Kanayama M, et al. Interleukin-17A deficiency accelerates unstable atherosclerotic plaque formation in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (2012) 32:273–80. doi: 10.1161/ATVBAHA.111.229997
104. Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. (2010) 106:1646–55. doi: 10.1161/CIRCRESAHA.109.213157
105. Wu L, Ong S, Talor MV, Barin JG, Baldeviano GC, Kass DA, et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med. (2014) 211:1449–64. doi: 10.1084/jem.20132126
106. Valaperti A, Marty RR, Kania G, Germano D, Mauermann N, Dirnhofer S, et al. CD11b+ monocytes abrogate Th17 CD4+ T cell-mediated experimental autoimmune myocarditis. J Immunol. (2008) 180:2686–95. doi: 10.4049/jimmunol.180.4.2686
107. Wantha S, Alard JE, Megens RT, van der Does AM, Doring Y, Drechsler M, et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ Res. (2013) 112:792–801. doi: 10.1161/CIRCRESAHA.112.300666
108. Liu Y, Zhu H, Su Z, Sun C, Yin J, Yuan H, et al. IL-17 contributes to cardiac fibrosis following experimental autoimmune myocarditis by a PKCbeta/Erk1/2/NF-kappaB-dependent signaling pathway. Int Immunol. (2012) 24:605–12. doi: 10.1093/intimm/dxs056
109. Yuan J, Cao AL, Yu M, Lin QW, Yu X, Zhang JH, et al. Th17 cells facilitate the humoral immune response in patients with acute viral myocarditis. J Clin Immunol. (2010) 30:226–34. doi: 10.1007/s10875-009-9355-z
110. Erbel C, Akhavanpoor M, Okuyucu D, Wangler S, Dietz A, Zhao L, et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J Immunol. (2014) 193:4344–55. doi: 10.4049/jimmunol.1400181
111. Eid RE, Rao DA, Zhou J, Lo SF, Ranjbaran H, Gallo A, et al. Interleukin-17 and interferon-gamma are produced concomitantly by human coronary artery-infiltrating T cells and act synergistically on vascular smooth muscle cells. Circulation. (2009) 119:1424–32. doi: 10.1161/CIRCULATIONAHA.108.827618
112. de Boer OJ, van der Meer JJ, Teeling P, van der Loos CM, Idu MM, van Maldegem F, et al. Differential expression of interleukin-17 family cytokines in intact and complicated human atherosclerotic plaques. J Pathol. (2010) 220:499–508. doi: 10.1002/path.2667
113. de Boer OJ, Li X, Teeling P, Mackaay C, Ploegmakers HJ, van der Loos CM, et al. Neutrophils, neutrophil extracellular traps and interleukin-17 associate with the organisation of thrombi in acute myocardial infarction. Thromb Haemost. (2013) 109:290–7. doi: 10.1160/TH12-06-0425
114. Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. (2010) 55:500–7. doi: 10.1161/HYPERTENSIONAHA.109.145094
115. de la Paz Sanchez-Martinez M, Blanco-Favela F, Mora-Ruiz MD, Chavez-Rueda AK, Bernabe-Garcia M, Chavez-Sanchez L. IL-17-differentiated macrophages secrete pro-inflammatory cytokines in response to oxidized low-density lipoprotein. Lipids Health Dis. (2017) 16:196. doi: 10.1186/s12944-017-0588-1
116. Taleb S, Romain M, Ramkhelawon B, Uyttenhove C, Pasterkamp G, Herbin O, et al. Loss of SOCS3 expression in T cells reveals a regulatory role for interleukin-17 in atherosclerosis. J Exp Med. (2009) 206:2067–77. doi: 10.1084/jem.20090545
117. Simon T, Taleb S, Danchin N, Laurans L, Rousseau B, Cattan S, et al. Circulating levels of interleukin-17 and cardiovascular outcomes in patients with acute myocardial infarction. Eur Heart J. (2013) 34:570–7. doi: 10.1093/eurheartj/ehs263
118. Rungapiromnan W, Yiu ZZN, Warren RB, Griffiths CEM, Ashcroft DM. Impact of biologic therapies on risk of major adverse cardiovascular events in patients with psoriasis: systematic review and meta-analysis of randomized controlled trials. Br J Dermatol. (2017) 176:890–901. doi: 10.1111/bjd.14964
119. Moreau KL, Deane KD, Meditz AL, Kohrt WM. Tumor necrosis factor-alpha inhibition improves endothelial function and decreases arterial stiffness in estrogen-deficient postmenopausal women. Atherosclerosis. (2013) 230:390–6. doi: 10.1016/j.atherosclerosis.2013.07.057
120. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
121. Straub RH. Concepts of evolutionary medicine and energy regulation contribute to the etiology of systemic chronic inflammatory diseases. Brain Behav Immun. (2011) 25:1–5. doi: 10.1016/j.bbi.2010.08.002
122. Schäffler A, Buechler C. CTRP family: linking immunity to metabolism. Trends Endocrinol Metab. (2012) 23:194–204. doi: 10.1016/j.tem.2011.12.003
123. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol. (2013) 4:52. doi: 10.3389/fendo.2013.00052
124. Chawla A, Nguyen KD, Goh YP. Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol. (2011) 11:738–49. doi: 10.1038/nri3071
125. Kahn SE, Cooper ME, Del Prato S. Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet. (2014) 383:1068–83. doi: 10.1016/S0140-6736(13)62154-6
126. Ouchi N, Parker JL, Lugus JJ, Walsh K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. (2011) 11:85–97. doi: 10.1038/nri2921
127. Zuniga LA, Shen WJ, Joyce-Shaikh B, Pyatnova EA, Richards AG, Thom C, et al. IL-17 regulates adipogenesis, glucose homeostasis, and obesity. J Immunol. (2010) 185:6947–59. doi: 10.4049/jimmunol.1001269
128. Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. (2011) 186:1162–72. doi: 10.4049/jimmunol.1002615
129. Fabbrini E, Cella M, McCartney SA, Fuchs A, Abumrad NA, Pietka TA, et al. (2013). Association between specific adipose tissue CD4+ T-cell populations and insulin resistance in obese individuals. Gastroenterology 145, 366-374 e361–363. doi: 10.1053/j.gastro.2013.04.010
130. Chuang HC, Sheu WH, Lin YT, Tsai CY, Yang CY, Cheng YJ, et al. HGK/MAP4K4 deficiency induces TRAF2 stabilization and Th17 differentiation leading to insulin resistance. Nat Commun. (2014) 5:4602. doi: 10.1038/ncomms5602
131. Neimann AL, Shin DB, Wang X, Margolis DJ, Troxel AB, Gelfand JM. Prevalence of cardiovascular risk factors in patients with psoriasis. J Am Acad Dermatol. (2006) 55:829–35. doi: 10.1016/j.jaad.2006.08.040
132. Sommer DM, Jenisch S, Suchan M, Christophers E, Weichenthal M. Increased prevalence of the metabolic syndrome in patients with moderate to severe psoriasis. Arch Dermatol Res. (2006) 298:321–8. doi: 10.1007/s00403-006-0703-z
133. Armstrong AW, Harskamp CT, Armstrong EJ. Psoriasis and metabolic syndrome: a systematic review and meta-analysis of observational studies. J Am Acad Dermatol. (2013) 68:654–62. doi: 10.1016/j.jaad.2012.08.015
134. Jin Y, Zhang F, Yang S, Kong Y, Xiao F, Hou Y, et al. Combined effects of HLA-Cw6, body mass index and waist-hip ratio on psoriasis vulgaris in Chinese Han population. J Dermatol Sci. (2008) 52:123–9. doi: 10.1016/j.jdermsci.2008.04.016
135. Langan SM, Seminara NM, Shin DB, Troxel AB, Kimmel SE, Mehta NN, et al. Prevalence of metabolic syndrome in patients with psoriasis: a population-based study in the United Kingdom. J Invest Dermatol. (2012) 132(3 Pt 1):556–62. doi: 10.1038/jid.2011.365
136. Chen YJ, Wu CY, Shen JL, Chu SY, Chen CK, Chang YT, et al. Psoriasis independently associated with hyperleptinemia contributing to metabolic syndrome. Arch Dermatol. (2008) 144:1571–5. doi: 10.1001/archderm.144.12.1571
137. Coto-Segura P, Eiris-Salvado N, Gonzalez-Lara L, Queiro-Silva R, Martinez-Camblor P, Maldonado-Seral C, et al. Psoriasis, psoriatic arthritis and type 2 diabetes mellitus: a systematic review and meta-analysis. Br J Dermatol. (2013) 169:783–93. doi: 10.1111/bjd.12473
138. Reich K. The concept of psoriasis as a systemic inflammation: implications for disease management. J Eur Acad Dermatol Venereol. (2012) 26(Suppl 2):3–11. doi: 10.1111/j.1468-3083.2011.04410.x
139. Chehimi M, Vidal H, Eljaafari A. Pathogenic role of IL-17-producing immune cells in obesity, and related inflammatory diseases. J Clin Med. (2017) 6:E68. doi: 10.3390/jcm6070068
140. Gaffen SL, Jain R, Garg AV, Cua DJ. The IL-23-IL-17 immune axis: from mechanisms to therapeutic testing. Nat Rev Immunol. (2014) 14:585–600. doi: 10.1038/nri3707
141. Shinjo T, Iwashita M, Yamashita A, Sano T, Tsuruta M, Matsunaga H, et al. IL-17A synergistically enhances TNFalpha-induced IL-6 and CCL20 production in 3T3-L1 adipocytes. Biochem Biophys Res Commun. (2016) 477:241–6. doi: 10.1016/j.bbrc.2016.06.049
142. Lee SJ, Lee EJ, Kim SH, Choi I, Lee DM, Lee HJ, et al. IL-17A promotes transdifferentiation of mouse myoblast cells (C2C12) into adipocytes by increasing the expression of peroxisome proliferator-activated receptor gamma through CAAT/enhancer binding protein beta signaling. Biotechnol Lett. (2011) 33:229–35. doi: 10.1007/s10529-010-0440-4
143. Shin JH, Shin DW, Noh M. Interleukin-17A inhibits adipocyte differentiation in human mesenchymal stem cells and regulates pro-inflammatory responses in adipocytes. Biochem Pharmacol. (2009) 77:1835–44. doi: 10.1016/j.bcp.2009.03.008
144. Lee SH, Jhun J, Byun JK, Kim EK, Jung K, Lee JE, et al. IL-17 axis accelerates the inflammatory progression of obese in mice via TBK1 and IKBKE pathway. Immunol Lett. (2017) 184:67–75. doi: 10.1016/j.imlet.2017.02.004
145. Dalmas E, Venteclef N, Caer C, Poitou C, Cremer I, Aron-Wisnewsky J, et al. T cell-derived IL-22 amplifies IL-1beta-driven inflammation in human adipose tissue: relevance to obesity and type 2 diabetes. Diabetes. (2014) 63:1966–77. doi: 10.2337/db13-1511
146. Ohshima K, Mogi M, Jing F, Iwanami J, Tsukuda K, Min LJ, et al. Roles of interleukin 17 in angiotensin II type 1 receptor-mediated insulin resistance. Hypertension. (2012) 59:493–9. doi: 10.1161/HYPERTENSIONAHA.111.183178
147. Suzuki T, Hirakawa S, Shimauchi T, Ito T, Sakabe J, Detmar M, et al. VEGF-A promotes IL-17A-producing gammadelta T cell accumulation in mouse skin and serves as a chemotactic factor for plasmacytoid dendritic cells. J Dermatol Sci. (2014) 74:116–24. doi: 10.1016/j.jdermsci.2013.12.013
148. Krueger JG, Brunner PM. Interleukin-17 alters the biology of many cell types involved in the genesis of psoriasis, systemic inflammation, and associated comorbidities. Exp Dermatol. (2017) 27:115–123. doi: 10.1111/exd.13467
149. Gisondi P, Dalle Vedove C, Girolomoni G. Efficacy and safety of secukinumab in chronic plaque psoriasis and psoriatic arthritis therapy. Dermatol Ther. (2014) 4:1–9. doi: 10.1007/s13555-014-0042-5
150. Toussirot E, Aubin F, Dumoulin G. Relationships between adipose tissue and psoriasis, with or without arthritis. Front Immunol. (2014) 5:368. doi: 10.3389/fimmu.2014.00368
151. Girolomoni G, Altomare G, Ayala F, Berardesca E, Calzavara Pinton P, Chimenti S, et al. Differential management of mild-to-severe psoriasis with biologic drugs: an Italian Delphi consensus expert panel. J Dermatolog Treat. (2015) 26:128–33. doi: 10.3109/09546634.2014.907466
152. Canavan TN, Elmets CA, Cantrell WL, Evans JM, Elewski BE. Anti-il-17 medications used in the treatment of plaque psoriasis and psoriatic arthritis: a comprehensive review. Am J Clin Dermatol. (2016) 17:33–47. doi: 10.1007/s40257-015-0162-4
153. Yiu ZZ, Griffiths CE. Interleukin 17-A inhibition in the treatment of psoriasis. Expert Rev Clin Immunol. (2016) 12:1–4. doi: 10.1586/1744666X.2016.1112739
154. Bhat RM, Leelavathy B, Aradhya SS, Gopal MG, Pratap DV, Mubashir M, et al. Secukinumab efficacy and safety in indian patients with moderate-to-severe plaque psoriasis: sub-analysis from FIXTURE, a randomized, placebo-controlled, phase 3 study. Indian Dermatol Online J. (2017) 8:16–24. doi: 10.4103/2229-5178.198765
155. Mease PJ, van der Heijde D, Ritchlin CT, Okada M, Cuchacovich RS, Shuler CL, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis. (2017) 76:79–87. doi: 10.1136/annrheumdis-2016-209709
156. Glatt S, Baeten D, Baker T, Griffiths M, Ionescu L, Lawson ADG, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis. (2018) 77:523–32. doi: 10.1136/annrheumdis-2017-212127
Keywords: IL-17A, psoriasis, comorbidities, cardiovascular, diabetes
Citation: von Stebut E, Boehncke W-H, Ghoreschi K, Gori T, Kaya Z, Thaci D and Schäffler A (2020) IL-17A in Psoriasis and Beyond: Cardiovascular and Metabolic Implications. Front. Immunol. 10:3096. doi: 10.3389/fimmu.2019.03096
Received: 08 September 2019; Accepted: 18 December 2019;
Published: 15 January 2020.
Edited by:
Joanna Cichy, Jagiellonian University, PolandReviewed by:
Christoph Baerwald, Leipzig University, GermanyGudrun Debes, Thomas Jefferson University, United States
Copyright © 2020 von Stebut, Boehncke, Ghoreschi, Gori, Kaya, Thaci and Schäffler. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esther von Stebut, esther.von-stebut@uk-koeln.de
†These authors have contributed equally to this work