Corrigendum: Consensus Recommendations for the Diagnosis and Management of X-Linked Hypophosphatemia in Belgium
 Michaël R. Laurent1*
Michaël R. Laurent1* Jean De Schepper2,3Dominique Trouet4,5
Jean De Schepper2,3Dominique Trouet4,5 Nathalie Godefroid6Emese Boros7Claudine Heinrichs7Bert Bravenboer8Brigitte Velkeniers8Johan Lammens9Pol Harvengt10Etienne Cavalier11Jean-François Kaux12Jacques Lombet13Kathleen De Waele3Charlotte Verroken14Koenraad van Hoeck4,5Geert R. Mortier15
Nathalie Godefroid6Emese Boros7Claudine Heinrichs7Bert Bravenboer8Brigitte Velkeniers8Johan Lammens9Pol Harvengt10Etienne Cavalier11Jean-François Kaux12Jacques Lombet13Kathleen De Waele3Charlotte Verroken14Koenraad van Hoeck4,5Geert R. Mortier15 Elena Levtchenko16†
Elena Levtchenko16† Johan Vande Walle17†
Johan Vande Walle17†- 1Centre for Metabolic Bone Diseases, University Hospitals Leuven, Leuven, Belgium
- 2Division of Pediatric Endocrinology, KidZ Health Castle, University Hospital Brussels, Vrije Universiteit Brussel (VUB), Brussels, Belgium
- 3Department of Pediatric Endocrinology, University Hospital Ghent, Ghent, Belgium
- 4Department of Pediatric Nephrology, Antwerp University Hospital, Antwerp, Belgium
- 5Laboratory of Experimental Medicine and Pediatrics, University of Antwerp, Antwerp, Belgium
- 6Pediatric Nephrology, Cliniques Universitaires St. Luc (UCL), Brussels, Belgium
- 7Paediatric Endocrinology Unit, Hôpital Universitaire des Enfants Reine Fabiola, Université Libre de Bruxelles, Brussels, Belgium
- 8Department of Endocrinology, University Hospital Brussels, Vrije Universiteit Brussel (VUB), Brussels, Belgium
- 9Department of Orthopaedic Surgery and Department of Development and Regeneration, Prometheus LRD Division of Skeletal Tissue Engineering, KU Leuven - University Hospitals Leuven, Leuven, Belgium
- 10XLH Belgium, Belgian X-Linked Hypophosphatemic Rickets (XLH) Patient Association, Waterloo, Belgium
- 11Department of Clinical Chemistry, University Hospital Center of Liège, University of Liège, Liège, Belgium
- 12Physical Medicine, Rehabilitation and Sports Traumatology, University and University Hospital of Liège, Liège, Belgium
- 13Division of Nephrology, Department of Pediatrics, University Hospital Center of Liège, Liège, Belgium
- 14Unit for Osteoporosis and Metabolic Bone Diseases, Department of Endocrinology and Metabolism, Ghent University Hospital, Ghent, Belgium
- 15Department of Medical Genetics, Antwerp University Hospital and University of Antwerp, Antwerp, Belgium
- 16Department of Pediatrics/Pediatric Nephrology, University Hospitals Leuven, Leuven, Belgium
- 17Department of Pediatric Nephrology, University Hospital Ghent, Ghent, Belgium
X-linked hypophosphatemia (XLH) is the most common genetic form of hypophosphatemic rickets and osteomalacia. In this disease, mutations in the PHEX gene lead to elevated levels of the hormone fibroblast growth factor 23 (FGF23), resulting in renal phosphate wasting and impaired skeletal and dental mineralization. Recently, international guidelines for the diagnosis and treatment of this condition have been published. However, more specific recommendations are needed to provide guidance at the national level, considering resource availability and health economic aspects. A national multidisciplinary group of Belgian experts convened to discuss translation of international best available evidence into locally feasible consensus recommendations. Patients with XLH may present to a wide array of primary, secondary and tertiary care physicians, among whom awareness of the disease should be raised. XLH has a very broad differential-diagnosis for which clinical features, biochemical and genetic testing in centers of expertise are recommended. Optimal care requires a multidisciplinary approach, guided by an expert in metabolic bone diseases and involving (according to the individual patient’s needs) pediatric and adult medical specialties and paramedical caregivers, including but not limited to general practitioners, dentists, radiologists and orthopedic surgeons. In children with severe or refractory symptoms, FGF23 inhibition using burosumab may provide superior outcomes compared to conventional medical therapy with phosphate supplements and active vitamin D analogues. Burosumab has also demonstrated promising results in adults on certain clinical outcomes such as pseudofractures. In summary, this work outlines recommendations for clinicians and policymakers, with a vision for improving the diagnostic and therapeutic landscape for XLH patients in Belgium.
Introduction
X-linked hypophosphatemia (XLH) is the most common genetic form of hypophosphatemic rickets and osteomalacia. Its incidence has been estimated at 3.9-5 cases per 100.000 live births, with no evidence of ethnic variation (1, 2). Belgium has an annual birth rate of ± 117.800 (year 2018), ± 1.93 million growing children (< 15 years) and 11.4 million total population (2019 census) (3). Extrapolation of the ~1:20.000 incidence and prevalence would imply that this rare disease has an incidence of less than 6 cases in newborns annually and a prevalence of 97 and 475 cases in the pediatric and adolescent/adult population, respectively. Other sources have reported prevalences lower than 1:20.000 e.g. 1.4 per 100.000 in the United Kingdom (4) to 1.7 per 100.000 in Norway (5). Possible reasons include gaps in diagnosis and referral of XLH patients from primary or secondary care to centers of expertise. This is certainly the case also in Belgium, where only recently efforts have been initiated to improve the care for patients suffering from rare/orphan diseases (6).
The pathophysiology of XLH has been reviewed extensively elsewhere (7, 8). In brief, mono-allelic mutations or chromosomal derangements affecting the Phosphate Regulating Endopeptidase Homolog, X-Linked (PHEX) gene on the X chromosome lead to elevated levels of the hormone fibroblast growth factor 23 (FGF23), resulting in renal phosphate wasting, impaired 1α-hydroxylation of 25-hydroxyvitamin D [25(OH)D] to the active hormone calcitriol (1,25-dihydroxyvitamin D [1,25(OH)2D]) and consequently, chronic hypophosphatemia, impaired skeletal mineralization and rickets (9). In children, the corresponding clinical features may include delayed growth and short stature, craniosynostosis and raised intracranial pressure, deformities of weight-bearing limbs, muscle weakness, gait abnormalities (10), tooth abscesses and excessive dental caries (9, 11, 12).
Following growth plate closure, a part of adolescent and young adult patients continue to experience debilitating symptoms while others may experience a “honeymoon” phase (similar to other metabolic bone diseases) with fewer musculoskeletal problems (except dental manifestations). During this phase, conventional therapy with phosphate supplements and active vitamin D analogues is often stopped, because subjective and skeletal benefits are thought to be lacking (13). During adolescence, the psychological burden increases (14), which may contribute to poor adherence and lack of follow-up. The historic perception of therapeutic futility in adults has probably contributed to the dearth of transitional care programs between pediatric and adult specialty care for XLH patients. Nevertheless, even adults with milder forms usually develop symptoms in their third or fourth decade, which may include bone and joint pain, fatigue, enthesopathy (commonly involving the hips and anterior spinal ligament), pseudofractures, dental complications and early osteoarthritis (15). These complications ultimately cause chronic pain, impaired mobility, loss of productivity and lower quality of life (9, 15–20). Extraskeletal complications include hearing loss, symptomatic Chiari malformations, arterial hypertension [(possibly induced by oral phosphate supplements (21, 22)]. An increased prevalence of overweight and obesity has also been observed in XLH (23). Recent data suggest that XLH may be associated with increased risk of mortality in older adults, but not in children (4). A recent population-based study also reported an increased risk of depression and socioeconomic deprivation (24).
Recently, international evidence-based guidelines for the diagnosis and management of XLH have been published (13, 25). However, efforts are also required to translate the principles outlined in these guidelines to more practical recommendations at the national level, considering local elements such available resources and health economic aspects (26). Towards this aim, and as part of an interdisciplinary effort to improve the diagnostic and therapeutic care pathway for XLH patients in Belgium, a multi-stakeholder panel gathered to develop national consensus recommendations.
Methods
First, two in-person meetings were held between several of the authors (ML, JS, NG, EB, CH, JLa, KH, EL and JV) to review the available evidence, facilitate discussion and to propose diagnostic and therapeutic criteria for Belgium. The international evidence-based guidelines (13, 25) as well as recent randomized trials were considered as the basis for practical recommendations applicable to the Belgian context. Further input was sought from all other co-authors through consecutive e-mail rounds. The writing panel is composed of national experts from all universities and involves specialists in pediatric nephrology, endocrinology, adult metabolic bone diseases, rheumatology, clinical genetics, orthopedic surgery, clinical chemistry and physical medicine and rehabilitation. Furthermore, a Belgian XLH patient who is a founding member of the Belgian patient organization (XLH Belgium) as well as of its French counterpart (RVRH-XLH) participated. This work was co-authored by members of and endorsed by the Belgian Society for Pediatric Nephrology, the Belgian Society for Pediatric Endocrinology and Diabetes, the Flemish Network on Rare Bone Diseases, the Belgian Bone Club, the Royal Belgian Society of Laboratory Medicine and the Royal Belgian Society of Physical and Rehabilitation Medicine.
Results
Diagnosis
The diagnosis of XLH relies on the combination of clinical, radiographic, biochemical and genetic features (25). More specifically, this involves signs of rickets and/or osteomalacia in association with hypophosphatemia and renal phosphate wasting in the absence of vitamin D deficiency. The diagnosis should be confirmed by genetic testing whenever possible.
The clinical features include those common for hypophosphatemic rickets, as outlined in the introduction i.e. short stature, waddling gait, and leg bowing in growing children, in addition to muscle weakness. Fatigue and chronic pain become more prevalent in older children and particularly adults. Growth delay usually becomes evident from 9-12 months of age in XLH children (27). Early diagnosis and treatment is associated with better outcomes in children. Even when plasma phosphate is measured, hypophosphatemia may be overlooked due to lack of attention, misinterpretation of reference values in children, or waxing and waning of phosphatemia. In adults, signs of prior rickets during childhood should be sought e.g. short stature and limb bowing, although these may be absent in patients with milder phenotypes or those having received appropriate treatment during childhood. Some clinical features distinctive for this form of hypophosphatemic rickets are dental abscesses and enthesopathy, which may present to rheumatologists and are sometimes mistaken for spondylarthropathies.
Hypophosphatemic rickets has a wide differential diagnosis (Table 1). Although XLH is the most common genetic form, both acquired and rarer inherited differential-diagnoses should be considered. Neither clinical, biochemical, radiographic or genetic examinations on their own can correctly distinguish XLH from other conditions. Therefore, we recommend a multimodal work-up of suspected XLH by an experienced clinician to exclude other diseases. Bone biopsy is not routinely recommended in XLH (13). Moreover, expertise in bone histomorphometry is still scarcely available in Belgium (mainly in collaboration with neighboring countries, although bone histomorphometry recently became reimbursed through the national health insurance).
TABLE 1
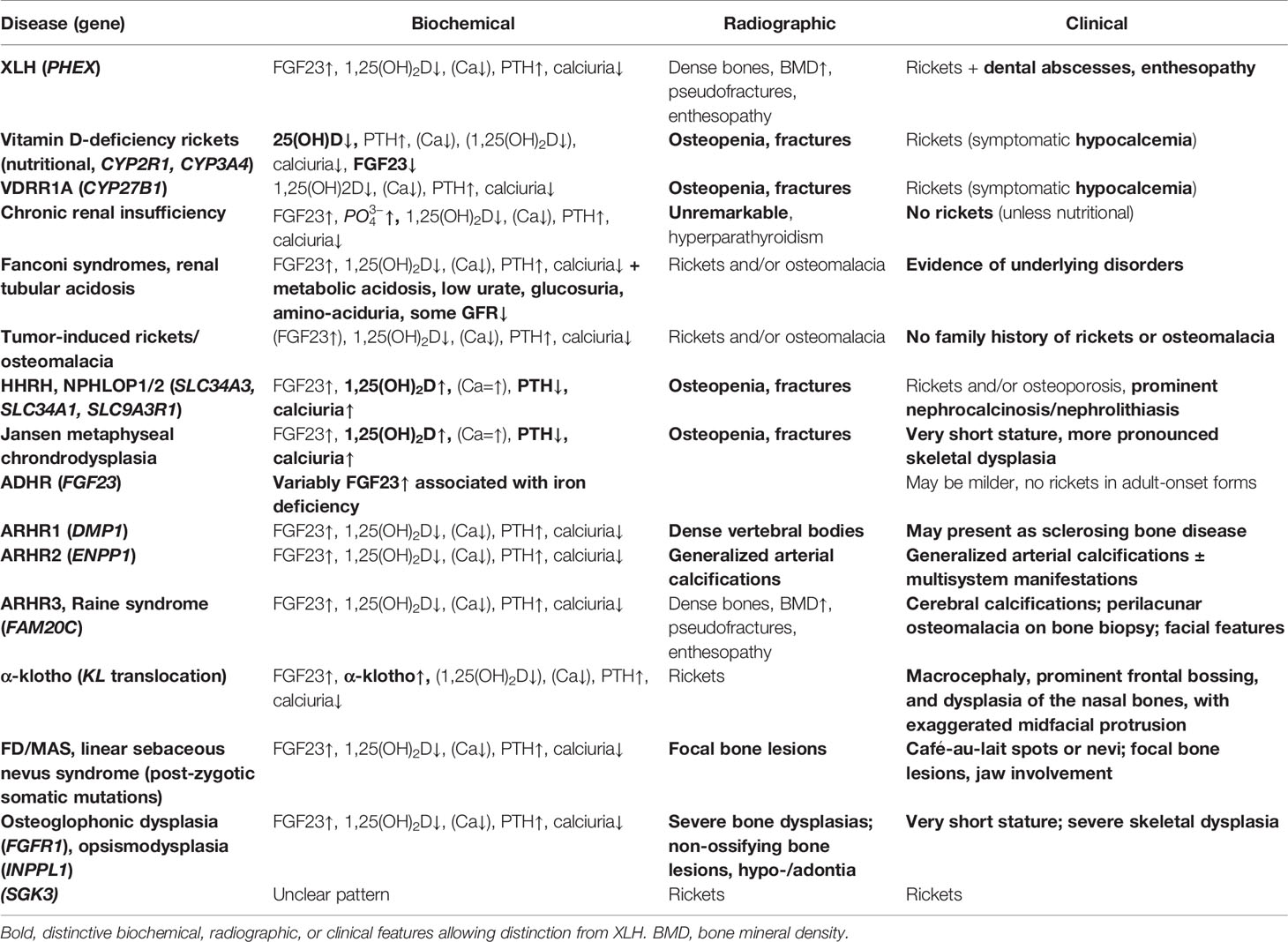
Table 1 Differential diagnoses of X-linked hypophosphatemia (XLH).
Biochemical Work-Up and Differential Diagnosis
Figure 1 shows a practical flowchart outlining the differential-diagnosis of hypophosphatemia in children or adults, according to biochemical features. The clinical, radiographic, biochemical and pathophysiological or genetic features of these causes are discussed in detail in this section.
FIGURE 1
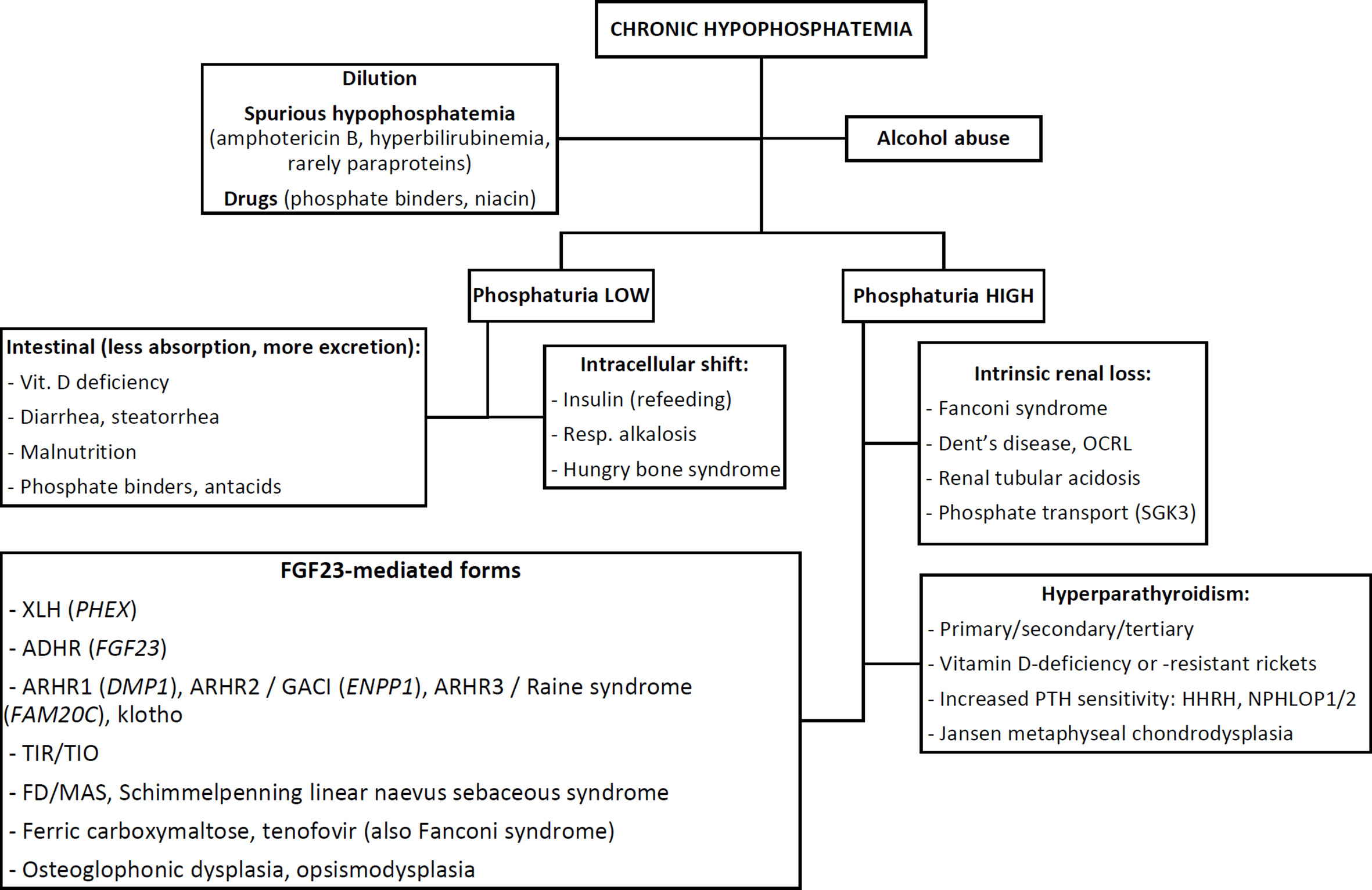
Figure 1 Flowchart outlining the differential-diagnosis of chronic hypophosphatemia based on biochemical features. See text for abbreviations.
As with the approach to any electrolyte disorder, the first step after careful history taking and clinical examination is to exclude obvious causes such as dilution (e.g. by massive fluid resuscitation, dialysis, plasmapheresis), spurious hypophosphatemia (from e.g. drug interference like amphotericin B, interference by bilirubin (28) or specific paraproteins), medication effects [e.g. excessive phosphate binders, niacin (29)] or alcohol abuse. Hypophosphatemia in alcoholics has a complex, multifactorial and incompletely understood pathophysiology. These causes should be considered first, since they can usually be diagnosed without further work-up.
Distinguishing Acquired vs. Genetic and Acute vs. Chronic Hypophosphatemia
Previously normal plasma phosphate levels suggest three possibilities: an acquired chronic cause, an acquired acute causes or a genetic, adult-onset cause. However, prior phosphate levels are often unavailable. Elevated alkaline phosphatase (ALP) is also indicative of chronic hypophosphatemia and consequent rickets/osteomalacia. Hypophosphatemia in the absence of rickets should raise suspicion for either an acute, transient cause (e.g. intracellular shift from hyperventilation, refeeding, hungry bone syndrome) or an acquired chronic cause such as alcohol abuse, tumor-induced rickets/osteomalacia (TIR/TIO) or certain medications such as tenofovir or frequent ferric carboxymaltose infusions (30). Notably, some genetic forms of hypophosphatemia may have an adult onset (notably, autosomal-dominant hypophosphatemic rickets, see below), in which case signs of rickets may be absent.
Chronic hypophosphatemia is believed to play a central role in the pathogenesis of almost all forms of rickets (31, 32). After confirming chronic hypophosphatemia, the next step is to assess phosphaturia i.e. whether hypophosphatemia is due to renal phosphate wasting or not (see Biochemical Work-Up: Pre-Analytic and Analytic Considerations below).
Once renal phosphate wasting has been confirmed, three mechanisms of renal phosphate loss remain: (i) defective intrinsic renal phosphate transport, (ii) parathyroid hormone (PTH)-mediated (and/or vitamin D-mediated) hyperphosphaturia, or (iii) FGF23-mediated causes.
Defective Intrinsic Renal Phosphate Reabsorption
The first category includes all causes of Fanconi syndrome i.e. a more generalized low molecular weight solute wasting at the proximal tubulus level. Low levels of uric acid and bicarbonate, glucosuria, aminoaciduria and low molecular weight (tubular) proteinuria should hint towards this category. At an older age, there may be slowly progressive renal insufficiency. However, some degree of renal tubular acidosis may be acquired during long-standing XLH, particularly when associated with nephrocalcinosis (33). The differential-diagnosis of Fanconi syndrome itself is broad and includes (non-exhaustively) mitochondrial respiratory chain diseases, metal intoxications, Wilson’s disease, cystinosis, multiple myeloma [especially when involving light chains (34)], Sjögren’s disease, medications like ifosfamide or tenofovir, genetics forms of renal tubular acidose, Dent’s disease and Lowe’s oculocerebrorenal syndrome (from OCRL1 or CLCN5 mutations).
Some genetic conditions may rather selectively impair renal phosphate reabsorption, including several undiagnosed diseases. Recently, mutations in SGK3 (serum/glucocorticoid-regulated kinase 3) were identified as a possible cause of autosomal-dominant hypophosphatemic rickets (35), although further study is needed to understand the underlying mechanisms.
Parathyroid Hormone-Mediated Hyperphosphaturia
The second category entails disorders of increased PTH or impaired vitamin D actions. Recent evidence shows elevated FGF23 in primary hyperparathyroidism (36–38). Both XLH and primary hyperparathyroidism may feature hypophosphatemia, renal stone formation, elevated PTH and FGF23 levels. Still, primary hyperparathyroidism should be easily distinguished by the presence of hypercalcemia, hypercalciuria, and high-normal to elevated 1,25(OH)2D as well as by the absence of rickety features.
Occasionally, it may be difficult to distinguish patients with non-familial XLH (i.e. probands without other affected family members) from children with nutritional, vitamin D-deficient- or -resistant rickets. Low 25(OH)D levels are common in XLH patients and should be corrected before the diagnosis is confirmed. In patients refractory to 25(OH)D replenishment, supervised supplementation may distinguish between poor compliance vs. resistance due to inactivating CYP2R1 (recessive) or activating CYP3A4 (autosomal-dominant) mutations (39).
Because FGF23 stimulates CYP24A1 and inhibits CYP27B1 (25-hydroxyvitamin D3-1α-hydroxylase) expression in both renal and extrarenal tissues (40), FGF23-mediated forms of hypophosphatemia are associated with low 1,25(OH)2D levels (41), secondary hyperparathyroidism (42), and low-normal to slightly decreased calcemia. However, 10-16.7% of adult XLH patients may exhibit tertiary (hypercalcemic) hyperparathyroidism, which is amenable to parathyroidectomy (42, 43). The levels of 1,25(OH)2D are also decreased in patients with very severe 25(OH)D deficiency, those with chronic renal insufficiency or in rare cases of CYP27B1 mutations (vitamin D-resistant rickets type 1A). In the latter three situations, FGF23 levels are low, high or unknown, respectively. Vitamin D-resistant rickets due to inactivating vitamin D receptor mutations is exquisitely rare and more easily distinguished from XLH due to elevated 1,25(OH)2D levels and because almost all patients display alopecia. Importantly, radiographic features prominently differ between XLH and nutritional or vitamin D-related forms of rickets, with dense bone cortices in XLH and radiolucent bones in vitamin D-related rickets. Thus, X-ray imaging as well as genetic testing may distinguish these disorders (see Table 1).
Hereditary hypophospatemic rickets with hypercalciuria (HHRH) is a group of disorders featuring increased renal sensitivity to PTH. Bi-allelic mutations in SLC34A3 cause HHRH, while heterozygous mutations in SLC34A1 and SLC9A3R1 cause the phenotypically similar hypophosphatemic nephrolithiasis/osteoporosis syndromes (NPHLOP1 and NPHLOP2, respectively). In all these conditions, patients may have hypophosphatemia, elevated FGF23 levels and nephrolithiasis/nephrocalcinosis (7). However, osteopenia/low bone mineral density, hypercalciuria, high-normal to elevated 1,25(OH)2D and low PTH point to the diagnosis of HHRH/NPHLOP. The same common and distinctive features apply to the ultrarare skeletal dysplasia Jansen metaphyseal chondrodysplasia, which is caused by activating PTH receptor 1 mutations (44). Additional clinical features in Jansen metaphyseal chondrodysplasia patients include severe short stature, brachycephaly, micrognathia, hypertelorism, and clinodactyly.
Fibroblast Growth Factor 23-Mediated Renal Phosphate Wasting
The third pathophysiological category involves FGF23-mediated forms of hypophosphatemic rickets. After XLH, the second most common genetic form of hypophosphatemic rickets is autosomal-dominant hypophosphatemic rickets (ADHR). Notably, it may be difficult to distinguish autosomal- and X-linked dominant inheritance in certain families. ADHR is caused by specific mutations in the FGF23 gene (mostly involving amino acid residues R176 and R179), making the intact (biologically active) hormone resistant to cleavage (45). Since iron deficiency increases FGF23 expression, this gene-environment interaction explains why adult-onset ADHR may be unmasked by iron deficiency and may be cured with (oral) iron supplements alone (46–50). Symptoms may wax and wane in parallel with iron loss during e.g. menstruation or pregnancy. As noted above, adults with ADHR may not have signs of rickets.
Ferric carboxymaltose (Injectafer®) infusions are a common drug-induced cause of transient (and sometimes symptomatic) hypophosphatemia, mostly in patients without chronic renal insufficiency. While the clinical significance in patients requiring sporadic infusions remains unknown, frequent infusions are a potential cause of acquired hypophosphatemic rickets and/or osteomalacia. The underlying mechanism involves specific carbohydrate moieties in the carboxymaltose apomolecule, which interfere with FGF23 cleavage. This explains why other i.v. iron formulations do not cause hypophosphatemia, and why intact (active) FGF23 increases more than c-terminal FGF23 in ferric carboxymaltose-induced hypophosphatemia (30, 51, 52). Interestingly, FGF23 may also be increased in tenofovir-induced Fanconi syndrome (53), via yet unknown mechanisms.
Autosomal-recessive forms of hypophosphatemic rickets (ARHR) may be caused by mutations in dentin matrix protein 1 (DMP1, as in ARHR1) or ectonucleotide pyrophosphatase/phosphodiesterase 1 (ENPP1, as in ARHR2). ARHR1 may present as a sclerosing bone dysplasia with hyperostosis and very dense vertebral bodies (54). DMP1 as well as PHEX are highly expressed in osteocytes, although it remains unknown how they alter the phosphate-FGF23 set point (55). ENPP1 is a critical enzyme in the generation of the mineralization inhibitor pyrophosphate. Loss-of-function mutations in ENPP1 lead to generalized arterial calcification of infancy (GACI). Many children with ENPP1 mutations who survive GACI will go on to develop ARHR2 (56, 57). The mechanism underlying raised FGF23 (58) and hypophosphatemia in GACI/ARHR2 remains incompletely understood. Extraskeletal features of GACI including hearing loss (59), thrombocytopenia, neurologic, cardiovascular, hepatic manifestations and hypoglycemia have recently been described (60). Recently, a case of ARHR was described with compound mutations in DMP1 and SPP1 (encoding osteopontin, another small integrin-binding ligand, N-linked glycoprotein [SIBLING] protein family member) (61).
FGF23 effects on the kidney are mediated by its co-receptor klotho. One case report described a balanced translocation affecting the KL gene, resulting in elevated α–klotho levels and hypophosphatemic rickets with elevated FGF23 (62).
FGF23 is believed to be cleaved at specific sites mainly by FURIN (PCSK3) (63), which also cleaves PTH and several other substrates. This cleavage is further regulated via phosphorylation by FAM20C (family with sequence similarity 20, member C, also known as DMP4; the gene mutated in Raine syndrome) and O-glycosylation by GALNT3 (polypeptide N-acetylgalactosaminyltransferase 3) (63–65). Raine syndrome (also called type 3 ARHR) was formerly believed to be a lethal osteosclerotic bone dysplasia, although now survivors into adulthood have been recognized (66–68). Clinical, biochemical and radiographic features of Raine syndrome may be very similar to sporadic XLH, but distinctive facial features, cerebral calcifications and osteomalacia surrounding the osteocyte lacunae on bone biopsy are a typical hallmark of this condition (69). In contrast, recessive, loss-of-function GALNT3 mutations impair FGF23 actions and thus, like in genetic or autoimmune FGF23 deficiency, lead to familial hyperphosphatemic tumoral calcinosis syndrome (70, 71).
In non-familial childhood- or adult-onset cases of hypophosphatemia, one must always bear in mind the possibility of tumor-induced hypophosphatemic rickets (TIR, in growing children) or TIO (72). In TIR and TIO, small mesenchymal tumors secrete FGF23 and/or other phosphatonins such as matrix extracellular phosphoglycoprotein (MEPE) (73). Thus, circulating FGF23 is usually but not always raised in TIR/TIO. Typical clinical features include chronic and progressive bone pain, muscle weakness and low bone mineral density. Diagnosis of TIO and TIR often remains extremely difficult, leading to extensive (and sometimes unnecessary) diagnostic procedures, since tumors may be too small for detection by conventional radiological methods (see Imaging Studies below).
Hypophosphatemic rickets in several sclerosing bone, skin or soft tissue disorders is believed to result from increased secretion of FGF23 or related phosphaturic hormones by the aberrant cells (74). Such may be the case in fibrous dysplasia/McCune-Albright syndrome (FD/MAS, which are caused by post-zygotic, somatic GNAS mutations) and Schimmelpenning-Feuerstein-Mims syndrome (which both feature bone lesions and café-au-lait spots), as well as in isolated linear nevus sebaceous syndrome (the latter two caused by post-zygotic somatic KRAS, HRAS or NRAS mutations). Finally, FGF23-mediated hypophosphatemia has been reported in two ultrarare skeletal dysplasias: osteoglophonic dysplasia (characterized by rhizomelic dwarfism, craniosynostosis, impacted teeth, hypodontia or anodontia, and multiple non-ossifying bone lesions) (75) and opsismodysplasia (a rare spondylo(epi)chondrodysplasia characterized by delayed skeletal maturation) (76).
Biochemical Work-Up: Pre-Analytic and Analytic Considerations
The measurement of plasma calcium (preferably ionized if feasible, or alternatively albumin-corrected), phosphate, ALP, creatinine, PTH and 25(OH)D are part of the standard work-up for any form of rickets or osteomalacia (77).
Alkaline Phosphatase
In the absence of liver disease, bone-specific ALP comprises ~90% of total ALP in children but only ~50% in adults (78, 79). Therefore, bone-specific ALP has been recommended for the monitoring of XLH in adults (25). Although bone-specific ALP is available in Belgium, it is still rarely used. In children, age-specific reference values should be used for correct interpretation of these analyses (particularly for ALP and phosphate). During the first months of life, plasma phosphate and ALP may be normal in XLH. ALP has no significant diurnal variation and can be used to monitor disease activity (taking age-specific changes during growth into account), as well as adherence to therapy (see Monitoring below).
Plasma and Urinary Phosphate
Since plasma phosphate fluctuates and is influenced by dietary intake, the international gold standard to assess phosphaturia is to determine the maximal tubular reabsorption of phosphorus per glomerular filtration rate (TmP/GFR), ideally from a fasted, second morning paired plasma and urine phosphate and creatinine sample (or 2-hour fasted morning urine collection) (80). Age-related reference ranges for TmP/GFR have been published (81). Coincidentally, the lower limit of normal for phosphate and TmP/GFR in children are numerically similar (82). The fractional tubular resorption of phosphate (TRP) may be within normal limits in children or adults with XLH. On the other hand, hypophosphatemia with a TmP/GFR < 0.85 points to a hyperphosphaturic mechanism. Other proposed definitions of hyperphosphaturia include 24h phosphaturia > 100 mg or hypophosphatemia with a fractional excretion of phosphate > 5%.
A 24h urine collection (or split 22h – 2h collection) may be useful to identify hypercalciuria, which may point to other diagnoses or excessive use of active vitamin D or calcium supplements. There are various definitions of hypercalciuria, either based on total excretion (> 200 to 250 mg/24h or 5.0 to 6.2 mmol/24h in adult women, > 250-300 mg/24h or 6.2 to 7.5 mg/24h in adult men (83), or > 4 mg/kg [0.1 mmol/kg] body weight/day in children) or based on urinary calcium concentration (> 200 mg/L) (84). Clearly, further work is needed to define optimal calciuria cut-points in different populations. The calcium/creatinine ratio allows to adjust for over- or undercollection in 24h urine collections, and may be a more practical (although less sensitive) method to allow the use of spot urine samples in children (85, 86). The addition of hydrochloric acid to the collection ensures dissolution of calcium crystals, thus preventing underestimation of calciuria (87).
1,25-Dihydroxyvitamin D
While the interpretation of calciuria and phosphaturia rely mostly on correct sample collection and interpretation of reference values, 1,25(OH)2D and FGF23 are more challenging laboratory analyses. Levels of 1,25(OH)2D are higher during childhood, and pediatric reference ranges have recently been proposed (88). International efforts to harmonize measurement of 1,25(OH)2D as well as PTH are underway (89).
Fibroblast Growth Factor 23
Like PTH, FGF23 is an unstable protein susceptible to decay in several fragments. Similar to phosphate, FGF23 shows diurnal variation; early-morning venous samples are recommended, while fasting appears to have little influence on FGF23 (90). Several FGF23 immunoassays are available: four methods for intact FGF23 (from Immutopics, Kainos, DiaSorin and Millipore) and one for c-terminal FGF23 (from Immutopics, which measures both intact molecules and c-terminal fragments). These methods differ not only in their marked use (only DiaSorin is marked for use in diagnostics), cost and compatibility with several automated laboratory platforms, but also in their reference ranges and even in their units of measurement (91, 92). First-generation assays required collection in protease inhibitor-coated tubes, but storage of samples on ice and immediate transport to the lab for prompt centrifugation is nowadays sufficient (92, 93). C-terminal but not intact FGF23 concentrations are much lower in serum than in EDTA plasma samples (93, 94). In treated patients with an unclear diagnosis, it is recommended to stop phosphate supplements (for at least two weeks) before measuring FGF23, because phosphate supplements may increase FGF23. Burosumab therapy (see Burosumab below) may cause analytical interference with certain FGF23 assays (95), but FGF23 measurements are not recommended during the follow-up of XLH patients (see Burosumab below). Thus, standardization and harmonization of FGF23 assays remain lacking, and results should always be interpreted cautiously. Recently, within- and between-subject biological variability for FGF23 have been published (96). Reference ranges are not universally established: while c-terminal FGF23 concentrations may be higher in children than in adults, they are rather similar for intact FGF23 (82). In Belgium, measurements of FGF23 and 1,25(OH)2D are reimbursed only once per year when requested by an internal medicine or pediatric specialist to evaluate abnormal calcemia or phosphatemia.
Imaging Studies
A skeletal survey using conventional radiography is useful in the work-up of XLH, to confirm rickets or distinguish it from other skeletal dysplasias (see Table 1), and to look for complications such as pseudofractures. X-rays of the knees or wrists are usually sufficient to confirm rickets in children. In adults, X-rays typically show enthesopathy, early spinal and extra-spinal osteoarthritis and/or pseudofractures (which often go clinically and biochemically undetected). However, radiation exposure limits the use of radiography during follow-up, particularly in children. In that regard, low-dose biplanar full-leg X-ray imaging using the EOS® system may be useful (97). In children, radiological signs of rickets in the hand, knees and lower limbs include long bone deformities and abnormal growth plates with widened and frayed metaphyses. Plain radiographs can confirm suspected rickets or can be useful pre-operatively, while clinical and biochemical evaluation (rather than routine repeated X-rays) is more important during follow-up.
Rickets of any cause can be graded using the Rickets Severity Scale (RSS), which has been validated in XLH (98). Higher RSS values (indicating more severe rickets) are associated with more severely impaired growth, walking ability, pain and physical disability (98), making this not only a radiographic but also a clinically relevant outcome. However, this score requires significant expertise [as evidenced by its moderate intra- and inter-rater reliability (98)] and is not yet widely available in Belgium.
Renal ultrasound can be used without radiation harm, to investigate the presence and/or severity of nephrocalcinosis and nephrolithiasis, although this requires an experienced operator. Panoramic dental X-rays may be required for stomatological work-up. Magnetic resonance imaging of the skull base may be indicated when there is concern for Chiari malformations (e.g., persistent headache, neurological or respiratory abnormalities) or to identify calcifications in suspected Raine syndrome (X-ray computed tomography may also be used in adults for the latter purpose). Bone scintigraphy may show increased metaphyseal uptake (99) in all forms of rickets and osteomalacia, which may be mistaken for other conditions e.g. avascular necrosis, transient migratory osteoporosis, etc. Routine technetium bone scans are not recommended in XLH and only useful to identify focal bone dysplasias such as FD/MAS.
Patients with XLH and several other sclerosing hypophosphatemic diseases usually have elevated bone mineral density Z- or T-scores on dual-energy X-ray absorptiometry (DXA). However, this can usually be appreciated well enough from available plain X-rays. Although low Z- or T-scores may point to other diagnoses such as HHRH/NPHLOP or TIR/TIO, DXA is not recommended in the work-up nor in the follow-up of XLH. Bone ultrasound and high-resolution quantitative computed tomography have also shown increased cortical and trabecular bone mass in XLH patients, although these techniques are still considered investigational (100) and not clinically useful.
Imaging plays a central role in the work-up of TIR/TIO, in order to determine whether the tumor can be surgically resected or requires alternative non-operative treatment. These tumors are often small and elusive, but they commonly arise in the lower limbs or head and neck area and may be located by whole-body magnetic resonance imaging, octreotide single photon emission computed tomography, positron emission tomography with 18fluorodeoxyglucose or DOTATOC/DOTATATE tracer, and/or by systemic venous sampling (101–103).
Genetic Testing
When the clinical, biochemical and radiographic findings suggest the diagnosis of XLH or another genetic disorder, genetic testing is recommended, both to confirm the diagnosis as well as for genetic counselling of the patient and his/her family members.
As the protean example of an X-linked dominant disorder, XLH is inherited from father to daughter and from mother to children of either sex. There is no convincing evidence that males (who are X-chromosome hemizygous) are more severely affected clinically or biochemically (13, 17, 104–107),. However, there may be a tendency that, compared to girls and women, affected boys and men may display more growth delay (5) as well as greater dysmorphic features such as larger head circumference, greater cranial height, shorter limbs and greater trunk length (108–110).
Genetic testing by Sanger sequencing or next-generation sequencing is readily available in several genetic centers in Belgium for PHEX as well as for other skeletal dysplasia genes using a whole-exome sequencing-based gene panel. Genetic testing also examines the 3’ untranslated region in which mutations associated with milder XLH phenotypes have been reported (111–113). In general however, mutations can affect any exon, without a clear genotype-phenotype correlation (114).
Up to 90% of patients clinically diagnosed with XLH will show a PHEX mutation (115–117). False-negative testing may occur in case of somatic mosaicism, large deletions, intronic and/or splice site mutations (118, 119). Quantitative polymerase chain reaction or multiplex ligation-dependent probe amplification may be useful to identify such cases (120). Lower diagnostic rates ~50% are obtained when applying PHEX sequencing to any unexplained hypophosphatemic rickets (121). Still, because it is the most common genetic form, single-gene PHEX sequencing may be the appropriate first step in FGF23-mediated hypophosphatemic rickets possibly due to XLH (122).
Genetic counselling is recommended before obtaining genetic testing, and afterwards to explain the results and implications. Especially in young adults and those planning pregnancy, counselling of the patient and his/her partner is warranted. If the underlying genetic defect is known, patients have a choice between natural conception or preimplantation genetic diagnosis (18). A genetic diagnosis in offspring can be performed, usually soon after birth.
A practical summary of the recommendations for the clinical, biochemical, radiographic and genetic work-up for suspected XLH in Belgium is shown in Table 2. These recommendations are generally consistent with the recent international guidelines (25).
TABLE 2

Table 2 Recommended diagnostic and monitoring tests for XLH in Belgium.
Multidisciplinary Care and Follow-Up
The recent international guidelines suggest follow-up of XLH patients by multidisciplinary teams (25). Individualized goals should be determined. Follow-up should focus on patient-centered outcomes and improving quality of life, by detecting and addressing complications early and monitoring compliance with treatment. Attention should be paid to the impact of the disease on other family members too.
We recommend that these multidisciplinary teams are organized by an expert in metabolic bone diseases and involve both pediatric and adult specialists, nurses, physiotherapists, social workers, psychologists, dietitians and occupational therapists (123). In Belgium, organization of multidisciplinary care can be facilitated by means of conventions e.g. for children with chronic kidney diseases or for metabolic diseases. The need for a specific convention for patients (children and adults) with a metabolic bone disease is stressed to finance these multi-disciplinary teams. Currently, children with XLH are mostly followed by pediatric endocrinologist and pediatric nephrologists, whereas adults are mostly followed by metabolic bone disease specialists and endocrinologists in Belgium.
We recommend the development of local protocols for transitional care between pediatric and adult metabolic bone disease specialists, as well as family-based outpatient clinics with pediatric-adult collaboration. In our experience, affected parents sometimes feel inappropriately guilty and/or neglect their own health to focus on their affected children (124). Several specialties should be available on a systematic or consulting basis including dentists, orthodontists and maxillofacial surgeons, pediatric and adult endocrinologists, nephrologists, rheumatologists, orthopedic surgeons, neurosurgeons, radiologists, geneticists, physical medicine and rehabilitation specialists, urologists, otolaryngolostist, ophthalmologists (to perform fundoscopy) etc. (Figure 2). Of note, since metabolic bone diseases is not a recognized separate specialty in Belgium, the lead specialist may differ by hospital. General practitioners in primary care play an important role in primary recognition of the disease and general follow-up e.g. with regards to compliance, extraskeletal manifestations and co-morbidities such as arterial hypertension or obesity, pain, side effects of treatment and psychosocial issues.
FIGURE 2
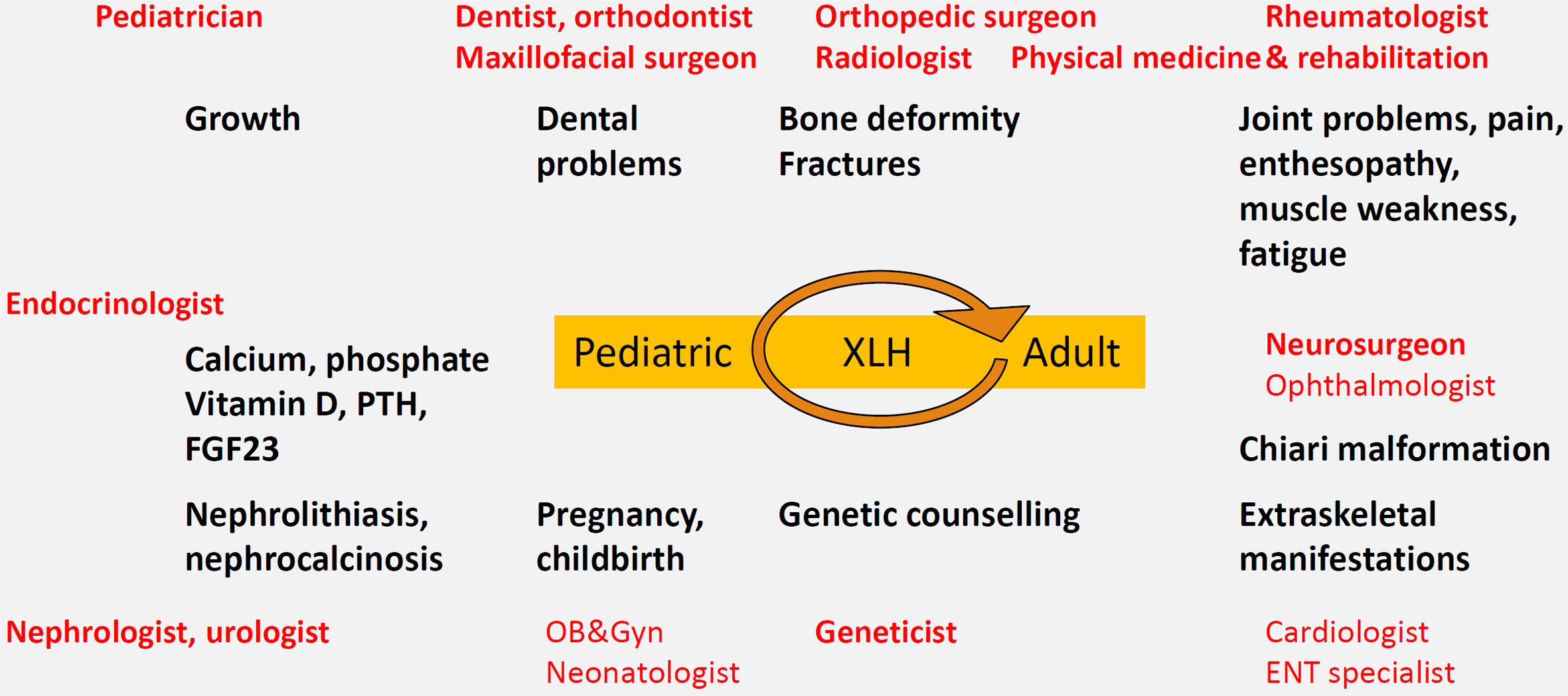
Figure 2 Multidisciplinary care model centered on the XLH patient, with attention to local protocols and transition from pediatric to adult metabolic bone specialist care.
The follow-up interval should be individualized to the patient, with more regular follow-up in young or growing children (on average every 3 months) compared to adults (who may be followed every 6 to 12 months, depending on their treatment, symptoms and needs) (25). XLH patients should see their dentist at least twice yearly. These intervals are however based on expert opinion (25).
Clinical assessment including height, weight, inter-malleolar and intercondylar distances, oral/dental and musculoskeletal examination and blood pressure measurement should be performed at every visit in children (see Table 2). Biochemical and/or radiological investigations should be evaluated only when clinically indicated.
In asymptomatic adult patients not receiving medical therapy, there is little need for repeated biochemical or radiological testing more than once a year. However, because vitamin D deficiency is common in Belgium and even more common in XLH patients, we recommend monitoring of 25(OH)D at least every twelve months, especially during the winter time, regardless of whether the patient receives medical treatment or not. ALP measurements are a useful indicator of skeletal complications such as progressive rickets/osteomalacia and/or pseudofractures, and therefore should be considered for the monitoring of patients not receiving medical therapy. In asymptomatic untreated patients, monitoring phosphate, 1,25(OH)2D or FGF23 levels is not useful.
Treatment
Non-Pharmacological Measures
The overarching goals of treatment should be patient-centered and focus on optimizing quality of life, mobility, pain and minimizing school and work absenteeism. The need for early treatment is stressed in children as it leads to better outcomes, such as improved linear growth, fewer bone deformities and better dental health. Patient education is essential at every visit, particularly in rare diseases and in adolescents. We encourage caregivers to provide patients with information about the nascent XLH patient association in Belgium. Multidisciplinary care should involve physiotherapists as well as psychological, nutritional and social support. In children, it might be necessary to liaise with school physicians to exchange information about e.g. growth and hearing problems, to ensure psychosocial wellbeing, and to ensure that medication or therapist care can be provided during school hours when necessary.
A healthy diet with sufficient nutritional calcium intake from dairy products is recommended in XLH, in contrast to calcium supplements, which are relatively contraindicated due to increased risk of kidney stone formation (25). Smoking should strongly be discouraged. Given the increased prevalence of arterial hypertension, overweight, obesity and metabolic syndrome in XLH, weight management and cardiovascular prevention should be an integral part of care. Notably, specialists should maintain a low index of suspicion for both obstructive as well as central sleep apnea in at-risk subjects e.g. those with Chiari malformations, those who are overweight or obese, and those with arterial hypertension, fatigue or sleep complaints.
Physical exercise has a myriad of health benefits and is strongly recommended although no formal guidance in XLH exists. Recommendations analogous to those in ankylosing spondylitis and osteoarthritis can be considered (125–127). Children and adults often display limited range of motion in the lower limbs and spine, with consequent gait abnormalities (128). We recommend that exercises and physiotherapy should aim to prevent or improve muscle weakness, back and joint pain, stiffness and limited mobility, by targeting muscle strength, core stability, joint range and general mobility, e.g. by resistance exercise training in combination with swimming, yoga, Pilates, dancing etc. Participation in leisure or professional sport activities is encouraged, with an emphasis on sports with lower risk of trauma. Targeted rehabilitation is often necessary following surgery and in case of enthesopathy or osteoarthritis. Given that XLH is a rare metabolic and growth disorder causing significant structural and functional musculoskeletal impairments, patients may be eligible for increased reimbursement of physiotherapy [“E-pathology list”, §L (129)]. In case of particularly disabling symptoms and functional impairment, multidisciplinary rehabilitation could be offered in general hospitals or rehabilitation centers under the supervision of a specialist in physical medicine and rehabilitation.
Analgesia
Chronic musculoskeletal pain is common in XLH adults and may be caused intrinsically by hypophosphatemia and osteomalacia/rickets (bone and muscle pain) and/or aggravated by (pseudo)fractures, enthesopathy, osteoarthritis etc. Based on history and clinical examinations, targeted imaging (using conventional X-ray and ultrasound imaging, MRI or scintigraphy if necessary) should always be considered to allow medical and/or surgical management of the underlying cause. Paracetamol has limited if any benefit, but may be considered for acute pain. Local and/or systemic non-steroidal anti-inflammatory drugs (NSAIDs; with gastroprotection if necessary) in combination with physical therapy (aiming for a good balance between exercise and rest, warmth or ice application, etc.) are considered first-line therapies. Although NSAIDs have been theorized to reduce phosphaturia, a randomized trial in children showed no improvement (130). Tramadol and strong opioids may be required for more advanced, otherwise untreatable musculoskeletal problems. Glucocorticoids or colchicine are not recommended. Given uncertainty regarding benefit and safety for calcific enthesopathy in general and lack of data in XLH, we recommend against the use of shockwave lithotripsy.
Phosphate and Active Vitamin D Analogs
The goal of medical treatment in children is to improve rickets, reduce skeletal deformities and avoid the need for surgery, improve height velocity and reduce bone pain (Figure 3). Early treatment in children is associated with improved outcomes (131). Although evidence was lacking previously, recent data suggest that conventional therapy may also reduce the burden of severe dental complications (132–135).
FIGURE 3
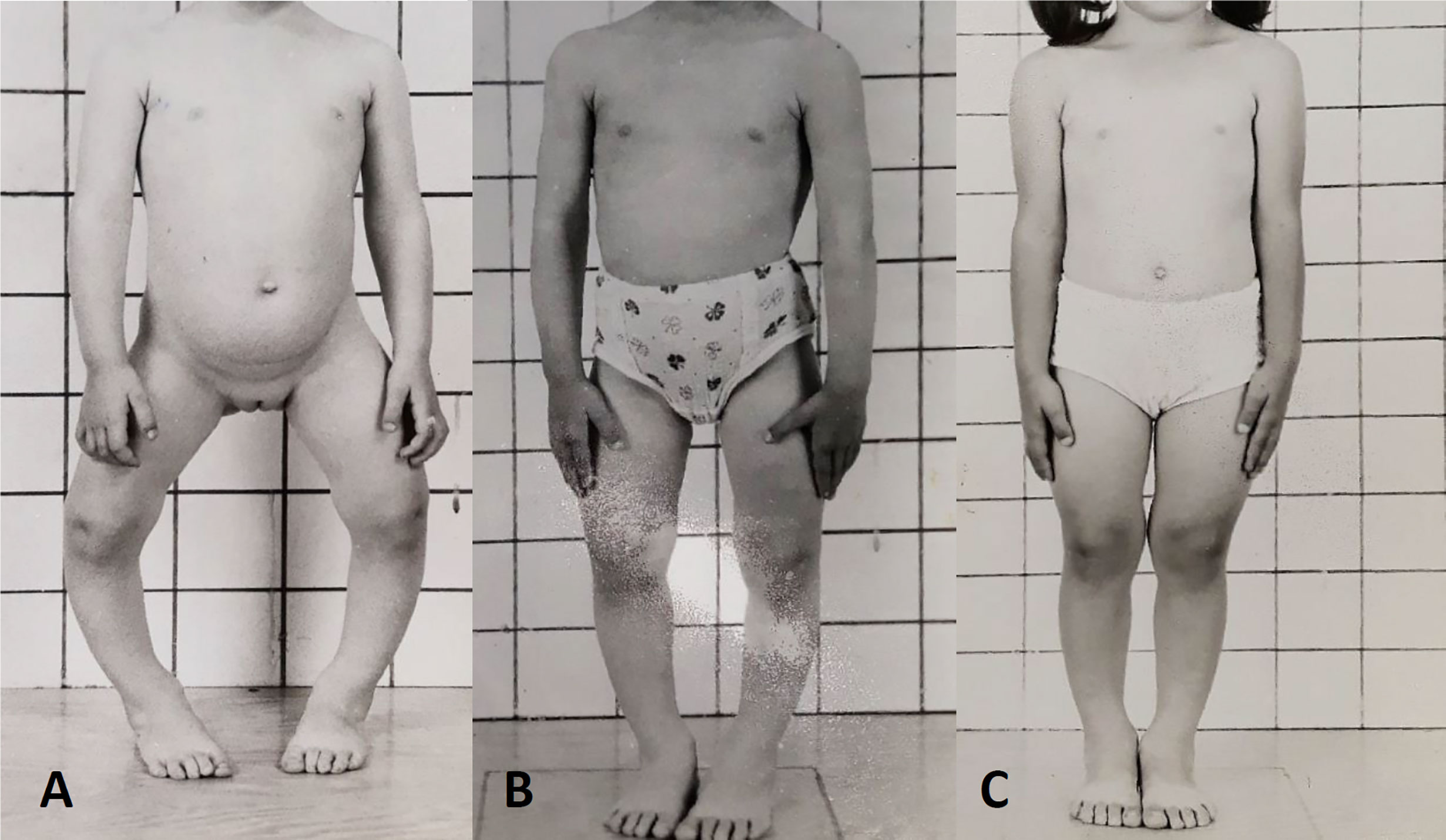
Figure 3 Response to conventional medical treatment with phosphate and active vitamin D supplements in XLH. (A) Clinical presentation at age 3 years. (B) One year, (C) 2 years after initiation of conventional therapy.
Conventional therapy in XLH consists of combination therapy with phosphate and active vitamin D analogs (preferably alfacalcidol, which has a wider therapeutic range and longer half-life than calcitriol and is available in liquid form for children). Phosphate supplements should not be prescribed without vitamin D analogues, since phosphate alone promotes secondary hyperparathyroidism and thereby renal phosphate wasting.
Conventional therapy is burdensome, since it requires multiple daily oral doses. Phosphate supplements are expensive, not reimbursed and generally cumbersome to obtain in Belgium. Financial provisions are available however via so-called “conventions” for tubulopathies in children or metabolic diseases in adults. Alfacalcidol is cheap, reimbursed in Belgium upon annual request by a specialist, but supply has occasionally been interrupted by stock breaches. Phosphate salts may be given in effervescent tablets imported from abroad, or in magistral capsules. In young children phosphate salts are usually given in several liquid formulations, such as galenic solutions (Joulie potio or others), i.v. phosphate solutions given orally, or commercial solutions. The choice of the formulation should be based on patient preferences and not solely to the habits of the prescriber. Side effects of oral phosphate include diarrhea, abdominal pain, bloating and secondary hyperparathyroidism. Spreading the dose throughout the day may alleviate side effects. Some patients prefer to dissolve phosphate in a bottle of drinking water, which facilitates spreading intake over the entire day and may reduce side effects. Furthermore, the high sodium and/or potassium load of phosphate supplements may predispose to arterial hypertension and/or hyperkaliemia, respectively. Phosphate complexes with calcium, therefore intake with milk should be avoided.
In adults, a small (n=16) uncontrolled prospective study reported reduced symptom scores following conventional treatment in 87% of patients (136). A trial of conventional treatment is therefore justified in symptomatic adults (137), although in many cases, symptoms of fatigue, low back pain, osteoarthritis, enthesopathy and spinal ligament calcifications may dominate, which generally do not respond to conventional therapy, nor does hearing loss (13). Furthermore, conventional medical therapy is often unpleasant, burdensome (14), and costly, requires monitoring and may be associated with side effects such as gastrointestinal symptoms, arterial hypertension or nephrolithiasis/nephrocalcinosis. There is consensus however that insufficiency fractures or pseudofractures, planned surgical procedures are indications for medical therapy in adults, while treatment may be considered in case of raised (bone) ALP and/or bone pain (13).
Concomitant vitamin D deficiency should be corrected with conventional vitamin D supplements e.g. cholecalciferol or ergocalciferol (138). Supraphysiological doses of active vitamin D analogs may be required to correct secondary hyperparathyroidism (139), but excessive vitamin D doses should be avoided due to risks of hypercalciuria, kidney stone formation and (rarely) hypercalcemia. Conventional treatment regimens with phosphate and vitamin D analogs improve intestinal phosphate absorption and paradoxically stimulate further FGF23 increases. Phosphate may exacerbate renal phosphate wasting (140) while active vitamin D attenuates phosphaturia (141). Of note, monotherapy with active vitamin D or CYP24A1 inhibition alone ameliorate rickets in preclinical mouse models (41, 142, 143) and may reduce the risk of secondary and tertiary hyperparathyroidism, supporting the use of the lowest possible dose of phosphate and higher doses of active vitamin D supplements.
In children, an initial dose of elemental phosphate between 20-40 (or 60) mg/kg body weight (0.7-1.3, up to 2.0 mmol/kg initial dose) is suggested, given as frequently as possible e.g. 4-6 daily divided doses in children (13). Doses > 80 mg/kg are to be avoided (25). Gradual titration may be used in the first weeks to avoid sudden gastrointestinal upset (13). Less frequent dosing e.g. 3-4 times daily may be used for maintenance or to improve compliance in adolescents and adults. In children, initial doses of alfacalcidol of 30-50 ng/kg (or calcitriol 20-30 ng/kg bodyweight) are suggested, which can usually be given once daily (evening dosage has been suggested) (25). Other active vitamin D analogs are not available in Belgium. Lower doses per body weight are recommended in adults. In general daily doses range between 750-1000 to 1600 mg of elementary phosphorus in 2-4 divided doses, and 0.75-1.5 µg of alfacalcidol (or 0.5 – 0.75 µg of calcitriol) (13, 25). However, dosage needs should always be individualized, since some patients need much higher or lower doses. With conventional therapy, phosphate dosages are not given with a goal of normalizing phosphatemia (keeping it at or above the lower limit is often not possible) but to improve growth and other symptoms and to normalize ALP, while avoiding secondary hyperparathyroidism and nephrocalcinosis (144). Active vitamin D analogs should be titrated to maintain PTH within the normal range but also to avoid hypercalcemia, hypercalciuria and kidney stone formation.
Although hypophosphatemia may exacerbate during pregnancy, the available evidence doesn’t clearly support a need for medical treatment in pregnant XLH mothers, since most will give birth uneventfully (145). Low phosphate levels in breast milk of XLH mothers has been reported in case reports, but adverse clinical outcomes have not been reported (146, 147). Similarly, no adverse effects of FGF23 excess on prenatal fetal-placental phosphate transport, breast milk phosphate concentrations or skeletal prenatal development are seen in mouse models (148, 149). Neonatal rickets is exceedingly rare, and there is currently insufficient evidence to formally recommend medical treatment for the sole indication of maternal or offspring skeletal health (18). Still, there is no international consensus on this topic (25). There is probably overuse of caesarean sections in this population, for no good reason (145).
Burosumab
In 2018, the European Medicines Agency (EMA) granted conditional market authorization to the fully humanized monoclonal anti-FGF23 antibody burosumab [formerly KRN23 (150)] for the treatment of XLH in children ≥ 1 year of age with a growing skeleton and radiographic evidence of bone disease (26). Late 2020, authorization was expanded for older adolescents and adults with radiographic evidence of bone disease, regardless of growth status.
In an open-label phase 2 trial, 52 children aged 5-12 years with XLH were randomized to receive burosumab without other conventional therapy, at a dose titrated to their phosphate level, every 2 or 4 weeks. The more frequent dosing led to more stable plasma phosphate concentrations, but the primary endpoint of the radiographic Rickets Severity Score was reduced in both groups (151). Most patients normalized their phosphate levels and TmP/GFR, and ALP levels declined. Greater height improvements were seen with the two-week dosing interval. Physical ability ameliorated and pain decreased in both groups (151). Another phase 2 trial confirmed the favorable efficacy and safety profile of burosumab in children aged 1-4 years (152).
Importantly, in an active-controlled open-label phase 3 trial, 61 children aged 1-12 years were randomized to receive conventional therapy or burosumab. Significantly greater improvements in radiographic healing of rickets (primary endpoint), growth, ALP and other biochemistries, lower-extremity deformities and mobility were observed with burosumab (153). Some patient-reported outcomes were significantly improved e.g. pain interference and physical health scores at week 40, although not for other outcomes or later time points (154). Some hypersensitivity and injection site reactions were noted in all trials, but there were no differences in serious treatment-related adverse events. Unfortunately, clinical trial data in adolescents remain lacking, despite the importance of the pubertal growth spurt. Nevertheless, the revised EMA market authorization would support continued use in adolescents when clinically indicated.
These data show that burosumab has the potential of improving clinical outcomes beyond current standard therapy. Notably, since it reduces the underlying renal phosphate wasting, burosumab has not been associated with complications of conventional therapy like nephrocalcinosis or secondary/tertiary hyperparathyroidism. However, burosumab is considerably more expensive than conventional medical therapy, and long-term outcomes as well as cost-effectiveness analyses are pending. Burosumab gained reimbursement in Belgium as of January 1st, 2021. Cost-sensitive criteria for use of burosumab in children in Belgium, proposed by the authors and approved by the competent authorities, are detailed in Table 3. Of note, these criteria still reflect the earlier EMA-approved indication (restricted to children with growing skeletons) and not the most recent version.
TABLE 3

Table 3 Reimbursement criteria for burosumab in children in Belgium as of January 2021.
The EMA-approved dose in children is a 2-weekly s.c. injection starting 0.8 mg/kg bodyweight, increased with 0.4 mg/kg dose increments (max. 2.0 mg/kg, cap at 90 mg dose) to achieve fasting plasma phosphate concentrations in the low-normal range for age. The average dose at this interval was 1.0 mg/kg in pediatric trials (25). Conventional treatment should be discontinued. Monitoring of peak phosphate levels at day 7-11 after injection, or before dosing after a three month period to achieve steady state is also suggested (25). In case of hyperphosphatemia, dosing should be withheld. The dose may need to be adjusted over time, but intervals for dose adjustment of 1-2 months are suggested (25). Creatinine levels should also be monitored, and treatment avoided in patients with incident renal insufficiency due to the theoretical risk of hyperphosphatemia and ectopic mineralization.
In adults, a phase 3 randomized trial (n=134) has compared placebo to a fixed 1 mg/kg bodyweight burosumab dose every four weeks. This normalized phosphatemia in almost 90% of patients. At 24 weeks, a significant decrease in joint stiffness and healing of active fractures was reported, with a safety profile similar to placebo (155). During the 24-week extension phase, all participants received open-label burosumab. Pseudofracture healing was confirmed in the group that switched from placebo to burosumab, and stiffness, pain and physical functioning and performance on the six-minute walking test improved significantly compared to baseline (156). Bone biopsies showed significant improvement in all osteomalacia indices (157). Thus, if approval is granted for use in adults by EMA (as has been done by its U.S. FDA counterpart, with a maximal dose of 90 mg), burosumab could represent an interesting treatment option for adult XLH patients suffering persistent bone and/or joint pain and disability, particularly from (pseudo-)fractures, despite a trial of optimal conventional therapy (25). Monitoring recommendations would be similar as in children (25).
Adjunctive Medical Therapies
Growth hormone is an off-label, yet theoretically attractive adjunctive therapy for XLH because it (transiently) increases phosphatemia and 1,25(OH)2D, lowers PTH and increases TmP/GFR, may improve height Z-scores without influencing body disproportion (158, 159) and might improve muscle strength. However, it doesn’t improve the underlying rickets and may increase ALP and exacerbate skeletal deformities (160). Moreover, it did not significantly improve adult height in long-term follow-up of a randomized controlled open-label study receiving conventional therapy (161). Clinical studies show that growth hormone therapy is more effective in prepubertal than in pubertal children (162). Although final height may be compromised despite conventional therapy in up to 60% of patients (163), extremely short children (Z-score ≤ -2.5) may be more likely to benefit (13, 164). Optimal control of rickets, PTH and ALP should be achieved before growth hormone therapy is considered in children with XLH (25). In Belgium, growth hormone therapy is not reimbursed for short stature related to XLH, although it has been obtained via medical need programs in the past (now no longer available).
Calcimimetics such as cinacalcet have been used to control secondary and tertiary hyperparathyroidism in XLH (165, 166). However, since they have been associated with severe side effects including hypocalcemia and QT-interval prolongation, their use should be limited (25).
Thiazide diuretics reduce hypercalciuria and may halt the progression of nephrocalcinosis in XLH (167). Thiazides should therefore be considered in case of arterial hypertension. However, adverse effects are common and require surveillance including hypotension, hypokalemia, hyponatremia, hypomagnesemia, hyperuricemia and increased insulin resistance. In contrast to hydrochlorothiazide, dipyridamole, which may reduce intrinsic renal phosphate leakage in some conditions, appears ineffective in XLH (168). The use of potassium citrate is not advised in XLH, because alkalinization may increase urinary phosphate precipitation (25).
Recently, a preclinical study in growing Hyp mice (the mouse model of Phex mutations) found that sclerostin inhibition increased phosphate and reduced FGF23 levels (169). Further evaluation of the efficacy and safety anti-sclerostin antibodies in the context of clinical trials is required. Other therapeutics targeting the FGF23 pathway e.g. suppressing the upregulation of osteopontin (170, 171) are also under development. Inadvertent treatment with bisphosphonates or other osteoporosis drugs, e.g. in case of misdiagnosis in patients with fractures, may induce deterioration and adverse skeletal effects (172).
Surgical Management
It is difficult to make firm evidence-based recommendations for orthopedic management in XLH (25). We recommend that care should be coordinated by an experienced orthopedic surgeon specialized in pediatric metabolic bone diseases. Insoles are not useful for pes planus in patients with varus or valgus knee deformity (25). In general, persistent or progressive severe deformity or disability despite ongoing optimal medical therapy may be an indication for surgery (25). Guided growth techniques by hemi-epiphysiodesis can be considered early as an alternative to more invasive options such as osteotomy (173), intramedullary nailing, external circular frames e.g. Ilizarov or combined techniques (174–177). Bed rest should be avoided as much as possible, and in those patients requiring it, careful monitoring for hypercalciuria is needed, which may warrant lowering of active vitamin D doses (25). Notably, medical treatment for at least 12 months in children and three to six months in adults, has been recommended before elective surgery, including dental implants (13, 25). Following surgery, prescribing rehabilitation is strongly recommended.
Repeated craniotomies may be necessary in symptomatic intracranial hypertension due to craniosynostosis (178). Neurosurgery may also be required for symptomatic Chiari malformation type 1, in which the cerebellar tonsils herniate through the foramen magnum and may compress the lower brainstem, upper spinal cord and/or cause syringomyelia (25). Up to 59% of children showed complete or partial sagittal suture fusion and 25% showed Chiari malformation type 1 in a recent large retrospective cohort study, however, most patients were asymptomatic (179). Rarely, symptomatic spinal enthesopathy or ossification of the posterior longitudinal spinal ligament may requiring laminoplasty, laminectomy or posterior decompression surgery (180–183).
Tertiary hyperparathyroidism with consequent hypercalcemia is a complication of longstanding high-dose phosphate supplements (184), although it has been noted in untreated patients too (18). It may be improved by partial or (sub)total parathyroidectomy, with or without ectopic parathyroid reimplantation. Clinicians should be aware that parathyroidectomy in these circumstances is usually followed by severe hungry bone syndrome with symptomatic hypocalcemia, which may require high doses of intravenous and oral calcium in combination with active vitamin D supplements (43, 185).
Monitoring
In patients receiving medical therapy, monitoring and adjustment of treatment doses should be based on measurements of calcium and phosphate in plasma and urine, creatinine, ALP and PTH at every visit (25). Notably, ALP may transiently increase during healing of rickets or signal the presence of pseudofractures. Increased ALP in otherwise well-controlled hypophosphatemia may signify poor compliance e.g. when patients improve their compliance shortly before clinic visits. If secondary hyperparathyroidism is present, alfacalcidol may be increased, phosphate doses decreased, or concomitant vitamin D deficiency may require replenishment.
Renal ultrasound to detect nephrocalcinosis or nephrolithiasis is recommended in medically treated XLH patients after one year and then every 1-2 years. Improvement of radiological signs of rickets can be seen after one year of treatment.
Patients receiving burosumab therapy may develop anti-drug antibodies, which may be accompanied by declining plasma phosphate levels and may require dosing increases when associated with clinical deterioration. Measuring anti-drug antibodies is not of clinical interest. The international guidelines suggest to measure 1,25(OH)2D every 3 to 6 months as a safety outcome in patients receiving burosumab therapy, together with monitoring for hypercalciuria (25). In patients receiving active vitamin D analogs and phosphate however, monitoring of 1,25(OH)2D is not recommended, because supraphysiological doses may be required to maintain PTH and calciuria within the desired range. Measuring FGF23 is not useful during follow-up of XLH patients, especially in patients treated with burosumab which may cause analytical interference (95).
Discussion and Conclusions
This consensus document provides a broad, detailed and practical overview of clinical aspects of diagnosis and management of XLH, which can guide specialists in Belgium. Key findings and policy recommendations are summarized in Table 4.
TABLE 4

Table 4 Summary of key policy recommendations.
Several key questions regarding XLH and other rare metabolic bone diseases remain unanswered (186): What are the best way to manage fatigue? What is the mechanism of pain? How can psychosocial support for patients and their families best be organized? How do rare metabolic bone diseases progress in ageing? Why do people with the same genetic mutation have different symptoms?
We recognize several limitations of these consensus recommendations. Most recommendations presented here, are based on expert opinion. We did not perform a systematic review nor used a formal GRADE or Delphi approach. However, recent international guidelines fulfill this need (25), and our recommendations can be considered a national translation of those guidelines. We believe national recommendations are useful, because Belgian policymakers have only recently initiated efforts to improve the care for patients with rare diseases, and no formal centers of expertise are officially recognized (currently, this task is delegated to all university hospitals). An important limitation is that not all specialties are represented among the authors. Specifically, a dentist or maxillofacial surgeon was not currently involved in XLH care pathways, highlighting the need to further liaise with other specialties in the care of XLH patients in our country. The Belgian burosumab reimbursement criteria as outlined in Table 3 are quite strict and not necessarily evidence-based, but they represent a consensus between several of the authors, within the strict Belgian reimbursement context. In several neighboring countries, burosumab is reimbursed when prescribed for children or adults treated in centers of excellence. Ideally, this would also preferable in Belgium. An alternative would be to use the existing framework of so-called “college of expert physicians” to peer-review reimbursement requests. Moreover, we chose not to present separate guidelines for children and adults, since we believe in an integrated, life course approach. Nevertheless, some recommendations are not relevant for either children or adults. Finally, several authors have a conflict of interest, although there was no sponsor involvement in the development, writing or publication of these guidelines. The authors would also like to emphasize that these recommendations are not intended as a substitute for expert clinical judgement.
Author Contributions
ML, JS, NG, EB, CH, JaL, KH, EL, and JV contributed to the conception of this work. ML, JS, EL, and JV wrote the first draft. All authors contributed to the article and approved the submitted version.
Conflict of Interest
ML has received lecture and consultancy fees from Alexion, Amgen, Kyowa Kirin, Menarini, Sandoz, Takeda, UCB and Will-Pharma. JS has received lecture, consultancy fees, and conference support from Kyowa Kirin, Alexion, Eli-Lily, Ferring, Ipsen, Menarini, Novo Nordisk, Pfizer, Sandoz, and Siemens Healthcare. DT has received conference support from Novo Nordisk. NG, JLa, and KH have received consultancy fees from Kyowa Kirin. EB has received conference support from Novo Nordisk and Pfizer. CH has received conference support from Novo Nordisk and Ferring. EC has received consultancy fees from bioMérieux, Diasorin, Fujirebio, IDS, and Menarini. PH is an employee of GlaxoSmithKline but participates in his own capacity. J-FK has received consultancy fees and conference support from Heel Belgium, Sanofi, and TRB Chemedica. KW has received conference support from Alexion, Ferring, Kyowa Kirin and Novo Nordisk. CV has received conference support from Boehringer Ingelheim. GM has received consultancy fees from Alexion, Biomarin, Kyowa Kirin, and Pfizer. EL has received consultancy fees and travel support from Kyowa Kirin, Chiesi, and Recordati. JV has received conference support and consultancy fees from Alexion, Bellco, Ferring, Medtronic, and Kyowa Kirin.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the patient for her written informed consent to use her clinical images, and Dr. H.G. Zmierczak for the useful discussions.
References
1. Beck-Nielsen SS, Brock-Jacobsen B, Gram J, Brixen K, Jensen TK. Incidence and prevalence of nutritional and hereditary rickets in southern Denmark. Eur J Endocrinol (2009) 160(3):491–7. doi: 10.1530/EJE-08-0818
2. Endo I, Fukumoto S, Ozono K, Namba N, Inoue D, Okazaki R, et al. Nationwide survey of fibroblast growth factor 23 (FGF23)-related hypophosphatemic diseases in Japan: prevalence, biochemical data and treatment. Endocr J (2015) 62(9):811–6. doi: 10.1507/endocrj.EJ15-0275
3. Statbel. (2020). https://statbel.fgov.be/nl/themas/bevolking/geboorten-en-vruchtbaarheid.
4. Hawley S, Shaw NJ, Delmestri A, Prieto-Alhambra D, Cooper C, Pinedo-Villanueva R, et al. Prevalence and Mortality of Individuals With X-LinkedHypophosphatemia: A United Kingdom Real-World Data Analysis. J ClinEndocrinol Metab (2020) 105(3):e871–8. doi: 10.1210/clinem/dgz203
5. Rafaelsen S, Johansson S, Raeder H, Bjerknes R. Hereditary hypophosphatemia in Norway: a retrospective population-based study of genotypes, phenotypes, and treatment complications. Eur J Endocrinol (2016) 174(2):125–36. doi: 10.1530/EJE-15-0515
6. Vlaams Agentschap Zorg en Gezondheid. Oprichting Vlaams Netwerk Zeldzame Ziekten. Vlaams Agentschap Zorg en Gezondheid (2017). Available at: https://www.zorg-en-gezondheid.be/oprichting-vlaams-netwerk-zeldzame-ziekten.
7. Bacchetta J, Bardet C, Prie D. Physiology of FGF23 and overview of genetic diseases associated with renal phosphate wasting. Metabolism (2020) 103S:153865. doi: 10.1016/j.metabol.2019.01.006
8. Gohil A, Imel EA. FGF23 and Associated Disorders of Phosphate Wasting. Pediatr Endocrinol Rev (2019) 17(1):17–34. doi: 10.17458/per.vol17.2019.gi.fgf23anddisordersphosphate
9. Beck-Nielsen SS, Mughal Z, Haffner D, Nilsson O, Levtchenko E, Ariceta G, et al. FGF23 and its role in X-linked hypophosphatemia-related morbidity. Orphanet J Rare Dis (2019) 14(1):58. doi: 10.1186/s13023-019-1014-8
10. Mindler GT, Kranzl A, Stauffer A, Haeusler G, Ganger R, Raimann A. Disease-specific gait deviations in pediatric patients with X-linked hypophosphatemia. Gait Posture (2020) 81:78–84. doi: 10.1016/j.gaitpost.2020.07.007
11. Veilleux LN, Cheung MS, Glorieux FH, Rauch F. The muscle-bone relationship in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab (2013) 98(5):E990–5. doi: 10.1210/jc.2012-4146
12. Veilleux LN, Cheung M, Ben Amor M, Rauch F. Abnormalities in muscle density and muscle function in hypophosphatemic rickets. J Clin Endocrinol Metab (2012) 97(8):E1492–8. doi: 10.1210/jc.2012-1336
13. Carpenter TO, Imel EA, Holm IA, Jan de Beur SM, Insogna KL. A clinician’s guide to X-linkedhypophosphatemia. J Bone Miner Res (2011) 26(7):1381–8. doi: 10.1002/jbmr.340
14. Ferizovic N, Marshall J, Williams AE, Mughal MZ, Shaw N, Mak C, et al. Exploring the Burden of X-Linked Hypophosphataemia: An OpportunisticQualitative Study of Patient Statements Generated During a Technology Appraisal. Adv Ther (2020) 37(2):770–84. doi: 10.1007/s12325-019-01193-0
15. Seefried L, Smyth M, Keen R, Harvengt P. Burden of disease associated with X-linked hypophosphataemia in adults: a systematic literature review. Osteoporos Int (2021) 32(1):7–22. doi: 10.1007/s00198-020-05548-0
16. Berndt M, Ehrich JH, Lazovic D, Zimmermann J, Hillmann G, Kayser C, et al. Clinical course of hypophosphatemic rickets in 23 adults. Clin Nephrol (1996) 45(1):33–41.
17. Che H, Roux C, Etcheto A, Rothenbuhler A, Kamenicky P, Linglart A, et al. Impaired quality of life in adults with X-linked hypophosphatemiaand skeletal symptoms. Eur J Endocrinol (2016) 174(3):325–33. doi: 10.1530/EJE-15-0661
18. Lecoq AL, Brandi ML, Linglart A, Kamenicky P. Management of X-linked hypophosphatemia in adults. Metabolism (2020) 103S:154049. doi: 10.1016/j.metabol.2019.154049
19. Skrinar A, Dvorak-Ewell M, Evins A, Macica C, Linglart A, Imel EA, et al. The Lifelong Impact of X-Linked Hypophosphatemia: Results From aBurden of Disease Survey. J Endocr Soc (2019) 3(7):1321–34. doi: 10.1210/js.2018-00365
20. Hughes M, Macica C, Meriano C, Doyle M. Giving Credence to the Experience of X-Linked Hypophosphatemia inAdulthood: An Interprofessional Mixed-Methods Study. J Patient Cent ResRev (2020) 7(2):176–88. doi: 10.17294/2330-0698.1727
21. Nehgme R, Fahey JT, Smith C, Carpenter TO. Cardiovascular abnormalities in patients with X-linkedhypophosphatemia. J Clin Endocrinol Metab (1997) 82(8):2450–4. doi: 10.1210/jcem.82.8.4181
22. Alon US, Monzavi R, Lilien M, Rasoulpour M, Geffner ME, Yadin O. Hypertension in hypophosphatemic rickets–role of secondaryhyperparathyroidism. Pediatr Nephrol (2003) 18(2):155–8. doi: 10.1007/s00467-002-1044-6
23. Zhukouskaya VV, Rothenbuhler A, Colao A, Di Somma C, Kamenicky P, Trabado S, et al. Increased prevalence of overweight and obesity in children withX-linked hypophosphatemia. Endocr Connect (2020) 9(2):144–53. doi: 10.1530/EC-19-0481
24. Hawley S, Shaw NJ, Delmestri A, Prieto-Alhambra D, Cooper C, Pinedo-Villanueva R, et al. Higher prevalence of non-skeletal comorbidity related to X-linkedhypophosphataemia: a UK parallel cohort study using CPRD. Rheumatol (Oxford) (2020). doi: 10.1093/rheumatology/keaa859
25. Haffner D, Emma F, Eastwood DM, Duplan MB, Bacchetta J, Schnabel D, et al. Clinical practice recommendations for the diagnosis and managementof X-linked hypophosphataemia. Nat Rev Nephrol (2019) 15(7):435–55. doi: 10.1038/s41581-019-0152-5
26. Padidela R, Cheung MS, Saraff V, Dharmaraj P. Clinical guidelines for burosumab in the treatment of XLH inchildren and adolescents: British paediatric and adolescent bone grouprecommendations. Endocr Connect (2020) 9(10):1051–6. doi: 10.1530/EC-20-0291
27. Mao M, Carpenter TO, Whyte MP, Skrinar A, Chen CY, San Martin J, et al. Growth Curves for Children with X-linkedHypophosphatemia. J Clin Endocrinol Metab (2020) 105(10):3243–9. doi: 10.1210/clinem/dgaa495
28. Duncanson GO, Worth HG. Pseudohypophosphataemia as a result of bilirubin interference. Ann Clin Biochem (1990) 27(Pt 3):253–7.doi: 10.1177/000456329002700312
29. Isakova T, Ix JH, Sprague SM, Raphael KL, Fried L, Gassman JJ, et al. Rationale and Approaches to Phosphate and Fibroblast Growth Factor23 Reduction in CKD. J Am Soc Nephrol (2015) 26(10):2328–39. doi: 10.1681/ASN.2015020117
30. Wolf M, Rubin J, Achebe M, Econs MJ, Peacock M, Imel EA, et al. Effects of Iron Isomaltoside vs Ferric Carboxymaltose onHypophosphatemia in Iron-Deficiency Anemia: Two Randomized Clinical Trials. JAMA (2020) 323(5):432–43. doi: 10.1001/jama.2019.22450
31. Tiosano D, Hochberg Z. Hypophosphatemia: the common denominator of allrickets. J Bone Miner Metab (2009) 27(4):392–401. doi: 10.1007/s00774-009-0079-1
32. Sabbagh Y, Carpenter TO, Demay MB. Hypophosphatemia leads to rickets by impairing caspase-mediatedapoptosis of hypertrophic chondrocytes. Proc Natl Acad Sci U S A(2005) 102(27):9637–42. doi: 10.1073/pnas.0502249102
33. Seikaly M, Browne R, Baum M. Nephrocalcinosis is associated with renal tubular acidosis in children with X-linked hypophosphatemia. Pediatrics (1996) 97(1):91–3.
34. Malhotra R, Guleria P, Barwad A, Pramanik R, Tandon N. A unique case of light chain (AL) amyloidosis masquerading ashypophosphatemic osteomalacia. Osteoporos Int (2021) 32(2):387–92. doi: 10.1007/s00198-020-05622-7
35. Cebeci AN, Zou M, BinEssa HA, Alzahrani AS, Al-Rijjal RA, Al-Enezi AF, et al. Mutation of SGK3, a Novel Regulator of Renal Phosphate Transport,Causes Autosomal Dominant Hypophosphatemic Rickets. J Clin EndocrinolMetab (2020) 105(6):1840–50. doi: 10.1210/clinem/dgz260
36. Witteveen JE, van Lierop AH, Papapoulos SE, Hamdy NA. Increased circulating levels of FGF23: an adaptive response inprimary hyperparathyroidism? Eur J Endocrinol (2012) 166(1):55–60. doi: 10.1530/EJE-11-0523
37. Nilsson IL, Norenstedt S, Granath F, Zedenius J, Pernow Y, Larsson TE. FGF23, metabolic risk factors, and blood pressure in patients withprimary hyperparathyroidism undergoing parathyroid adenomectomy. Surgery (2016) 159(1):211–7. doi: 10.1016/j.surg.2015.06.057
38. Kawata T, Imanishi Y, Kobayashi K, Miki T, Arnold A, Inaba M, et al. Parathyroid hormone regulates fibroblast growth factor-23 in a mousemodel of primary hyperparathyroidism. J Am Soc Nephrol (2007) 18(10):2683–8. doi: 10.1681/ASN.2006070783
39. Roizen JD, Li D, O’Lear L, Javaid MK, Shaw NJ, Ebeling PR, et al. CYP3A4 mutation causes vitamin D-dependent rickets type3. J Clin Invest (2018) 128(5):1913–8. doi: 10.1172/JCI98680
40. Bacchetta J, Sea JL, Chun RF, Lisse TS, Wesseling-Perry K, Gales B, et al. Fibroblast growth factor 23 inhibits extrarenal synthesis of1,25-dihydroxyvitamin D in human monocytes. J Bone Miner Res (2013) 28(1):46–55. doi: 10.1002/jbmr.1740
41. Bai X, Miao D, Xiao S, Qiu D, St-Arnaud R, Petkovich M, et al. CYP24 inhibition as a therapeutic target in FGF23-mediated renalphosphate wasting disorders. J Clin Invest (2016) 126(2):667–80. doi: 10.1172/JCI81928
42. Lecoq AL, Chaumet-Riffaud P, Blanchard A, Dupeux M, Rothenbuhler A, Lambert B, et al. Hyperparathyroidism in Patients With X-LinkedHypophosphatemia. J Bone Miner Res (2020) 35(7):1263–73. doi: 10.1002/jbmr.3992
43. DeLacey S, Liu Z, Broyles A, El-Azab SA, Guandique CF, James BC, et al. Hyperparathyroidism and parathyroidectomy in X-linkedhypophosphatemia patients. Bone (2019) 127:386–92. doi: 10.1016/j.bone.2019.06.025
44. Saito H, Noda H, Gatault P, Bockenhauer D, Loke KY, Hiort O, et al. Progression of Mineral Ion Abnormalities in Patients With JansenMetaphyseal Chondrodysplasia. J Clin Endocrinol Metab (2018) 103(7):2660–9. doi: 10.1210/jc.2018-00332
45. Liu C, Zhao Z, Wang O, Li M, Xing X, Hsieh E, et al. Earlier Onset in Autosomal Dominant Hypophosphatemic Rickets of R179than R176 Mutations in Fibroblast Growth Factor 23: Report of 20 Chinese Cases and Review of theLiterature. Calcif Tissue Int (2019) 105(5):476–86. doi: 10.1007/s00223-019-00597-y
46. Imel EA, Peacock M, Gray AK, Padgett LR, Hui SL, Econs MJ. Iron modifies plasma FGF23 differently in autosomal dominanthypophosphatemic rickets and healthy humans. J Clin Endocrinol Metab (2011) 96(11):3541–9. doi: 10.1210/jc.2011-1239
47. Farrow EG, Yu X, Summers LJ, Davis SI, Fleet JC, Allen MR, et al. Iron deficiency drives an autosomal dominant hypophosphatemicrickets (ADHR) phenotype in fibroblast growth factor-23 (Fgf23) knock-in mice. Proc Natl Acad Sci U S A (2011) 108(46):E1146–55. doi: 10.1073/pnas.1110905108
48. Imel EA, Gray AK, Padgett LR, Econs MJ. Iron and fibroblast growth factor 23 in X-linked hypophosphatemia. Bone (2014) 60:87–92. doi: 10.1016/j.bone.2013.12.001
49. Kapelari K, Kohle J, Kotzot D, Hogler W. Iron Supplementation Associated With Loss of Phenotype in Autosomal Dominant Hypophosphatemic Rickets. J Clin Endocrinol Metab (2015) 100(9):3388–92. doi: 10.1210/jc.2015-2391
50. Imel EA, Liu Z, Coffman M, Acton D, Mehta R, Econs MJ. Oral Iron Replacement Normalizes Fibroblast Growth Factor 23 in Iron-Deficient Patients With Autosomal Dominant Hypophosphatemic Rickets. J Bone Miner Res (2020) 35(2):231–8. doi: 10.1002/jbmr.3878
51. Wolf M, Koch TA, Bregman DB. Effects of iron deficiency anemia and its treatment on fibroblast growth factor 23 and phosphate homeostasis in women. J Bone Miner Res (2013) 28(8):1793–803. doi: 10.1002/jbmr.1923
52. Wolf M, Chertow GM, Macdougall IC, Kaper R, Krop J, Strauss W. Randomized trial of intravenous iron-inducedhypophosphatemia. JCI Insight (2018) 3(23):e124486. doi: 10.1172/jci.insight.124486
53. Saeedi R, Jiang SY, Holmes DT, Kendler DL. Fibroblast growth factor 23 is elevated in tenofovir-related hypophosphatemia. Calcif Tissue Int (2014) 94(6):665–8. doi: 10.1007/s00223-014-9854-7
54. Saito T, Shimizu Y, Hori M, Taguchi M, Igarashi T, Fukumoto S, et al. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone (2011) 49(4):913–6. doi: 10.1016/j.bone.2011.06.029
55. Guirado E, Chen Y, Ross RD, Zhang Y, Chaussain C, George A. Disrupted Protein Expression and Altered Proteolytic Events in Hypophosphatemic Dentin Can Be Rescued by Dentin Matrix Protein 1. Front Physiol (2020) 11:82. doi: 10.3389/fphys.2020.00082
56. Kotwal A, Ferrer A, Kumar R, Singh RJ, Murthy V, Schultz-Rogers L, et al. Clinical and Biochemical Phenotypes in a Family With ENPP1 Mutations. J Bone Miner Res (2020) 35(4):662–70. doi: 10.1002/jbmr.3938
57. Oheim R, Zimmerman K, Maulding ND, Sturznickel J, von Kroge S, Kavanagh D, et al. Human Heterozygous ENPP1 Deficiency Is Associated With Early Onset Osteoporosis, a Phenotype Recapitulated in a Mouse Model of Enpp1 Deficiency. J Bone Miner Res (2020) 35(3):528–39. doi: 10.1002/jbmr.3911
58. Ferreira CR, Hackbarth ME, Ziegler SG, Pan KS, Roberts MS, Rosing DR, et al. Prospective phenotyping of long-term survivors of generalizedarterial calcification of infancy (GACI). Genet Med (2021) 23(2):396–407. doi: 10.1038/s41436-020-00983-0
59. Brachet C, Mansbach AL, Clerckx A, Deltenre P, Heinrichs C. Hearing loss is part of the clinical picture of ENPP1 loss of function mutation. Horm Res Paediatr (2014) 81(1):63–6. doi: 10.1159/000354661
60. Staretz-Chacham O, Shukrun R, Barel O, Pode-Shakked B, Pleniceanu O, Anikster Y, et al. Novel homozygous ENPP1 mutation causes generalized arterial calcifications of infancy, thrombocytopenia, and cardiovascular and central nervous system syndrome. Am J Med Genet A (2019) 179(10):2112–8. doi: 10.1002/ajmg.a.61334
61. Whyte MP, Amalnath SD, McAlister WH, McKee MD, Veis DJ, Huskey M, et al. Hypophosphatemic osteosclerosis, hyperostosis, and enthesopathy associated with novel homozygous mutations of DMP1 encoding dentin matrix protein 1 and SPP1 encoding osteopontin: The first digenic SIBLING protein osteopathy? Bone (2020) 132:115190. doi: 10.1016/j.bone.2019.115190
62. Brownstein CA, Adler F, Nelson-Williams C, Iijima J, Li P, Imura A, et al. A translocation causing increased alpha-klotho level results in hypophosphatemic rickets and hyperparathyroidism. Proc Natl Acad Sci U S A (2008) 105(9):3455–60. doi: 10.1073/pnas.0712361105
63. Tagliabracci VS, Engel JL, Wiley SE, Xiao J, Gonzalez DJ, Nidumanda Appaiah H, et al. Dynamic regulation of FGF23 by Fam20C phosphorylation, GalNAc-T3 glycosylation, and furin proteolysis. Proc Natl Acad Sci U S A (2014) 111(15):5520–5. doi: 10.1073/pnas.1402218111
64. de Las Rivas M, Paul Daniel EJ, Narimatsu Y, Companon I, Kato K, Hermosilla P, et al. Molecular basis for fibroblast growth factor 23 O-glycosylation by GalNAc-T3. Nat Chem Biol (2020) 16(3):351–60. doi: 10.1038/s41589-019-0444-x
65. Takashi Y, Kosako H, Sawatsubashi S, Kinoshita Y, Ito N, Tsoumpra MK, et al. Activation of unliganded FGF receptor by extracellular phosphate potentiates proteolytic protection of FGF23 by its O-glycosylation. Proc Natl Acad Sci U S A (2019) 116(23):11418–27. doi: 10.1073/pnas.1815166116
66. Takeyari S, Yamamoto T, Kinoshita Y, Fukumoto S, Glorieux FH, Michigami T, et al. Hypophosphatemic osteomalacia and bone sclerosis caused by a novel homozygous mutation of the FAM20C gene in an elderly man with a mild variant of Raine syndrome. Bone (2014) 67:56–62. doi: 10.1016/j.bone.2014.06.026
67. Acevedo AC, Poulter JA, Alves PG, de Lima CL, Castro LC, Yamaguti PM, et al. Variability of systemic and oro-dental phenotype in two families with non-lethal Raine syndrome with FAM20C mutations. BMC Med Genet (2015) 16:8. doi: 10.1186/s12881-015-0154-5
68. Rolvien T, Kornak U, Schinke T, Amling M, Oheim R. A novel FAM20C mutation causing hypophosphatemic osteomalacia withosteosclerosis (mild Raine syndrome) in an elderly man with spontaneous osteonecrosis of theknee. Osteoporos Int (2019) 30(3):685–9. doi: 10.1007/s00198-018-4667-6
69. Whyte MP, McAlister WH, Fallon MD, Pierpont ME, Bijanki VN, Duan S, et al. Raine Syndrome (OMIM #259775), Caused By FAM20C Mutation, IsCongenital Sclerosing Osteomalacia With Cerebral Calcification (OMIM 259660). J Bone Miner Res (2017) 32(4):757–69. doi: 10.1002/jbmr.3034
70. Roberts MS, Burbelo PD, Egli-Spichtig D, Perwad F, Romero CJ, Ichikawa S, et al. Autoimmune hyperphosphatemic tumoral calcinosis in a patient withFGF23 autoantibodies. J Clin Invest (2018) 128(12):5368–73. doi: 10.1172/JCI122004
71. Dauchez A, Souffir C, Quartier P, Baujat G, Briot K, Roux C. Hyperphosphatemic Familial Tumoral Calcinosis With Galnt3 Mutation:Transient Response to Anti-Interleukin-1 Treatments. JBMR Plus (2019) 3(7):e10185. doi: 10.1002/jbm4.10185
72. Jiang Y, Xia WB, Xing XP, Silva BC, Li M, Wang O, et al. Tumor-induced osteomalacia: an important cause of adult-onsethypophosphatemic osteomalacia in China: Report of 39 cases and review of theliterature. J Bone Miner Res (2012) 27(9):1967–75. doi: 10.1002/jbmr.1642
73. Imanishi Y, Hashimoto J, Ando W, Kobayashi K, Ueda T, Nagata Y, et al. Matrix extracellular phosphoglycoprotein is expressed in causative tumors of oncogenic osteomalacia. J Bone Miner Metab (2012) 30(1):93–9. doi: 10.1007/s00774-011-0290-8
74. Hoffman WH, Jain A, Chen H, Fedarko NS. Matrix extracellular phosphoglycoprotein (MEPE) correlates with serum phosphorus prior to and during octreotide treatment and following excisional surgery in hypophosphatemic linear sebaceous nevus syndrome. Am J Med Genet A (2008) 146A(16):2164–8. doi: 10.1002/ajmg.a.32395
75. Kuthiroly S, Yesodharan D, Ghosh A, White KE, Nampoothiri S. Osteoglophonic Dysplasia: Phenotypic and Radiological Clues. J Pediatr Genet (2017) 6(4):247–51. doi: 10.1055/s-0037-1602816
76. Zeger MD, Adkins D, Fordham LA, White KE, Schoenau E, Rauch F, et al. Hypophosphatemic rickets in opsismodysplasia. J Pediatr Endocrinol Metab (2007) 20(1):79–86. doi: 10.1515/JPEM.2007.20.1.79
77. Laurent MR, Bravenboer N, Van Schoor NM, Bouillon R, Pettifor JM, Lips P. Rickets and Osteomalacia. In: Bilezikian JP, editor. Primer on the Metabolic Bone Diseases and Disorders of Mineral Metabolism, Ninth Edition. Hoboken, NJ, USA: American Society for Bone and Mineral Research (2018). p. 684–94.
78. Rosalki SB, Foo AY, Burlina A, Prellwitz W, Stieber P, Neumeier D, et al. Multicenter evaluation of Iso-ALP test kit for measurement of bone alkaline phosphatase activity in serum and plasma. Clin Chem (1993) 39(4):648–52. doi: 10.1093/clinchem/39.4.648
79. Bouman AA, de Ridder CM, Nijhof JH, Netelenbos JC, Delemarre-vd Waal HA. Immunoadsorption assay for bone alkaline phosphatase compared with wheat germ agglutinin precipitation assay in serum from (pre)pubertal girls. Clin Chem (1996) 42(12):1970–4.
80. Walton RJ, Bijvoet OL. Nomogram for derivation of renal threshold phosphateconcentration. Lancet (1975) 2(7929):309–10. doi: 10.1016/s0140-6736(75)92736-1
81. Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications andinterpretation. Ann Clin Biochem (1998) 35(Pt 2):201–6. doi: 10.1177/000456329803500203
82. Imel EA. Congenital Conditions of Hypophosphatemia inChildren. Calcif Tissue Int (2021) 108(1):74–90. doi: 10.1007/s00223-020-00692-5
83. Curhan GC, Willett WC, Speizer FE, Stampfer MJ. Twenty-four-hour urine chemistries and the risk of kidney stonesamong women and men. Kidney Int (2001) 59(6):2290–8. doi: 10.1046/j.1523-1755.2001.00746.x
84. Pak CY, Sakhaee K, Moe OW, Poindexter J, Adams-Huet B, Pearle MS, et al. Defining hypercalciuria in nephrolithiasis. Kidney Int (2011) 80(7):777–82. doi: 10.1038/ki.2011.227
85. So NP, Osorio AV, Simon SD, Alon US. Normal urinary calcium/creatinine ratios in African-American andCaucasian children. Pediatr Nephrol (2001) 16(2):133–9. doi: 10.1007/s004670000510
86. Metz MP. Determining urinary calcium/creatinine cut-offs for the paediatricpopulation using published data. Ann Clin Biochem (2006) 43(Pt 5):398–401. doi: 10.1258/00045630677852010
87. Goldfarb DS. Metabolic Investigations: When and in Whom. In: Rao N, PG, Kavanagh J, editors. Urinary Tract Stone Disease. London: Springer (2010). p. 659–65.
88. Higgins V, Truong D, White-Al Habeeb NMA, Fung AWS, Hoffman B, Adeli K. Pediatric reference intervals for 1,25-dihydroxyvitamin D using theDiaSorin LIAISON XL assay in the healthy CALIPER cohort. Clin Chem LabMed (2018) 56(6):964–72. doi: 10.1515/cclm-2017-0767
89. Vanderschueren D, Pye SR, O’Neill TW, Lee DM, Jans I, Billen J, et al. Active vitamin D (1,25-dihydroxyvitamin D) and bone health inmiddle-aged and elderly men: the European Male Aging Study (EMAS). J ClinEndocrinol Metab (2013) 98(3):995–1005. doi: 10.1210/jc.2012-2772
90. Smith ER, Cai MM, McMahon LP, Holt SG. Biological variability of plasma intact and C-terminal FGF23 measurements. J Clin Endocrinol Metab (2012) 97(9):3357–65. doi: 10.1210/jc.2012-1811
91. Heijboer AC, Levitus M, Vervloet MG, Lips P, ter Wee PM, Dijstelbloem HM, et al. Determination of fibroblast growth factor 23. Ann Clin Biochem (2009) 46(Pt 4):338–40. doi: 10.1258/acb.2009.009066
92. Dirks NF, Smith ER, van Schoor NM, Vervloet MG, Ackermans MT, de Jonge R, et al. Pre-analytical stability of FGF23 with the contemporary immunoassays. Clin Chim Acta (2019) 493:104–6. doi: 10.1016/j.cca.2019.02.032
93. El-Maouche D, Dumitrescu CE, Andreopoulou P, Gafni RI, Brillante BA, Bhattacharyya N, et al. Stability and degradation of fibroblast growth factor 23 (FGF23): the effect of time and temperature and assay type. Osteoporos Int (2016) 27(7):2345–53. doi: 10.1007/s00198-016-3543-5
94. Souberbielle JC, Prié D, Piketty ML, Rothenbuhler A, Delanaye P, Chanson P, et al. Evaluation of a New Fully Automated Assay for Plasma Intact FGF23. Calcif Tissue Int (2017) 101(5):510–8. doi: 10.1007/s00223-017-0307-y
95. Piketty ML, Brabant S, Souberbielle JC, Maruani G, Audrain C, Rothenbuhler A, et al. FGF23 measurement in burosumab-treated patients: an emerging treatment may induce a new analytical interference. Clin Chem Lab Med (2020) 58(11):e267–e9. doi: 10.1515/cclm-2020-0460
96. Cavalier E, Lukas P, Bottani M, Aarsand AK, Ceriotti F, Coskun A, et al. European Biological Variation Study (EuBIVAS): within- and between-subject biological variation estimates of beta-isomerized C-terminal telopeptide of type I collagen (beta-CTX), N-terminal propeptide of type I collagen (PINP), osteocalcin, intact fibroblast growth factor 23 and uncarboxylated-unphosphorylated matrix-Gla protein-a cooperation between the EFLM Working Group on Biological Variation and the International Osteoporosis Foundation-International Federation of Clinical Chemistry Committee on Bone Metabolism. Osteoporos Int (2020) 31(8):1461–70. doi: 10.1007/s00198-020-05362-8
97. Melhem E, Assi A, El Rachkidi R, Ghanem I. EOS((R)) biplanar X-ray imaging: concept, developments, benefits, and limitations. J Child Orthop (2016) 10(1):1–14. doi: 10.1007/s11832-016-0713-0
98. Thacher TD, Pettifor JM, Tebben PJ, Creo AL, Skrinar A, Mao M, et al. Rickets severity predicts clinical outcomes in children with X-linked hypophosphatemia: Utility of the radiographic Rickets Severity Score. Bone (2019) 122:76–81. doi: 10.1016/j.bone.2019.02.010
99. Van de Wiele C, Dierckx RA, Weynants L, Simons M, Kaufman JM. Whole-body bone scan findings in X-linked hypophosphatemia. Clin Nucl Med (1996) 21(6):483. doi: 10.1097/00003072-199606000-00013
100. Raimann A, Mehany SN, Feil P, Weber M, Pietschmann P, Boni-Mikats A, et al. Decreased Compressional Sound Velocity Is an Indicator for Compromised Bone Stiffness in X-Linked Hypophosphatemic Rickets (XLH). Front Endocrinol (Lausanne) (2020) 11:355. doi: 10.3389/fendo.2020.00355
101. Chong WH, Andreopoulou P, Chen CC, Reynolds J, Guthrie L, Kelly M, et al. Tumor localization and biochemical response to cure in tumor-induced osteomalacia. J Bone Miner Res (2013) 28(6):1386–98. doi: 10.1002/jbmr.1881
102. Breer S, Brunkhorst T, Beil FT, Peldschus K, Heiland M, Klutmann S, et al. 68Ga DOTA-TATE PET/CT allows tumor localization in patients with tumor-induced osteomalacia but negative 111In-octreotide SPECT/CT. Bone (2014) 64:222–7. doi: 10.1016/j.bone.2014.04.016
103. El-Maouche D, Sadowski SM, Papadakis GZ, Guthrie L, Cottle-Delisle C, Merkel R, et al. (68)Ga-DOTATATE for Tumor Localization in Tumor-Induced Osteomalacia. J Clin Endocrinol Metab (2016) 101(10):3575–81. doi: 10.1210/jc.2016-2052
104. Whyte MP, Schranck FW, Armamento-Villareal R. X-linked hypophosphatemia: a search for gender, race, anticipation, or parent of origin effects on disease expression in children. J Clin Endocrinol Metab (1996) 81(11):4075–80. doi: 10.1210/jcem.81.11.8923863
105. Zhang C, Zhao Z, Sun Y, Xu L, JiaJue R, Cui L, et al. Clinical and genetic analysis in a large Chinese cohort of patientswith X-linked hypophosphatemia. Bone (2019) 121:212–20. doi: 10.1016/j.bone.2019.01.021
106. Ichikawa S, Gray AK, Bikorimana E, Econs MJ. Dosage effect of a Phex mutation in a murine model of X-linkedhypophosphatemia. Calcif Tissue Int (2013) 93(2):155–62. doi: 10.1007/s00223-013-9736-4
107. Chesher D, Oddy M, Darbar U, Sayal P, Casey A, Ryan A, et al. Outcome of adult patients with X-linked hypophosphatemia caused byPHEX gene mutations. J Inherit Metab Dis (2018) 41(5):865–76. doi: 10.1007/s10545-018-0147-6
108. Pronicka E, Popowska E, Rowinska E, Arasimowicz E, Syczewska M, Jurkiewicz D, et al. Anthropometric characteristics of X-linkedhypophosphatemia. Am J Med Genet A (2004) 126A(2):141–9. doi: 10.1002/ajmg.a.20572
109. Beck-Nielsen SS, Brusgaard K, Rasmussen LM, Brixen K, Brock-Jacobsen B, Poulsen MR, et al. Phenotype presentation of hypophosphatemic rickets inadults. Calcif Tissue Int (2010) 87(2):108–19. doi: 10.1007/s00223-010-9373-0
110. Gjorup H, Kjaer I, Sonnesen L, Haubek D, Beck-Nielsen SS, Hintze H, et al. Craniofacial morphology in patients with hypophosphatemic rickets: a cephalometric study focusing on differences between bone of cartilaginous and intramembranous origin. Am J Med Genet A (2011) 155A(11):2654–60. doi: 10.1002/ajmg.a.34242
111. Ichikawa S, Traxler EA, Estwick SA, Curry LR, Johnson ML, Sorenson AH, et al. Mutational survey of the PHEX gene in patients with X-linked hypophosphatemic rickets. Bone (2008) 43(4):663–6. doi: 10.1016/j.bone.2008.06.002
112. Mumm S, Huskey M, Cajic A, Wollberg V, Zhang F, Madson KL, et al. PHEX 3’-UTR c.*231A>G near the polyadenylation signal is a relatively common, mild, American mutation that masquerades as sporadic or X-linked recessive hypophosphatemic rickets. J Bone Miner Res (2015) 30(1):137–43. doi: 10.1002/jbmr.2307
113. Smith PS, Gottesman GS, Zhang F, Cook F, Ramirez B, Wenkert D, et al. X-Linked Hypophosphatemia: Uniquely Mild Disease Associated With PHEX 3’-UTR Mutation c.*231A>G (A Retrospective Case-Control Study). J Bone Miner Res (2020) 35(5):920–31. doi: 10.1002/jbmr.3955
114. Zheng B, Wang C, Chen Q, Che R, Sha Y, Zhao F, et al. Functional Characterization of PHEX Gene Variants in Children With X-Linked Hypophosphatemic Rickets Shows No Evidence of Genotype-Phenotype Correlation. J Bone Miner Res (2020) 35(9):1718–25. doi: 10.1002/jbmr.4035
115. Gaucher C, Walrant-Debray O, Nguyen TM, Esterle L, Garabedian M, Jehan F. PHEX analysis in 118 pedigrees reveals new genetic clues in hypophosphatemic rickets. Hum Genet (2009) 125(4):401–11. doi: 10.1007/s00439-009-0631-z
116. Beck-Nielsen SS, Brixen K, Gram J, Brusgaard K. Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and CLCN5 in patients with hypophosphatemic rickets. J Hum Genet (2012) 57(7):453–8. doi: 10.1038/jhg.2012.56
117. Capelli S, Donghi V, Maruca K, Vezzoli G, Corbetta S, Brandi ML, et al. Clinical and molecular heterogeneity in a large series of patients with hypophosphatemic rickets. Bone (2015) 79:143–9. doi: 10.1016/j.bone.2015.05.040
118. Christie PT, Harding B, Nesbit MA, Whyte MP, Thakker RV. X-linked hypophosphatemia attributable to pseudoexons of the PHEX gene. J Clin Endocrinol Metab (2001) 86(8):3840–4. doi: 10.1210/jcem.86.8.7730
119. Thiele S, Werner R, Stubbe A, Hiort O, Hoeppner W. Validation of a next-generation sequencing (NGS) panel to improve the diagnosis of X-linked hypophosphataemia (XLH) and other genetic disorders of renal phosphate wasting. Eur J Endocrinol (2020) 183(5):497–504. doi: 10.1530/EJE-20-0275
120. Lin Y, Xu J, Li X, Sheng H, Su L, Wu M, et al. Novel variants and uncommon cases among southern Chinese children with X-linked hypophosphatemia. J Endocrinol Invest (2020) 43(11):1577–90. doi: 10.1007/s40618-020-01240-6
121. Ruppe MD, Brosnan PG, Au KS, Tran PX, Dominguez BW, Northrup H. Mutational analysis of PHEX, FGF23 and DMP1 in a cohort of patients with hypophosphatemic rickets. Clin Endocrinol (Oxf) (2011) 74(3):312–8. doi: 10.1111/j.1365-2265.2010.03919.x
122. Kinoshita Y, Saito T, Shimizu Y, Hori M, Taguchi M, Igarashi T, et al. Mutational analysis of patients with FGF23-related hypophosphatemic rickets. Eur J Endocrinol (2012) 167(2):165–72. doi: 10.1530/EJE-12-0071
123. Raimann A, Mindler GT, Kocijan R, Bekes K, Zwerina J, Haeusler G, et al. Multidisciplinary patient care in X-linked hypophosphatemic rickets: one challenge, many perspectives. Wien Med Wochenschr (2020) 170(5-6):116–23. doi: 10.1007/s10354-019-00732-2
124. Lo SH, Lachmann R, Williams A, Piglowska N, Lloyd AJ. Exploring the burden of X-linked hypophosphatemia: a European multi-country qualitative study. Qual Life Res (2020) 29(7):1883–93. doi: 10.1007/s11136-020-02465-x
125. Pecourneau V, Degboe Y, Barnetche T, Cantagrel A, Constantin A, Ruyssen-Witrand A. Effectiveness of Exercise Programs in Ankylosing Spondylitis: A Meta-Analysis of Randomized Controlled Trials. Arch Phys Med Rehabil (2018) 99(2):383–9 e1. doi: 10.1016/j.apmr.2017.07.015
126. Millner JR, Barron JS, Beinke KM, Butterworth RH, Chasle BE, Dutton LJ, et al. Exercise for ankylosing spondylitis: An evidence-based consensus statement. Semin Arthritis Rheum (2016) 45(4):411–27. doi: 10.1016/j.semarthrit.2015.08.003
127. Rausch Osthoff AK, Niedermann K, Braun J, Adams J, Brodin N, Dagfinrud H, et al. 2018 EULAR recommendations for physical activity in people with inflammatory arthritis and osteoarthritis. Ann Rheum Dis (2018) 77(9):1251–60. doi: 10.1136/annrheumdis-2018-213585
128. Steele A, Gonzalez R, Garbalosa JC, Steigbigel K, Grgurich T, Parisi EJ, et al. Osteoarthritis, Osteophytes, and Enthesophytes Affect BiomechanicalFunction in Adults With X-linked Hypophosphatemia. J Clin EndocrinolMetab (2020) 105(4):e1798–814. doi: 10.1210/clinem/dgaa064
129. RIZIV/INAMI. Voor welke zware aandoeningen geniet uw patiënt van een voorkeurstarief voor zijn kinesitherapie behandeling? (2020). Available at: https://www.riziv.fgov.be/nl/professionals/individuelezorgverleners/kinesitherapeuten/Paginas/zware-aandoeningen-lijst-minder-betalen.aspx.
130. Seikaly MG, Waber PG, Baum M. Urinary prostaglandins and the effect of indomethacin on phosphate excretion in children with hypophosphatemic rickets. Pediatr Res (2008) 64(2):210–2. doi: 10.1203/PDR.0b013e318175d788
131. Mäkitie O, Doria A, Kooh SW, Cole WG, Daneman A, Sochett E. Early treatment improves growth and biochemical and radiographic outcome in X-linked hypophosphatemic rickets. J Clin Endocrinol Metab (2003) 88(8):3591–7. doi: 10.1210/jc.2003-030036
132. Chaussain-Miller C, Sinding C, Wolikow M, Lasfargues JJ, Godeau G, Garabedian M. Dental abnormalities in patients with familial hypophosphatemic vitamin D-resistant rickets: prevention by early treatment with 1-hydroxyvitamin D. J Pediatr (2003) 142(3):324–31. doi: 10.1067/mpd.2003.119
133. Chaussain-Miller C, Sinding C, Septier D, Wolikow M, Goldberg M, Garabedian M. Dentin structure in familial hypophosphatemic rickets: benefits of vitamin D and phosphate treatment. Oral Dis (2007) 13(5):482–9. doi: 10.1111/j.1601-0825.2006.01326.x
134. Biosse Duplan M, Coyac BR, Bardet C, Zadikian C, Rothenbuhler A, Kamenicky P, et al. Phosphate and Vitamin D Prevent Periodontitis in X-Linked Hypophosphatemia. J Dent Res (2017) 96(4):388–95. doi: 10.1177/0022034516677528
135. Connor J, Olear EA, Insogna KL, Katz L, Baker S, Kaur R, et al. Conventional Therapy in Adults With X-Linked Hypophosphatemia: Effects on Enthesopathy and Dental Disease. J Clin Endocrinol Metab (2015) 100(10):3625–32. doi: 10.1210/JC.2015-2199
136. Sullivan W, Carpenter T, Glorieux F, Travers R, Insogna K. A prospective trial of phosphate and 1,25-dihydroxyvitamin D3 therapy in symptomatic adults with X-linked hypophosphatemic rickets. J Clin Endocrinol Metab (1992) 75(3):879–85. doi: 10.1210/jcem.75.3.1517380
137. Suzuki E, Yamada M, Ariyasu D, Izawa M, Miyamoto J, Koto S, et al. Patients with Hypophosphatemic Osteomalacia Need Continuous Treatment during Adulthood. Clin Pediatr Endocrinol (2009) 18(1):29–33. doi: 10.1297/cpe.18.29
138. Glorieux FH, Marie PJ, Pettifor JM, Delvin EE. Bone response to phosphate salts, ergocalciferol, and calcitriol in hypophosphatemic vitamin D-resistant rickets. N Engl J Med (1980) 303(18):1023–31. doi: 10.1056/NEJM198010303031802
139. Harrell RM, Lyles KW, Harrelson JM, Friedman NE, Drezner MK. Healing of bone disease in X-linked hypophosphatemic rickets/osteomalacia. Induction and maintenance with phosphorus and calcitriol. J Clin Invest (1985) 75(6):1858–68. doi: 10.1172/JCI111900
140. Costa T, Marie PJ, Scriver CR, Cole DE, Reade TM, Nogrady B, et al. X-linked hypophosphatemia: effect of calcitriol on renal handling of phosphate, serum phosphate, and bone mineralization. J Clin Endocrinol Metab (1981) 52(3):463–72. doi: 10.1210/jcem-52-3-463
141. Martins JS, Liu ES, Sneddon WB, Friedman PA, Demay MB. 1,25-Dihydroxyvitamin D Maintains Brush Border Membrane NaPi2a and Attenuates Phosphaturia in Hyp Mice. Endocrinology (2019) 160(10):2204–14. doi: 10.1210/en.2019-00186
142. Liu ES, Martins JS, Raimann A, Chae BT, Brooks DJ, Jorgetti V, et al. 1,25-Dihydroxyvitamin D Alone Improves Skeletal Growth, Microarchitecture, and Strength in a Murine Model of XLH, Despite Enhanced FGF23 Expression. J Bone Miner Res (2016) 31(5):929–39. doi: 10.1002/jbmr.2783
143. Barratt KR, Sawyer RK, Atkins GJ, St-Arnaud R, Anderson PH. Vitamin D supplementation improves bone mineralisation independent of dietary phosphate in male X-linked hypophosphatemic (Hyp) mice. Bone (2020) 115767. doi: 10.1016/j.bone.2020.115767
144. Verge CF, Lam A, Simpson JM, Cowell CT, Howard NJ, Silink M. Effects of therapy in X-linked hypophosphatemic rickets. N Engl J Med (1991) 325(26):1843–8. doi: 10.1056/NEJM199112263252604
145. Reid IR, Hardy DC, Murphy WA, Teitelbaum SL, Bergfeld MA, Whyte MP. X-linked hypophosphatemia: a clinical, biochemical, and histopathologic assessment of morbidity in adults. Med (Baltimore) (1989) 68(6):336–52. doi: 10.1097/00005792-198911000-00002
146. Reade TM, Scriver CR. Hypophosphatemic rickets and breast milk. N Engl J Med (1979) 300(24):1397. doi: 10.1056/NEJM197906143002415
147. Jonas AJ, Dominguez B. Low breast milk phosphorus concentration in familial hypophosphatemia. J Pediatr Gastroenterol Nutr (1989) 8(4):541–3. doi: 10.1097/00005176-198905000-00021
148. Ma Y, Samaraweera M, Cooke-Hubley S, Kirby BJ, Karaplis AC, Lanske B, et al. Neither absence nor excess of FGF23 disturbs murine fetal-placental phosphorus homeostasis or prenatal skeletal development and mineralization. Endocrinology (2014) 155(5):1596–605. doi: 10.1210/en.2013-2061
149. Delzer PR, Meyer RA Jr. Normal milk composition in lactating X-linked hypophosphatemic mice despite continued hypophosphatemia. Calcif Tissue Int (1983) 35(6):750–4. doi: 10.1007/BF02405118
150. Carpenter TO, Imel EA, Ruppe MD, Weber TJ, Klausner MA, Wooddell MM, et al. Randomized trial of the anti-FGF23 antibody KRN23 in X-linked hypophosphatemia. J Clin Invest (2014) 124(4):1587–97. doi: 10.1172/JCI72829
151. Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N Engl J Med (2018) 378(21):1987–98. doi: 10.1056/NEJMoa1714641
152. Whyte MP, Carpenter TO, Gottesman GS, Mao M, Skrinar A, San Martin J, et al. Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: a multicentre, open-label, phase 2 trial. Lancet Diabetes Endocrinol (2019) 7(3):189–99. doi: 10.1016/S2213-8587(18)30338-3
153. Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet (2019) 393(10189):2416–27. doi: 10.1016/S0140-6736(19)30654-3
154. Padidela R, Whyte MP, Glorieux FH, Munns CF, Ward LM, Nilsson O, et al. Patient-Reported Outcomes from a Randomized, Active-Controlled, Open-Label, Phase 3 Trial of Burosumab Versus Conventional Therapy in Children with X-Linked Hypophosphatemia. Calcif Tissue Int (2021). doi: 10.1007/s00223-020-00797-x
155. Insogna KL, Briot K, Imel EA, Kamenicky P, Ruppe MD, Portale AA, et al. A Randomized, Double-Blind, Placebo-Controlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J Bone Miner Res (2018) 33(8):1383–93. doi: 10.1002/jbmr.3475
156. Portale AA, Carpenter TO, Brandi ML, Briot K, Cheong HI, Cohen-Solal M, et al. Continued Beneficial Effects of Burosumab in Adults with X-Linked Hypophosphatemia: Results from a 24-Week Treatment Continuation Period After a 24-Week Double-Blind Placebo-Controlled Period. Calcif Tissue Int (2019) 105(3):271–84. doi: 10.1007/s00223-019-00568-3
157. Insogna KL, Rauch F, Kamenicky P, Ito N, Kubota T, Nakamura A, et al. Burosumab Improved Histomorphometric Measures of Osteomalacia in Adults with X-Linked Hypophosphatemia: A Phase 3, Single-Arm, International Trial. J Bone Miner Res (2019) 34(12):2183–91. doi: 10.1002/jbmr.3843
158. Zivicnjak M, Schnabel D, Staude H, Even G, Marx M, Beetz R, et al. Three-year growth hormone treatment in short children with X-linked hypophosphatemic rickets: effects on linear growth and body disproportion. J Clin Endocrinol Metab (2011) 96(12):E2097–105. doi: 10.1210/jc.2011-0399
159. Baroncelli GI, Bertelloni S, Ceccarelli C, Saggese G. Effect of growth hormone treatment on final height, phosphate metabolism, and bone mineral density in children with X-linked hypophosphatemic rickets. J Pediatr (2001) 138(2):236–43. doi: 10.1067/mpd.2001.108955
160. Mäkitie O, Toiviainen-Salo S, Marttinen E, Kaitila I, Sochett E, Sipila I. Metabolic control and growth during exclusive growth hormone treatment in X-linked hypophosphatemic rickets. Horm Res (2008) 69(4):212–20. doi: 10.1159/000113021
161. Meyerhoff N, Haffner D, Staude H, Wuhl E, Marx M, Beetz R, et al. Effects of growth hormone treatment on adult height in severely short children with X-linked hypophosphatemic rickets. Pediatr Nephrol (2018) 33(3):447–56. doi: 10.1007/s00467-017-3820-3
162. Rothenbuhler A, Esterle L, Gueorguieva I, Salles JP, Mignot B, Colle M, et al. Two-year recombinant human growth hormone (rhGH) treatment is more effective in pre-pubertal compared to pubertal short children with X-linked hypophosphatemic rickets (XLHR). Growth Horm IGF Res (2017) 36:11–5. doi: 10.1016/j.ghir.2017.08.001
163. Linglart A, Biosse-Duplan M, Briot K, Chaussain C, Esterle L, Guillaume-Czitrom S, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect (2014) 3(1):R13–30. doi: 10.1530/EC-13-0103
164. Saggese G, Baroncelli GI, Bertelloni S, Perri G. Long-term growth hormone treatment in children with renal hypophosphatemic rickets: effects on growth, mineral metabolism, and bone density. J Pediatr (1995) 127(3):395–402. doi: 10.1016/S0022-3476(95)70070-6
165. Alon US, Levy-Olomucki R, Moore WV, Stubbs J, Liu S, Quarles LD. Calcimimetics as an adjuvant treatment for familial hypophosphatemic rickets. Clin J Am Soc Nephrol (2008) 3(3):658–64. doi: 10.2215/CJN.04981107
166. Raeder H, Shaw N, Netelenbos C, Bjerknes R. A case of X-linked hypophosphatemic rickets: complications and the therapeutic use of cinacalcet. Eur J Endocrinol (2008) 159(Suppl 1):S101–5. doi: 10.1530/EJE-08-0383
167. Seikaly MG, Baum M. Thiazide diuretics arrest the progression of nephrocalcinosis in children with X-linked hypophosphatemia. Pediatrics (2001) 108(1):E6. doi: 10.1542/peds.108.1.e6
168. Seikaly MG, Quigley R, Baum M. Effect of dipyridamole on serum and urinary phosphate in X-linked hypophosphatemia. Pediatr Nephrol (2000) 15(1-2):57–9. doi: 10.1007/s004670000425
169. Carpenter KA, Ross RD. Sclerostin Antibody Treatment Increases Bone Mass and Normalizes Circulating Phosphate Levels in Growing Hyp Mice. J Bone Miner Res (2020) 35(3):596–607. doi: 10.1002/jbmr.3923
170. Hoac B, Ostergaard M, Wittig NK, Boukpessi T, Buss DJ, Chaussain C, et al. Genetic Ablation of Osteopontin in Osteomalacic Hyp Mice PartiallyRescues the Deficient Mineralization Without Correcting Hypophosphatemia. J Bone Miner Res (2020) 35(10):2032–48. doi: 10.1002/jbmr.4101
171. Fratzl-Zelman N, Gamsjaeger S, Blouin S, Kocijan R, Plasenzotti P, Rokidi S, et al. Alterations of bone material properties in adult patients with X-linked hypophosphatemia (XLH). J Struct Biol (2020) 211(3):107556. doi: 10.1016/j.jsb.2020.107556
172. Cundy T, Que L, Hassan IM, Hughes L. Bisphosphonate-Induced Deterioration of Osteomalacia in Undiagnosed Adult Fanconi Syndrome. JBMR Plus (2020) 4(8):e10374. doi: 10.1002/jbm4.10374
173. Li J, Rai S, Ze R, Tang X, Liu R, Hong P. Rotational and translational osteotomy for treatment of severe deformity in hypophosphatemic rickets: A case report. Med (Baltimore) (2020) 99(3):e18425. doi: 10.1097/MD.0000000000018425
174. Sharkey MS, Grunseich K, Carpenter TO. Contemporary Medical and Surgical Management of X-linked Hypophosphatemic Rickets. J Am Acad Orthop Surg (2015) 23(7):433–42. doi: 10.5435/JAAOS-D-14-00082
175. Horn A, Wright J, Bockenhauer D, Van’t Hoff W, Eastwood DM. The orthopaedic management of lower limb deformity in hypophosphataemic rickets. J Child Orthop (2017) 11(4):298–305. doi: 10.1302/1863-2548.11.170003
176. Popkov A, Aranovich A, Popkov D. Results of deformity correction in children with X-linked hereditary hypophosphatemic rickets by external fixation or combined technique. Int Orthop (2015) 39(12):2423–31. doi: 10.1007/s00264-015-2814-7
177. Gizard A, Rothenbuhler A, Pejin Z, Finidori G, Glorion C, de Billy B, et al. Outcomes of orthopedic surgery in a cohort of 49 patients with X-linked hypophosphatemic rickets (XLHR). Endocr Connect (2017) 6(8):566–73. doi: 10.1530/EC-17-0154
178. Vega RA, Opalak C, Harshbarger RJ, Fearon JA, Ritter AM, Collins JJ, et al. Hypophosphatemic rickets and craniosynostosis: a multicenter case series. J Neurosurg Pediatr (2016) 17(6):694–700. doi: 10.3171/2015.10.PEDS15273
179. Rothenbuhler A, Fadel N, Debza Y, Bacchetta J, Diallo MT, Adamsbaum C, et al. High Incidence of Cranial Synostosis and Chiari I Malformation in Children With X-Linked Hypophosphatemic Rickets (XLHR). J Bone Miner Res (2019) 34(3):490–6. doi: 10.1002/jbmr.3614
180. Soehle M, Casey AT. Cervical spinal cord compression attributable to a calcified intervertebral disc in a patient with X-linked hypophosphatemic rickets: case report and review of the literature. Neurosurgery (2002) 51(1):239–42; discussion 42-3. doi: 10.1097/00006123-200207000-00038
181. Shiba M, Mizuno M, Kuraishi K, Suzuki H. Cervical ossification of posterior longitudinal ligament in x-linked hypophosphatemic rickets revealing homogeneously increased vertebral bone density. Asian Spine J (2015) 9(1):106–9. doi: 10.4184/asj.2015.9.1.106
182. Riccio AR, Entezami P, Giuffrida A, Dowling J, Forrest G, German JW. Minimally Invasive Surgical Management of Thoracic Ossification of the Ligamentum Flavum Associated with X-linked Hypophosphatemia. World Neurosurg (2016) 94:580 e5– e10. doi: 10.1016/j.wneu.2016.07.076
183. Nakagawa K, Galati AN. Ultra-early onset of ossification of the posterior longitudinal ligament with congenital rickets. Neurology (2016) 87(16):1738–40. doi: 10.1212/WNL.0000000000003237
184. Mäkitie O, Kooh SW, Sochett E. Prolonged high-dose phosphate treatment: a risk factor for tertiary hyperparathyroidism in X-linked hypophosphatemic rickets. Clin Endocrinol (Oxf) (2003) 58(2):163–8. doi: 10.1046/j.1365-2265.2003.01685.x
185. Savio RM, Gosnell JE, Posen S, Reeve TS, Delbridge LW. Parathyroidectomy for tertiary hyperparathyroidism associated withX-linked dominant hypophosphatemic rickets. Arch Surg (2004) 139(2):218–22. doi: 10.1001/archsurg.139.2.218
Keywords: burosumab, fibroblast growth factor 23 (FGF23), osteomalacia, rickets, vitamin D, X-linked hypophosphatemia
Citation: Laurent MR, De Schepper J, Trouet D, Godefroid N, Boros E, Heinrichs C, Bravenboer B, Velkeniers B, Lammens J, Harvengt P, Cavalier E, Kaux J-F, Lombet J, De Waele K, Verroken C, van Hoeck K, Mortier GR, Levtchenko E and Vande Walle J (2021) Consensus Recommendations for the Diagnosis and Management of X-Linked Hypophosphatemia in Belgium. Front. Endocrinol. 12:641543. doi: 10.3389/fendo.2021.641543
Received: 14 December 2020; Accepted: 19 February 2021;
Published: 19 March 2021.
Edited by:
Gudrun Stenbeck, Brunel University London, United KingdomReviewed by:
Alberto Falchetti, Istituto Auxologico Italiano (IRCCS), ItalyMelissa Orlandin Premaor, Federal University of Minas Gerais, Brazil
Copyright © 2021 Laurent, De Schepper, Trouet, Godefroid, Boros, Heinrichs, Bravenboer, Velkeniers, Lammens, Harvengt, Cavalier, Kaux, Lombet, De Waele, Verroken, van Hoeck, Mortier, Levtchenko and Vande Walle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Michaël R. Laurent, michael.laurent@uzleuven.be
†These authors share senior authorship