 Fatemeh Nasiri1,2
Fatemeh Nasiri1,2 Mehrasa Kazemi3†Seyed Mohamad Javad Mirarefin4†
Mehrasa Kazemi3†Seyed Mohamad Javad Mirarefin4† Maral Mahboubi Kancha5Milad Ahmadi Najafabadi1
Maral Mahboubi Kancha5Milad Ahmadi Najafabadi1 Faeze Salem1
Faeze Salem1 Setareh Dashti Shokoohi1
Setareh Dashti Shokoohi1 Sahar Evazi Bakhshi6
Sahar Evazi Bakhshi6 Pouya Safarzadeh Kozani7
Pouya Safarzadeh Kozani7 Pooria Safarzadeh Kozani1*
Pooria Safarzadeh Kozani1*- 1Department of Medical Biotechnology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
- 2Department of Production Platforms & Analytics, Human Health Therapeutics Research Centre, National Research Council Canada, Montreal, QC, Canada
- 3Department of Laboratory Medicine, Thalassemia Research Center, Hemoglobinopathy Institute, Mazandaran University of Medical Sciences, Sari, Iran
- 4Department of Immunology, Faculty of Medical Sciences, Tarbiat Modares University, Tehran, Iran
- 5Department of Medical Nanotechnology, School of Medicine, Shahroud University of Medical Sciences, Shahroud, Iran
- 6Department of Anatomical Sciences, School of Medicine, Guilan University of Medical Sciences, Rasht, Iran
- 7Department of Medical Biotechnology, Faculty of Paramedicine, Guilan University of Medical Sciences, Rasht, Iran
Triple-negative breast cancer (TNBC) is known as the most intricate and hard-to-treat subtype of breast cancer. TNBC cells do not express the well-known estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 (HER2) expressed by other breast cancer subtypes. This phenomenon leaves no room for novel treatment approaches including endocrine and HER2-specific antibody therapies. To date, surgery, radiotherapy, and systemic chemotherapy remain the principal therapy options for TNBC treatment. However, in numerous cases, these approaches either result in minimal clinical benefit or are nonfunctional, resulting in disease recurrence and poor prognosis. Nowadays, chimeric antigen receptor T cell (CAR-T) therapy is becoming more established as an option for the treatment of various types of hematologic malignancies. CAR-Ts are genetically engineered T lymphocytes that employ the body’s immune system mechanisms to selectively recognize cancer cells expressing tumor-associated antigens (TAAs) of interest and efficiently eliminate them. However, despite the clinical triumph of CAR-T therapy in hematologic neoplasms, CAR-T therapy of solid tumors, including TNBC, has been much more challenging. In this review, we will discuss the success of CAR-T therapy in hematological neoplasms and its caveats in solid tumors, and then we summarize the potential CAR-T targetable TAAs in TNBC studied in different investigational stages.
Introduction
Breast cancer is the malignancy arising from the breast tissues. It is the most common malignancy worldwide and is estimated to be detected in more than 280,000 women in the United States in 2022 (1). According to the estimations, more than 43,000 women will die because of breast cancer only in 2022 (1). The molecular process that leads to the emergence of breast cancer entails the active proliferation of epithelial cells of the breast tissue resulting in the formation of malignant cells in the ductal or lobular compartment of the breast (2, 3). There are various types of classifications for categorizing breast cancer. Histologic grade, disease stage, and expression of classic hormone and growth factor receptors are three well-known criteria used for breast cancer classification and determining the aggressive capacity of a breast tumor (2–7). The hormone and growth factor receptors employed for breast cancer classification include estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) (2–7). Based on the expression or lack of expression of these receptors and the expression rate of Ki-67, there are four subtypes of breast cancer including Luminal A, Luminal B, HER2 amplified, and triple-negative breast cancer (TNBC) (2–7). The first three subtypes are easier to manage since the patients with these subtypes are eligible for antibody-based therapies such as anti-HER2 monoclonal antibodies (mAbs) or antibody-drug conjugates (ADCs), tyrosine kinase inhibitors, and endocrine therapies (including ER degraders) (8–10). Dissimilar from these subtypes, TNBC is a heterogeneous type of basal-like tumor that does not express the abovementioned receptors and it represents about 15 to 20% of all breast cancer cases (11).
Due to the unresponsiveness of TNBC patients to anti-HER2 mAb-based and endocrine therapies, traditional anticancer treatments including surgery, radiotherapy, and chemotherapy continue to be the commonly available therapies for these patients (12, 13). Of note, these traditional cancer treatment modalities manage to eradicate malignant cells in a nonspecific manner resulting in mild to severe treatment-related side effects (13). Additionally, poly (ADP-ribose) polymerase (PARP) inhibitors are a class of pharmacological inhibitors that have been approved by the US Food and Drug Administration (FDA) for the treatment of TNBCs with BRCA mutations (14). Recently, many bodies of research have indicated that the immune system pathways are remarkably involved in the emergence and progression of TNBC (15, 16). In this case, researchers have utilized immune checkpoint blockade therapies such as programmed death-1 (PD-1)- or programmed death-ligand 1 (PD-L1)-specific antibodies for suppressing the outgrowth of TNBC (16, 17). Moreover, T-cell-redirecting bispecific antibodies (TRBAs) have also been under investigation as candidates for the treatment of TNBC (16, 17). Recently, the US FDA approved the PD-L1-specific mAb atezolizumab in combination with the chemotherapeutic agent nab-paclitaxel for the treatment of patients with PD-L1-proficient unresectable TNBC (18). Such progress in the field of TNBC immunotherapy has encouraged researchers to employ other types of cancer immunotherapy such as cancer vaccines and adoptive cell therapy using genetically engineered immune cells for a more selective and successful fight against TNBC (16, 19, 20).
The importance of cancer-specific immune cells in TNBC was elucidated when researchers discovered that the presence of tumor-infiltrating lymphocytes (TILs) in residual disease following neoadjuvant chemotherapy is correlated with superior prognosis in TNBC patients (21). In a population of TILs, there are tumor-reactive T cells that recognize cancer-specific antigens and are responsible for the antitumor activity of a TIL population (22, 23). However, there are several limitations in regards to the application of TILs for adoptive cell therapy of various types of tumors. These caveats include the exhausted phenotype of TILs resulting from multiple target antigen encountering, and difficulties in regards to the isolation and ex vivo expansion of TILs (24, 25). Genetically engineered T cells expressing chimeric antigen receptors (CAR-Ts) have recently changed the face of immune cell-based cancer immunotherapy (26, 27). CAR-Ts possess antibody-derived targeting domains linked to T-cell activating domains which grant them the ability to recognize cancer cells via target cell membrane-expressed antigens independent of major histocompatibility complexes (MHCs) (26, 27). Moreover, CAR-Ts can be easily expanded to clinically relevant scales; therefore, they are considered an ideal alternative to TILs (26, 27). The major purpose of this review is to discuss CAR-T therapy hurdles in solid tumors with a specific focus on TNBC, shine a light on the crucial stratagems to tackle these caveats, and discuss the potential and promising target antigens under investigation in various stages, from early developmental stages to clinical settings, for CAR-T therapy of TNBC (Figure 1).
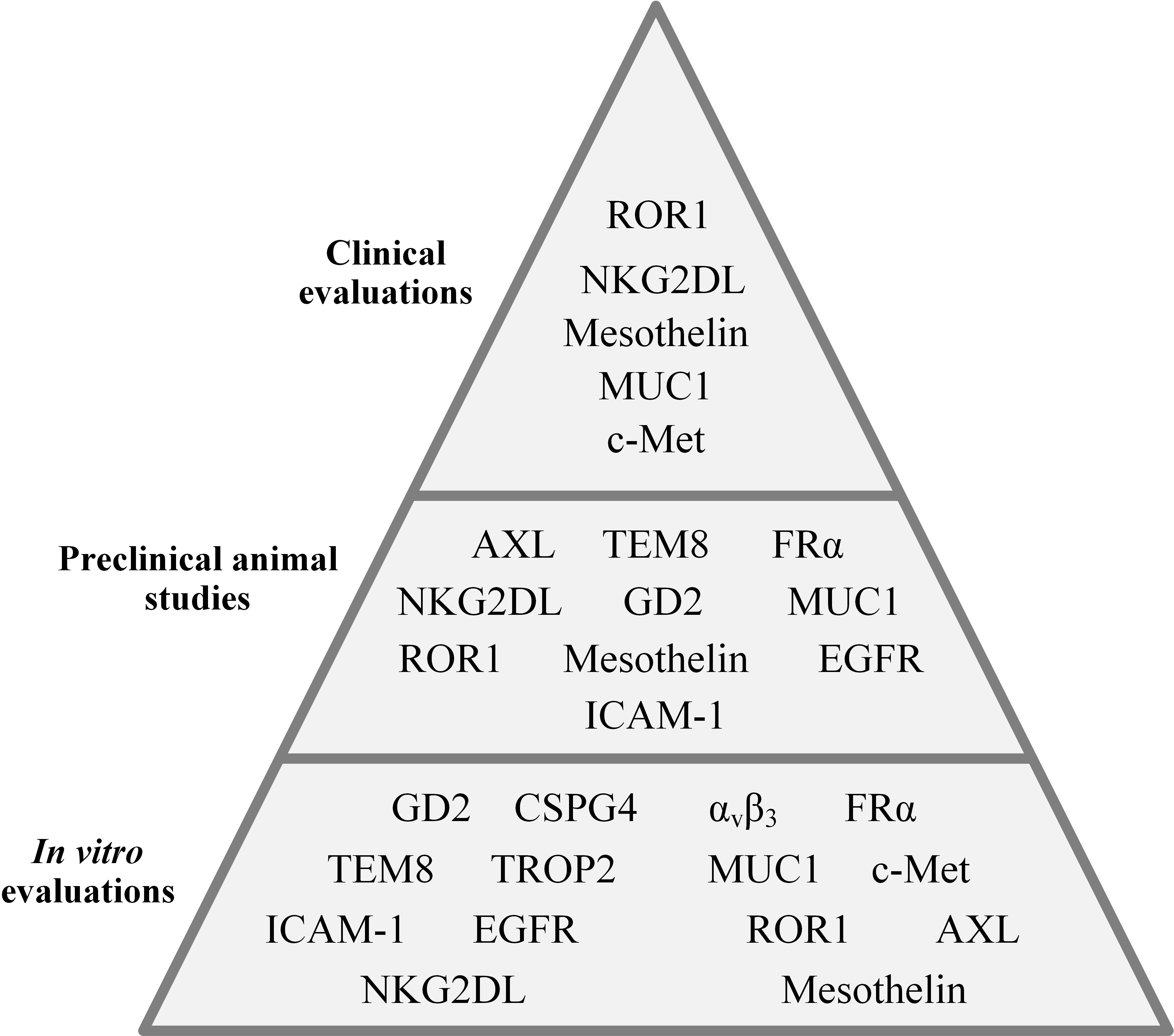
Figure 1 An overall presentation of the CAR-T-targeted TNBC-associated antigens evaluated in different investigational stages.
CAR-T therapy fundamentals
About twenty years ago, when a person was diagnosed with cancer, surgery, chemotherapy, and radiotherapy were the only available treatment options. However, these treatment modalities could not mediate complete disease remission in most cases (28–30). Additionally, disease relapse, shortly after the completion of the treatment, was always a challenge (28–30). Alongside the mentioned limitations, the severity of the treatment-related side effects was another factor affecting the patients’ general well-being (28–30). Years later, with the emergence of immunotherapy and the development of novel treatment modalities, cancer treatment became more selective and efficient resulting in prolonged survival and reduced adverse events in patients.
CAR-Ts are the ultimate result of years of research and experience in various aspects of immunotherapy and cell therapy. The process of CAR-T generation starts with obtaining peripheral blood mononuclear cells (PBMCs) from patients (in the case of autologous CAR-T therapy) or third-party healthy donors (in the case of allogeneic CAR-T therapy) (31, 32). From the population of the isolated PBMCs, T lymphocytes are isolated. These T cells undergo activation and genetic manipulation steps, usually using retroviruses, to express a synthetic chimeric antigen receptor (CAR) on their surface (32). After expansion to a number of cells required for clinical applications, the engineered T cells are infused into recipients. It is worth mentioning that before CAR-T administration, patients usually undergo lymphodepletion chemotherapy (32).
A CAR construct is made of three important sections. An extracellular domain, a transmembrane domain, and an endodomain (32). The ectodomain of CARs is derived from a tumor antigen-specific fragment of a mAb such as a single-chain fragment variable (scFv) or single variable domain on a heavy chain (VHH) (32, 33). This targeting domain is linked to a flexible spacer, called a hinge, acting as a connector between the ectodomain and the transmembrane domain (32). Recently, it has been demonstrated that both hinge and transmembrane domains have remarkable impacts on CAR expression and signaling (34–36). In detail, the CAR expression rate on T cells and its stability are highly impacted by the type of the transmembrane domain (rather than hinge) (34). The CAR transmembrane domain can control the extent of CAR signaling by regulating the level of CAR surface expression (34). On the other hand, the hinge domain can control the membrane transport efficiency of CARs in T cells as well as the modality of CAR expression (34). Muller et al. demonstrated that CAR constructs with CD28 (but not CD8) as the transmembrane domain can form heterodimers with the endogenous CD28 present in human T cells which can result in more robust signal transductions enhancing CAR-T activation in the context of confronting low levels of a particular target antigen (35). They added that the CD28 transmembrane domain is capable of regulating various CAR-T functionalities by employing endogenous pathways (35). Additionally, June et al. reported that CAR-Ts with CD28 transmembrane domain exhibited prolonged in vitro expansion (up to 3 months) after a single TCR stimulation and without any IL-2 supplementation (37). Other researchers have added that CAR-Ts whose hinge and transmembrane domain are both based on CD28 are capable of producing target antigen-dependent inflammatory cytokines more than CAR-Ts whose hinge and transmembrane domain are both based on CD8 (38). Moreover, it has been demonstrated that these CAR-Ts require a lower antigen density for activation (studied only in CD19-redirected CAR-Ts with 4-1BB as the co-stimulatory domain) (39). Conclusively, broader investigation into the impact of CAR hinge and transmembrane domains on the persistence, tumoricidal efficacy, and phenotypic characteristics of CAR-Ts can further help the development of CAR-Ts with greater therapeutic benefits.
The endodomain of CARs is responsible for activating the effector cell upon target antigen encountering. This part of CARs has a CD3ζ signaling domain derived from the CD3 complex of the T-cell receptor (TCR) fused to intracellular costimulatory domains such as CD27, CD28, 4–1BB, OX40, and/or ICOS (32). Of note, dissimilar from first-generation CAR-Ts which did not possess any intracellular costimulatory domains, second- and third-generation CAR-Ts have one and two intracellular costimulatory domains, respectively (40–43). According to scientific evidence, the addition of intracellular costimulatory domains to the construct of CARs resulted in superior post-infusion expansion and persistence of second- and third-generation CAR-Ts in comparison with their first-generation counterparts (40–43). Recently, the construct of CARs has been further modified to achieve certain aims during CAR-T therapy. For instance, fourth-generation CAR-Ts possess an intracellular expression inducer of a cytokine of interest resulting in CAR-T-mediated tumor site delivery of a cytokine of interest leading to more enhanced and safer antitumor responses, especially in solid tumor CAR-T therapy (44–46). In detail, one of the well-known characteristics of solid tumors is the heterogeneity of tumor cells. Various populations of tumor cells might express different antigens in which case a single type of CAR-Ts redirected against a particular type of target antigen may not be clinically beneficial. However, expanding the antitumor responses by triggering the immune responses by other types of endogenous immune cells can help mediate more efficient tumoricidal reactions. In this regard, CAR-Ts have been engineered to specifically deliver a transgenic product, which can be chemokines or cytokines, to the targeted tumor sites (44, 46, 47). In detail, Chmielewski et al. generated CAR-Ts harboring an engineered intracellular expression inducer of IL-12 which can recruit macrophages (47). These CAR-Ts, which are also regarded as “T-cell redirected for universal cytokine-mediated killing (TRUCKs)” or “armored CAR-Ts”, are engineered to produce and release inducible IL-12 upon encountering CAR target antigen (47). This mechanism results in simultaneous antitumor attacks against both tumor cells expressing the CAR-redirected target antigen and tumor cells deficient in its expression (47). This strategy demonstrated that local delivery of IL-12 by CAR-Ts in the solid tumor tissues can be applied for targeting both target antigen-negative and hard-to-reach tumor cells through recruiting and activating innate immune components without the risk of systemic cytokine administration-related toxicities (47). Such CAR-Ts can be clinically valuable for targeting different target antigens of TNBC.
Moreover, fifth-generation CAR-Ts have also been developed which harbor an intracellular domain of a cytokine receptor, for example, interleukin (IL) 2 receptor subunit beta (IL-2Rβ) (44–46). In detail, proper activation and expansion of T cells are highly dependent on various signals including T-cell receptor (TCR), co-stimulatory, and cytokine-mediated signals (48). In the case of CAR-Ts, T cells can only benefit from TCR signaling which is provided through their CD3ζ domain and co-stimulatory domains (48). Therefore, researchers have generated CAR-Ts with novel constructs which are capable of inducing cytokine signaling once the effector cells encounter the target antigen (48). Fifth-generation CAR-Ts harbor truncated cytoplasmic domain of the IL-2Rβ and a STAT3-binding tyrosine-X-X-glutamine (YXXQ) motif alongside a primary activation domain and a co-stimulatory domain (48). Kagoya et al. reported that these CAR-Ts demonstrated target antigen-specific activation of the JAK kinase and the STAT3 and STAT5 transcription factor signaling pathways (48). These signaling pathways prevented terminal phenotypic differentiation of the effector cells and mediated their efficient expansion in vitro (48). Moreover, in vivo assessments proved that these CAR-Ts function efficiently in terms of persistence and tumoricidal reactions in comparison with their conventional counterparts, both in hematologic and solid tumors (48). Such novel CAR constructs can be evaluated in the case of TNBC CAR-T therapy to help achieve more efficient and less toxic antitumor responses. The constructs of different generations of CARs have been illustrated in Figure 2.
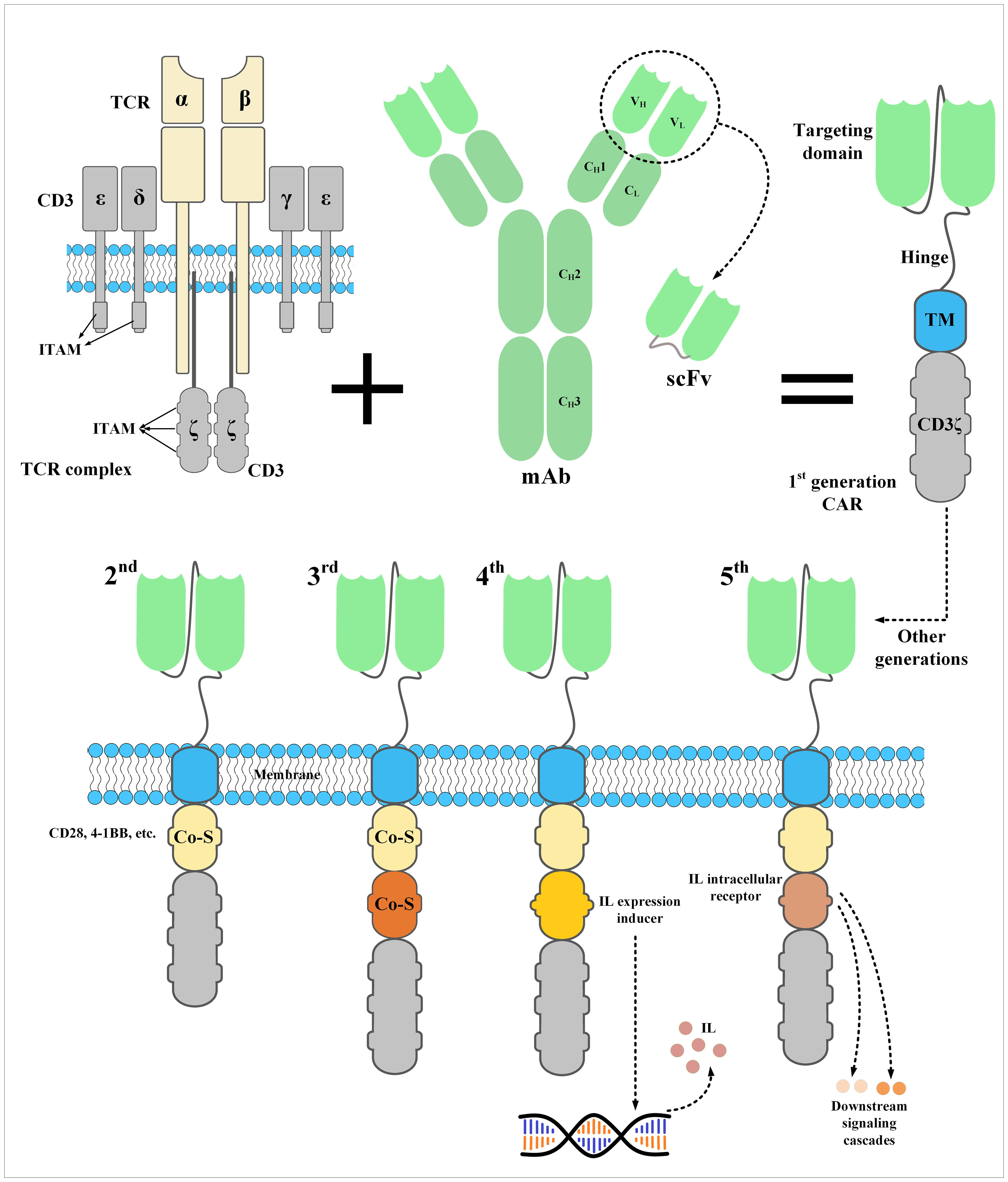
Figure 2 The structure of a CAR and its five generations. CARs are the result of meticulous protein engineering. The targeting domain of CARs is usually derived from the single-chain fragment variable (scFv) of a monoclonal antibody. scFvs are made from the variable light chain (VL) and variable heavy chain (VH) of a monoclonal antibody fused together through a synthetic linker peptide. A spacer called hinge connects the targeting domain of CARs to their transmembrane domain, which connects the ectodomain to the endodomain. Currently, the endodomain of CARs consists of one or two costimulatory domains and an activation domain. An interleukin expression inducer domain and an interleukin intracellular receptor are also located on the endodomain of the fourth- and fifth-generation CARs, respectively. Of note, the first-generation CARs lacked a costimulatory domain which led to their inadequate in vivo persistence and weak antitumor responses. CAR, chimeric antigen receptor; Co-S, costimulatory domain; IL, interleukin; ITAM, immunoreceptor tyrosine-based activation motif; mAb, monoclonal antibody; scFv, single-chain fragment variable; TCR, T-cell receptor; TM, transmembrane domain.
In regards to the application of scFvs as the targeting domain of CARs, it has been discovered that target antigen-independent CAR scFv aggregation occurs which is a result of CAR CD3ζ phosphorylation and/or the intrinsic instability of the VH and VL chains of the scFv (49). This phenomenon, known as “tonic signaling”, can mediate premature CAR-T activation (before encountering the intended target antigen) and render these effector cells exhausted leading to their impaired antitumor activity (49). Long et al. have demonstrated that the 4-1BB co-stimulatory domain in CAR-Ts better manages to minimize the phenotypic characteristics of CAR-T exhaustion mediated by the aggregation of CAR scFvs in comparison with CAR-Ts harboring CD28 as their co-stimulatory domains (49). These findings are consistent with the reports by Frigault and colleagues who reported that antigen-independent signaling cascades in CAR-T with CD28 as their co-stimulatory domains culminate in their exhaustion (based on animal studies) (37). Such findings might explain why CAR-Ts with the 4-1BB co-stimulatory domain demonstrate superior post-infusion persistence over their counterparts with the CD28 co-stimulatory domain (49).
Alongside the mentioned findings, other researchers have proposed the replacement of particular framework residues of the CAR scFv as an attempt to minimize the aggregation tendency of these scFvs and circumvent spontaneous tonic signaling (50). In detail, Landoni et al. employed in silico techniques to find the framework residues of a CSPG4-specific scFv (known as 763.74 scFv) that contributed to its stability and whose substations would result in decreasing the tendency of scFv aggregation, and consequently tonic signaling and CAR-T exhaustion (50). Moreover, the importance of scFv is also accentuated in regard to the on-target off-tumor effects of CAR-Ts. In the context of solid tumors, target antigens are usually expressed by healthy tissues as well (however, at physiological levels), which results in the CAR-T-mediated cytolytic reactions against healthy cells (51). Since the discovery of tumor-specific antigens is not always a feasible task, researchers have suggested targeting the tumor-specific glycoforms (called T, Tn, or sialyl Tn glycoforms) of commonly known TAAs (such as MUC1) (51). mAbs against such glycoforms are called cancer-specific mAbs (CasMabs) and scFvs derived from these CasMabs could be applied as the targeting domain of CAR-Ts for the development of cancer-specific CAR-Ts (Cas-CAR-Ts) (51). Such CAR-Ts have been developed and assessed in different investigational stages, and have proven to be efficient and safe (51). As another scFv-related strategy to minimize off-tumor toxicities, researchers have demonstrated that CAR-Ts equipped with targeting domains whose affinity towards their target antigen is moderate (micromolar affinity range), rather than high (nanomolar affinity range), manage to efficiently target tumor cells with high target antigen density while sparing normal cells which express the target antigen at a lower level (52). All of the mentioned strategies accentuate the importance of CAR targeting domains, particularly scFvs, and how their engineering can culminate in the development of safer and more efficient CAR-Ts (52).
CAR-T therapy challenges in solid tumors
The tremendous success of CAR-T therapy in relapsed/refractory (R/R) B-cell hematologic malignancies has rendered this type of cancer treatment approach a considerable treatment option (53–55). Based on such favorable clinical outcomes especially in patients non-responsive to the other types of treatments, the US FDA has approved six CAR-T products for the treatment of patients with failed previous lines of treatments. The list of the FDA-approved CAR-T products has been summarized in Table 1. However, patients with several hematologic malignancies, including T-cell neoplasms, as well as solid tumor patients have not yet benefited from the anticancer capability of this unique type of therapy (68–70). There are various limitations attributed to the inability of CAR-Ts to mediate tumor rejection and disease remission in solid tumors. In detail, CAR-Ts are redirected towards TAAs or TSAs (51, 71). Therefore, ideal CAR-T targets are antigens that are absent or have a low-level expression on the healthy cells of normal tissues but are overexpressed by malignant cells (51, 71). Targeting such target antigens can result in minimal unwanted off-tumor toxicities toward healthy tissues. However, finding target antigens with such characteristics is hardly feasible in solid tumors (51, 71). Additionally, another limitation of solid tumor CAR-T therapy is intratumor antigen heterogeneity described as tumor cells of a particular type of tumor expressing different levels of a CAR-redirected target antigen on their surface or not expressing the target antigen at all (72–74). This phenomenon results in impaired detection of malignant cells by CAR-Ts further leading to disease escape and relapse (72–74).
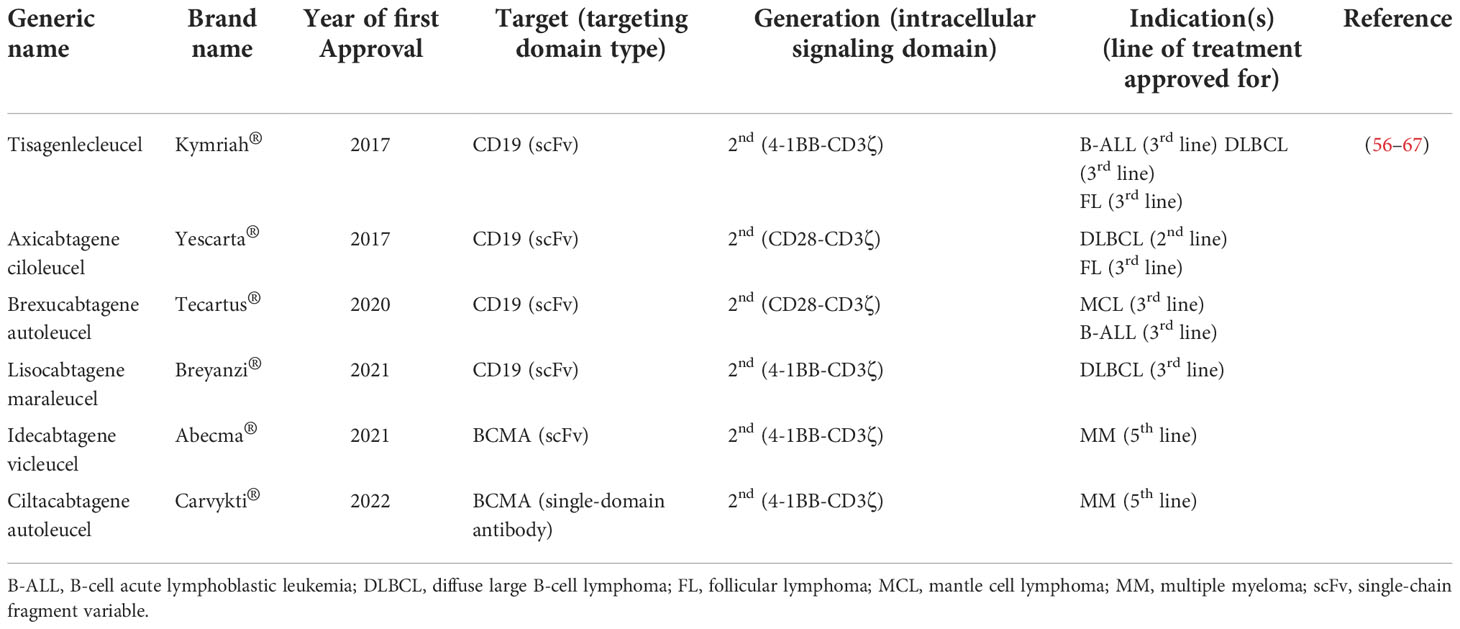
Table 1 A list of CAR-T products approved by the US FDA for the treatment of different hematologic malignancies.
Another limitation of CAR-T therapy in solid tumors is associated with the immunosuppressive tumor microenvironment (TME). The TME consists of various types of immune suppressor cells such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), cancer-associated fibroblasts (CAFs), and tumor-associated macrophages (TAMs) (75–78). There are also various suppressive factors such as chemokines, cytokines, and extracellular matrix (ECM) present in the TME. The immune suppressor cells residing in the TME are beneficial for tumors in terms of supporting their progression, angiogenesis, and metastasis by providing them with various types of growth factors, chemokines, and cytokines such as ILs, transforming growth factor beta (TGF-β), indoleamine 2,3-dioxygenase (IDO), and vascular endothelial growth factor (VEGF) (75–78). Moreover, the expression of immune checkpoint molecules (such as CTLA-4 and PD-1) on tumor-residing T lymphocytes is another factor that hampers antitumor reactions (79–82). Overall, the mentioned immunosuppressive factors in the TME remarkably suppress CAR-T-mediated antitumor reactions in solid tumor CAR-T therapy.
In addition to all of the abovementioned hurdles, the inadequate trafficking and infiltration of CAR-Ts into the tumor tissues is known as another major limitation impairing the functionality and tumoricidal activity of CAR-Ts (83–86). Dissimilar to CAR-T therapy in hematologic malignancies where CAR-Ts encounter target cells in the bloodstream and the lymph nodes, in the case of solid tumors, CAR-Ts must cross the vascular endothelium and penetrate the tumor tissues (86). There are various tumor-associated action mechanisms that remarkably limit the accessibility of CAR-Ts to tumor cells (83–86). In brief, CAR-T tumor site penetration and trafficking are substantially correlated with the presence of various types of chemokines (87). Tumor tissues downregulate the expression of such chemokines leading to restricted tumor tissue CAR-T infiltration (87). Additionally, researchers demonstrated that the dense nature of the ECM of the tumor tissues is an important obstacle to CAR-T penetration and infiltration into the TME (88). Tumor ECM also protects the TME from the antitumor activity of CAR-Ts as it has been demonstrated that its degradation can result in superior CAR-T-mediated tumor cell eradication (89). In a nutshell, profound knowledge of solid tumor mechanisms can better help us develop counterstrategies to overcome the mentioned hurdles for a more efficient and safer CAR-T therapy in patients with various types of solid tumors. A simplified view of the principal limitations of CAR-T therapy in solid tumors and examples of counterstrategies for tackling these hurdles have been brought together in Figure 3 and Table 2, respectively.
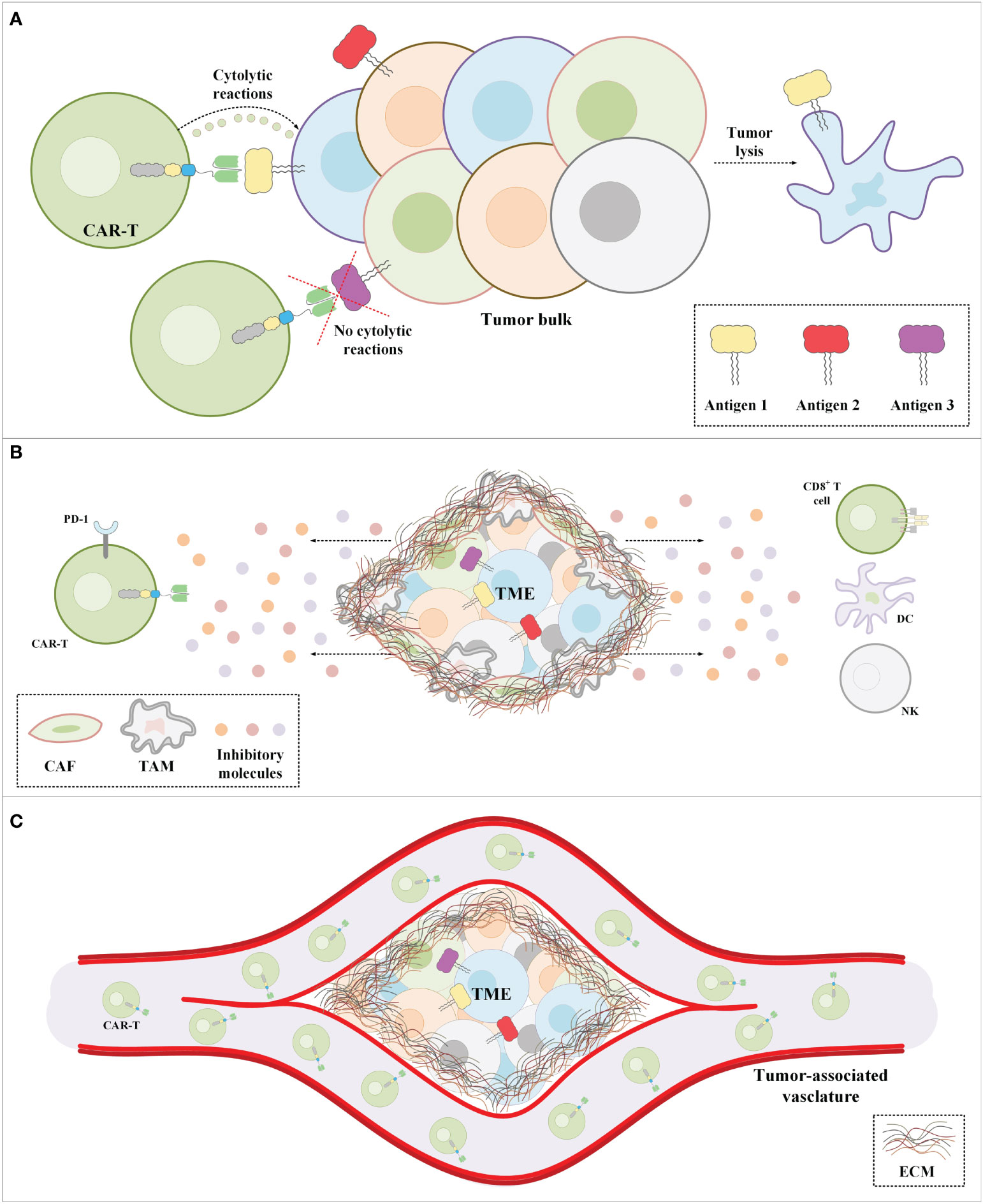
Figure 3 The major impediments of CAR-T therapy in solid tumors. (A) CAR-Ts encounter tumor cells only a proportion of which express the CAR-redirected target antigen. Moreover, in a population of tumor cells, there might be malignant cells not expressing any known target antigens. (B) The immunosuppressive nature of the TME suppresses CAR-T antitumor activity. (C) The extracellular matrix of solid tumors is the most important physical barrier between the tumor cells and CAR-Ts in CAR-T therapy of solid tumors. Cancer-associated fibroblasts are among the mediators responsible for the formation of the stroma extracellular matrix. CAF, cancer-associated fibroblast; CAR-T, chimeric antigen receptor T-cell; DC, dendritic cell; EMC, extracellular matrix; NK, natural killer cell; PD-1, programmed death-1; TAM, tumor-associated macrophage; TME, tumor microenvironment.

Table 2 Significant limitations related to CAR-T therapy of solid tumors and examples of strategies to tackle these challenges.
Targets antigens investigated for the CAR-T therapy of TNBC
Chondroitin sulfate proteoglycan 4 (CSPG4)
CSPG4 is a cell membrane-spanning protein with a high level of glycosylation (137). Various roles have been proposed for CCSPG4 which include involvement in the regulation of neuronal networks, replacement of epithelial keratinocytes, and homeostasis of epidermal stem cells (137). CSPG4 has been studied as a target antigen in cancer immunotherapy based on its limited expression level in normal tissues and overexpression and supporting roles in cancer progression and invasion in various types of neoplasms, including TNBC (137). Researchers demonstrated that antibody-mediated targeting of CSPG4 using an scFv fused to Tau, which is a negative regulator of protein translation, results in efficient cytotoxicity against CSPG4-proficient TNBC-derived cell lines including MDA-MB-231 and Hs 578T (138).
In 2014, Geldres et al. generated second-generation CSPG4-redirected CAR-Ts, and reported that these cells significantly suppressed the growth of various CSPG4-expressing cell lines (including SENMA, CLB, UACC-812, MILL, MDA-MB-231, PHI, and PCI-30) (139). Moreover, these researchers reported that these CAR-Ts suppressed tumor cell growth in human melanoma, head and neck squamous cell carcinoma, and breast carcinoma preclinical mouse models established using SENMA, PCI-30, and UACC-812 cell lines, respectively (139). In the same year, Beard et al. generated second-generation CSPG4-redirected CAR-Ts using murine-based scFvs and reported target antigen-dependent cytotoxicity and cytokine secretion of these CAR-Ts against glioblastoma, breast cancer, mesothelioma, osteosarcoma, and melanoma CSPG4-expressing cell lines (A1207, MDAMB231, Mill, MgG-63, and mel938, respectively) and glioblastoma stem cells (140). Such data may support the applicability of CSPG4-redirected CAR-Ts in TNBC and various other neoplasms. However, more preclinical and clinical data are critically required for more reliable conclusions in this regard.
Intracellular adhesion molecule-1 (ICAM-1)
ICAM-1 is a transmembrane glycoprotein that mediates the transmigration of leukocytes through the endothelium of various cell types (141). In 2014, Guo et al. reported that ICAM-1 is upregulated and overexpressed in TNBC cell lines and tissues and it might act as a possible biomarker and target antigen for the diagnosis and treatment of TNBC (142). These researchers demonstrated that antibody-assisted targeting of ICAM-1 is feasible and efficient even after systemic administration into xenograft TNBC tumor models (142). In 2017, Park et al. generated ICAM-1-redirected CAR-Ts with micromolar affinity against ICAM-1 (instead of nanomolar affinity) to prevent CAR-T-mediated cytotoxicity in non-malignant cells with normal ICAM-1 expression levels (52). These researchers demonstrated that these affinity-tuned CAR-Ts demonstrated superior tumoricidal efficacy and safety index compared to their higher affinity counterparts (52). Moreover, in 2019, using preclinical models, it was demonstrated that micromolar affinity-tuned ICAM-1-redirected CAR-Ts can target tumors with a high level of ICAM-1 expression while sparing normal tissues with lower and basal ICAM-1 expression levels with significant efficacy (143). The researchers of this report also suggested the investigation of micromolar affinity-tuned ICAM-1-redirected CAR-Ts in a Phase I clinical trial for assessing the safety and possible efficacy of these cells against R/R thyroid cancers (143). Moreover, Yang et al. generated ICAM-1-redirected CAR-Ts and investigated the efficacy of these cells in vitro and in vivo (144). These researchers demonstrated that in vitro CAR-T-mediated cytotoxicity against HeLa and MDA-MB-231 cell lines was specific and ICAM-1 expression-dependent (144). However, the ongoing preclinical assessments of this study, from which no results have yet been reported, will further elucidate the applicability and safety index of targeting ICAM-1 using CAT-Ts for the treatment of TNBC (144).
Natural killer group 2, member D ligand (NKG2DL)
Malignant cells, such as TNBC cells, exhibit upregulated levels of stress-induced ligands, which are naturally recognized by molecules such as natural killer group 2, member D (NKG2D) (145). NKG2DL has been considered an immunotherapy target antigen in various studies (146). In 2005, Zhang et al. generated CARs by fusing the full-length NKG2D to the cytoplasmic domain of CD3ζ and endogenous DAP10 costimulation (146). These researchers demonstrated that T cells expressing these CARs reacted to NKG2DL-expressing tumor cells by secreting cytokines and chemokines and exhibiting cytotoxicity (146). Moreover, in vivo results also supported the tumor suppression ability of these CAR-Ts (146). Other researchers have also reported that NKG2DL-redirected CAR-Ts mediated disease remission in a patient with acute myeloid leukemia (AML) (NCT02203825) (147, 148).
In 2018, Han et al. evaluated the antitumor activity of NKG2DL-redirected CAR-Ts against TNBC cell lines and cell line-established preclinical TNBC mouse models (149). These researchers generated different NKG2DL-redirected CAR-Ts by fusing the extracellular domain of human NKG2D to the TCR CD3ζ alone or with CD27 or 4-1BB co-stimulatory domains (149). They reported that the in vitro expansion of CAR-Ts without any co-stimulatory domain was dependent on the high CD25 expression and the presence of IL-2 (149). Moreover, it was reported that NKG2DL-redirected CAR-Ts efficiently recognized TNBC NKG2DL-expressing MDA-MB-231 and MDA-MB-468 cell lines and eliminated them (149). In vivo experiments demonstrated tumor growth suppression ability of NKG2DL-redirected CAR-Ts in MDA-MB-231-derived TNBC preclinical mouse models paving the way for more preclinical and early phase clinical evaluations of this potential TNBC treatment approach (149). It is worth mentioning that a Phase I clinical trial (NCT04107142) has investigated the safety and tolerability of NKG2DL-redirected CAR-Ts in patients with R/R solid tumors including TNBC; however, the results are yet to be reported.
Receptor tyrosine kinase AXL
AXL is a well-known member of the TAM family of receptor tyrosine kinases. Growth arrest-specific protein 6 (GAS6) is known as a high-affinity ligand for AXL (150, 151). AXL signaling operates as a critical pathway mediating tumor cell survival, expansion, migration, and invasion suggesting the potential of AXL as a suitable cancer treatment target antigen (150, 151). Normally, AXL has a low expression level during adulthood; however, its abnormal expression has been observed in various types of neoplasms including breast cancer (152).
Ye et al. developed an anti-AXL mAb with the ability to target both human and murine AXL (153). These researchers demonstrated that this mAb suppressed tumor outgrowth, reduced distant organ tumor cell metastasis, and had enhancing effects on anti-VEGF treatment in MDA-MB-231-established breast cancer xenograft models (153). In 2018, Wei et al. reported the overexpression of AXL in multiple tumor cell lines (including MDA-MB-231, but not MCF-7) and patient-derived samples (154). These researchers also generated AXL-redirected CAR-Ts using a novel AXL-specific scFv, and demonstrated that these CAR-Ts exhibit target antigen-dependent cytotoxicity and cytokine secretion against the AXL-expressing TNBC cell line MDA-MB-231 (154). In vivo evaluations of these CAR-Ts in MDA-MB-231-established xenograft models also indicated significant tumoricidal reactions and in vivo persistence (154). Moreover, Zhao et al. designed AXL-redirected CAR-Ts co-expressing a constitutively activated IL-7 receptor (C7R), and reported that these CAR-Ts established significant tumoricidal capability, which was higher than that of conventional AXL-redirected CAR-Ts, against TNBC MDA-MB-231 and MDA-MB-468 cell lines (155). These researchers added that the co-expressed C7R significantly enhanced the activation, expansion, and antitumor activity of the developed CAR-Ts (155). Additionally, Zhao et al. indicated that according to in vivo results, C7R-co-expressing AXL-redirected CAR-Ts resulted in prolonged survival and a reduced rate of tumor relapse in preclinical mouse models bearing subcutaneous MDA-MB-231 cells (155). Even though such data may underline the applicability of AXL as a CAR-T therapy target antigen in TNBC, further investigations are required.
Tumor endothelial marker 8 (TEM8)
TEM8, alternatively known as anthrax toxin receptor 1 (ANTXR1), is an integrin-like transmembrane protein with roles in the migration and invasion of endothelial cells (156). TEM8 is overexpressed in invasive and TNBC breast cancer tissue correlating with elevated possibility and potential of disease relapse in basal breast cancer (157–159). Researchers have demonstrated that overexpressing TEM8 in preclinical breast cancer models resulted in the amplified capability of the tumor for expansion and metastasis whereas blocking or knocking out TEM8 expression hindered tumor progression in various preclinical models (159–162). Byrd et al. generated TEM8-redirected CAR-Ts and assessed the cytotoxicity of these cells in vitro and in vivo (162). They demonstrated that TEM8-redirected CAR-Ts managed to efficiently inhibit the growth of various TNBC lines (Hs 578T, MDA-MB-231, MDA-MB-436, and MDA-MB-468), a human breast tumor-associated endothelial cell line (HC 6020), and murine tumor-associated endothelial cell lines (2H11 and bEND.3) (162). It is worth mentioning that all of these tumor-associated endothelial cell lines tested positive for variable levels of TEM8 expression (162). Moreover, according to in vivo experiments, these researchers indicated that their TEM8-redirected CAR-Ts mediated tumor growth suppression and prolonged survival in localized patient-derived xenograft (PDX) and lung metastatic TNBC cell line LMD231-established xenograft preclinical models and via eliminating TEM8-expressing TNBC tumor cells and targeting the tumor endothelium to prevent further tumor neovascularization (162). Moreover, Petrovic et al. generated different types of TEM8-redirected CAR-Ts including one type generated using the same antibody utilized in the discussed study by Byrd et al. (163). The in vivo results reported by Petrovic and co-workers indicated that these CAR-Ts rapidly and selectively disappeared from the circulation and caused rapid toxicity after administration into healthy C57BL6 and NSG mice (163). Using TEM8-knockout mouse models, Petrovic et al. demonstrated that the selective clearance of these CAR-Ts from the circulation was because of their cytotoxicity toward normal tissue-expressed TEM8 (163). Such data may speculate critical safety concerns regarding the potential on-target off-tumor toxicity in CAR-T-mediated TEM8 targeting in TNBC (163). Therefore, more comprehensive investigations are required before moving on to clinical trials.
Integrin alpha V beta 3 (αvβ3)
Integrins are adhesion receptors with critical roles in intercellular signal transduction; however, they are also involved in tumor cell migration, tissue invasion, and survival (164). αvβ3 is a well-known integrin in neoplasm-related studies. The expression of this integrin is normally observed in newly forming endothelial cells; however, accumulating evidence confirms its expression in various types of malignancies (165–168). Targeting αvβ3 using antagonists has been evaluated in clinical settings but has not been found effective (169). Moreover, antibody-assisted targeting of this integrin also did not result in beneficial therapeutic effects (170–172). In this regard, researchers have recently contemplated targeting αvβ3 using CAR-Ts.
In 2018, Wallstabe et al. investigated the applicability of targeting αvβ3 using CAR-Ts (173). These researchers used two codon-optimized scFvs as the targeting domain of two different CARs. Of note, these scFvs had been previously humanized (174). The αvβ3-redirected CAR-Ts developed by Wallstabe et al. were also equipped with a truncated epidermal growth factor receptor (EGFRt) enabling the eradication of the infused CAR-Ts by administering the anti-EGFR mAb cetuximab (173). In terms of detecting the expression of αvβ3 on human cell lines, these researchers confirmed the high-level expression of this target antigen on the human TNBC cell line MDA-MB-231 as well as on various melanoma cell lines (173). Their data further confirmed the suitability of integrin αvβ3 as a target antigen by being only overexpressed on malignant cells in hematologic and non-hematologic cancers (173). In vitro functionality assessments demonstrated that αvβ3-redirected CAR-Ts exhibited potent and exclusive tumoricidal activity by producing IFN-γ and IL-2 upon encountering αvβ3-expressing tumor cells (173). It is worth mentioning that the in vivo preclinical assessments of this study did not involve mouse models of TNBC, and the preclinical models in which αvβ3-redirected CAR-Ts demonstrated promising tumoricidal activity were A-375 cell line-established melanoma models (173). Overall, these data only demonstrate that αvβ3 might be a target antigen for CAR-T therapy of TNBC and they cannot guarantee that targeting αvβ3 is safe and effective, and does not result in unwanted toxicities towards normal tissues (173). It is worth noting that CAR-Ts redirected against αvβ3 have also been evaluated for targeting other malignancies such as glioma and it has been indicated that this target antigen holds promising immunotherapeutic value with a low risk of on-target off-tumor toxicity due to its restricted expression on normal tissues (175).
Receptor tyrosine kinase-like orphan receptor 1 (ROR1)
ROR1 is a type 1 membrane-spanning tyrosine kinase receptor with significant roles in embryonic and fetal development (176, 177). Normally, ROR1 expression is observed during embryogenesis but not in normal adult tissues except for adipocytes and a subset of immature B-cell precursors (176, 177). Malignancy-associated expression of ROR1 has been observed in B-cell chronic lymphocytic leukemia (B-CLL), mantle cell lymphoma (MCL), breast cancer, and ovarian cancer (178).
Various researchers have generated ROR-1-redirected CAR-Ts, and have investigated their various aspects in solid tumors and hematologic malignancies (179–182). In 2019, Wallstabe et al. developed microphysiologic three-dimensional (3D) MDA-MB-231-established breast cancer and A549-established lung cancer models, and reported that ROR1-redirected CAR-Ts efficiently infiltrated into the tumor tissues and mediated tumoricidal reactions against multiple layers of malignant cells (183). These researchers suggested that such 3D models may act as reliable platforms for evaluating the safety and efficacy of CAR-Ts before preclinical and clinical assessments (183). Hudecek et al. are also among other researchers who developed ROR1-redirected CAR-Ts and demonstrated their antitumor activity against various solid tumor cell lines including the breast cancer cell lines MDA-MB-231 and MDA-MB-468 (178). Moreover, Srivastava et al. claimed that ROR1-redirected CAR-Ts mediated lethal bone marrow failure since these cells attack ROR1-expressing stromal cells alongside targeting ROR1-expressing malignant cells (182). In this case, these researchers developed ROR1-redirected CAR-Ts harboring synthetic Notch (synNotch) receptors specific for EpCAM or B7-H3 (expressed by ROR1-expressing tumor cells but not ROR1-expressing stromal cells) and reported that these CAR-Ts safely mediated efficient tumoricidal activity without toxicity (182).
Moreover, a Phase I clinical trial (NCT02706392) has investigated the safety of second generation ROR1-redirected CAR-Ts in patients with various types of neoplasms including metastatic TNBC (184). So far, the reported results of 4 enrolled TNBC patients indicated that dose-limiting toxicities, severe neurotoxicity, or severe cytokine release syndrome (CRS) were not detected at dose levels 1 and 2 (184). It is worth mentioning that half of the patients experienced grade 1 CRS (184). Also, post-CAR-T administration tumor tissue biopsy demonstrated infiltration of CD3-positive T cells and macrophages proposing efficient tumor site CAR-T trafficking (184). In regards to clinical outcomes, 2 out of 4 patients achieved stable disease (one at 15 weeks and the other at 19 weeks post infusion) (184). One patient achieved stable disease after the first CAR-T administration and established partial response following the second CAR-T administration which has prolonged for 14 weeks as of the time of the report (184). Of note, the results of this trial are expected to be updated (184).
Receptor tyrosine kinase c-Met
c-Met, alternatively known as MET or hepatocyte growth factor receptor (HGFR), is a membrane-bound tyrosine kinase known to have critical roles in organogenesis and cancer development (185). c-Met is involved in the development of TNBC (186). c-Met inhibitors have been studied in various solid tumors and these inhibitors have resulted in encouraging results in lung and ovarian cancer (187, 188). c-Met overexpression has been observed in more than 50% of TNBC patients correlating with unfavorable overall survival (OS) of these patients (185, 186). In this regard, Kim et al. reported that c-Met expression is high in TNBC cell lines, and siRNA-mediated silencing of c-Met decreases the proliferation and migration capacity of certain TNBC cell lines (186). Such data can support the critical role and applicability of c-Met as a target for cancer immunotherapy.
Tchou et al. developed mRNA electroporation-generated second generation c-Met-redirected CAR-Ts and demonstrated the antitumor activity of these cells in in vitro killing assays against BT20 (a TNBC-derived breast cancer cell line) and TB129, both with similar c-Met expression levels (189). These CAR-Ts also managed to suppress tumor growth in xenograft preclinical mouse models established using the ovarian cancer cell line SK-OV-3 (189). These researchers also conducted a Phase I clinical trial (NCT01837602) to study the safety and feasibility of intratumoral delivery of these CAR-Ts for treating metastatic breast cancer (189). The results of this trial demonstrated that CAR-T administration was well-tolerated as no CAR-T-related adverse events (>grade 1) were observed (189). Moreover, immunohistochemical analysis of tumors treated with intratumoral CAR-T delivery demonstrated significant administration site tumor necrosis, cellular debris, and the presence of macrophages around the necrotic areas (189). It is worth mentioning that Tchou et al. used mRNA-based CAR-Ts based on the concerns regarding the tumoricidal effects of c-Met-redirected CAR-Ts for targeting non-malignant c-Met-expressing cells (189). Moreover, Shah et al. reported the results from a Phase I clinical trial (NCT03060356) evaluating the safety and feasibility of intravenously delivered mRNA electroporation-generated second generation c-Met-redirected CAR-Ts in patients with metastatic or unresectable melanoma or TNBC with more than 30% c-Met expression (190). According to this report, 5 patients experienced grade 1 or 2 CAR-T infusion-related toxicity (no grade 3 or CRS was observed) but one patient terminated the therapy course due to these toxicities (190). Among the patients with TNBC who received the c-Met-redirected CAR-Ts (4 patients), 2 (50%) achieved stable disease and 2 (50%) experienced partial disease (190). Ultimately, these researchers suggested using lentiviral-generated CAR-Ts alongside lymphodepleting chemotherapy in future clinical studies (190).
Folate receptor alpha (FRα)
FRα, alternatively termed FOLR1 or folate binding protein (FBP), is a glycosylphosphatidylinositol (GPI)-anchored transmembrane protein with a high affinity for binding the active folate form and managing its transportation (191, 192). The overexpression of FRα has been detected in various solid tumors such as ovarian and breast cancers remarkably correlating with disease grade and stage (193, 194). TNBC is also among solid malignancies in which FRα upregulation has been observed (195). It has been demonstrated that overexpression of FRα provides malignant cells with the privilege of proliferation (195). Song et al. generated FRα-redirected CAR-Ts, and reported target antigen engagement-dependent proinflammatory cytokine secretion by these cells after co-culturing with FRα-expressing human ovarian cell lines in culture (SKOV3, A1847, and OVCAR3) (196). Moreover, these researchers reported that these CAR-Ts induced tumor growth suppression in cell line-established FRα-positive human ovarian cancer preclinical models (196). Years later, Song et al. evaluated FRα-redirected CAR-Ts in breast cancer cell lines and preclinical mouse models (197). These researchers reported that their FRα-redirected CAR-Ts secreted significant levels of IFN-γ upon co-cultivation with TNBC cell lines expressing FRα (197). These researchers also added that their engineered effector cells also mediated remarkable tumor outgrowth suppression in cell line-established preclinical xenograft mouse models of TNBC (197). However, Song et al. concluded that the tumoricidal activity of their FRα-redirected CAR-Ts was not as robust as it was in the case of ovarian cancer xenograft models, where the expression levels of the target antigen were higher (197). Moreover, they demonstrated the same CAR-Ts induced improved tumor suppression in preclinical models established using MDA-MB-231 cells engineered to overexpress FRα (197).
Lanitis et al. used a strategy for generating FRα-redirected CAR-Ts with a reduced level of capability for mediating on-target off-tumor toxicity (198). In detail, they generated a trans-signaling CAR in which the activation domain is physically separated from the co-stimulatory domain in two separate CARs, one set of which targets mesothelin and the other set targets FRα (198). These CAR-Ts exhibited ineffective cytokine secretion upon encountering target cells expressing only one of the target antigens but exhibited significantly improved cytokine secretion against tumor cells expressing both target antigens in vitro (198). Furthermore, these CAR-Ts demonstrated effective tumoricidal activity and persistence in vivo (198). Conclusively, these researchers suggested this method as a potent strategy for reducing the possibility of on-target off-tumor activity of CAR-Ts towards non-malignant tissues (198). In addition to this strategy, other researchers have also demonstrated that folate-FITC bispecific molecules can manage to mediate the redirection of FITC-redirected CAR-Ts activity against folate receptor (FR)-positive malignant cells (199).
Epidermal growth factor receptor (EGFR)
EGFR is a membrane-spanning glycoprotein that is a member of the ERBB receptor tyrosine kinase family (200). It has been known to be involved in malignant cell proliferation and metastasis (200). TNBC is among solid tumors in which EGFR overexpression has been detected (201). EGFR targeting using CAR-Ts has been investigated in TNBC as well as other solid tumors (202–204). In detail, Li et al. generated EGFR-redirected CAR-Ts using the non-viral piggyBac transposon system, and reported that these cells demonstrated tumoricidal activity against EGFR-positive cells in vitro and suppressed tumor growth in human lung cancer xenografts (203). Moreover, a Phase I clinical trial (NCT03182816) has investigated PiggyBac transposon-generated EGFR-redirected CAR-Ts in patients with advanced R/R non-small cell lung cancer (202).
In the case of TNBC, Liu et al. investigated EGFR-redirected CAR-Ts in vitro and in vivo (205). These researchers reported the overexpression of EGFR in TNBC Hs 578T, MDA-MB-468, and MDA-MB-231 cell lines in comparison with the non-TNBC cell line MCF-7 (205). In vitro co-cultivation assay of EGFR-redirected CAR-Ts with the mentioned TNBC cell lines demonstrated the target antigen-dependent cytokine secretion and antitumor activity of these CAR-Ts (205). Moreover, these researchers reported tumor growth suppression by EGFR-redirected CAR-Ts in cell line-established and PDX preclinical mouse models (205). Recently, Xia et al. reported the overexpression of EGFR in TNBC cell lines MDA-MB-231, MDA-MB-468, Hs 578T, and HCC1860 (201). These researchers generated third-generation EGFR-redirected CAR-Ts and reported that these effector cells demonstrated specific cytokine secretion, antitumor activity, and upregulation of T-cell activation markers (including CD69 and CD25) upon co-cultivation with EGFR-positive TNBC cells lines (201). In vivo assessments using severe combined immunodeficient (SCID) mice subcutaneously implanted with the MDA-MB-231 cell line demonstrated the capability of these CAR-Ts to inhibit TNBC tumorigenesis in preclinical mouse models with “minimal” levels of off‐tumor cytotoxicity (201). Overall, both of the discussed studies proposed that EGFR may be a potent CAR-T target antigen for TNBC treatment; however, more preclinical and clinical investigations are critically required (201).
Tumor-restricted variants of EGFR such as EGFR variant III (EGFRvIII) can be utilized in case of safety concerns regarding the expression of EGFR on normal tissues (206). However, so far, such variants have only been investigated in a limited number of solid tumors such as glioma (206). Moreover, modulating the immunosuppressive nature of the TME using immune checkpoint blockade approaches has been shown to improve CAR-T functioning in solid tumors (207). Several clinical trials (NCT03182816, NCT02873390, NCT02862028, and NCT03170141), some of which are completed and some are still ongoing, have aimed to investigate the effects of EGFR-redirected CAR-Ts with the ability to secrete anti-CTLA-4, anti-PD-1, or anti-PD-L1 antibodies in EGFR-positive advanced solid tumors (207). However, no reports regarding the results of such trials in patients with TNBC have yet been published (207).
Mesothelin
Mesothelin is a tumor differentiation glycoprotein involved in cell adhesion (208, 209). It has a normally restricted expression on the mesothelial surfaces of the body, but it is remarkably overexpressed in a wide range of solid cancers including TNBC (208, 209). There is scientific evidence that mesothelin is engaged in oncogenesis through various cellular signaling pathways including NF-κB, PI3K, and MAPK (208–210). TNBC is one of the solid tumors in which mesothelin overexpression has been detected. In detail, one study has demonstrated that the majority of TNBC cases (67%) exhibit mesothelin overexpression as confirmed using immunohistochemical analysis (211). The limited normal expression of mesothelin and its high-level expression in a large proportion of TNBC cases have rendered it an appealing target antigen for various types of cancer immunotherapy. In 2019, Del Bano et al. investigated bispecific antibodies with mesothelin targeting and CD16 engagement domains, and demonstrated that this construct mediated the recruitment and penetration of natural killer (NK) cells into tumor spheroids and provoked robust dose-dependent cell-mediated cytotoxicity of mesothelin-expressing TNBC cell lines (212). CAR-T-mediated mesothelin targeting has also been studied in the context of TNBC. In detail, Hu et al. assessed the expression of mesothelin on three TNBC cell lines including MDA-MB-231, BT-549, and Hs 578T (107). They reported that only BT-549 cells (but not MDA-MB-231 and Hs 578T cells) expressed mesothelin as verified by both Western blot and flow cytometry (107). These researchers generated second-generation mesothelin-redirected CAR-Ts and evaluated their performance in vitro and in vivo (107). Of note, Hu et al. disrupted the PD-1 gene locus in T cells before CAR transgene introduction. In detail, these CAR-Ts demonstrated remarkably increased cytokine production and antitumor activity against PD-L1-expressing cancer cells in culture (107). Accumulating evidence suggests that PD-L1 is remarkably overexpressed in TNBC cells (213). Therefore, the strategy proposed by Hu et al. might overcome the suppressive effects of PD-1-PD-L1 interaction on CAR-Ts (107). Moreover, Hu et al. added that PD-1-deficient mesothelin-redirected CAR-Ts exhibited improved tumor outgrowth suppression and disease recurrence prevention in BT-549-established preclinical TNBC mouse models in comparison with CAR-Ts with or without PD-1-specific antibody blockade (107). Overall, even though more preclinical and clinical data are required for safe conclusions on the applicability of using mesothelin as a CAR-T therapy target antigen for TNBC, the study by Hu et al. highlights the potential of checkpoint blockade alongside mesothelin-redirected CAR-T therapy for targeting TNBC. It is worth mentioning that a Phase I clinical trial (NCT02792114) is currently investigating the safety and tolerability of mesothelin-redirected CAR-Ts in patients with metastatic/advanced mesothelin-proficient breast cancer including TNBC. Moreover, another Phase I/II clinical trial (NCT02414269) is also investigating second generation mesothelin-redirected CAR-Ts in patients with lung cancer or breast cancer. Additionally, two other clinical trials (NCT01355965 and NCT02580747) have been completed but no official reports in regards to the results of these trials in TNBC patients have been published yet.
Disialoganglioside GD2
GD2 is a surface antigen with normal expression limited to peripheral pain fibers, neurons, and melanocytes (214). GD2 expression has been documented in neuroectoderm-originated neoplasms such as neuroblastoma and melanoma (214). The prevalent tumor cell-restricted expression of GD2 has made it a suitable target antigen for various types of cancer immunotherapy. GD2 is mostly known as a target antigen for the treatment of neuroblastoma. In 2011, Louis et al. conducted a Phase I clinical trial (NCT00085930) by generating GD2-redirected CAR-Ts using Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes or blood T cells for investigating the effectiveness and prolonged persistence of these cells (215). Back in 2008, these researchers also reported the results of a Phase I clinical trial in 11 pediatric patients with neuroblastoma (216). In 2015, the US FDA approved the anti-GD2 mAb dinutuximab (also known as ch14.18) for the treatment of pediatric patients with high-risk neuroblastoma (217). This mAb has recently been investigated in TNBC (218, 219). Ly et al. investigated dinutuximab application for targeting GD2-expressing breast cancer stem cells (BCSCs) and suppression of cancer progression (219). BCSCs are a type of cells in the early tumor that are chemotherapy resistant, capable of metastases, and remarkably tumorigenic (218). Therefore, BCSC targeting is known as a critical approach for precluding cancer metastases and rendering tumors susceptible to chemotherapy (219, 220). Ly et al. first demonstrated the upregulation of GD2 in TNBC cell lines, PDX preclinical models, and primary TNBC samples (219). In detail, Hs 578T and HCC1395 were two TNBC cell lines in which more than 90% of cells were GD2-proficient (219). Moreover, GD2 expression was also documented in about 60% of primary TNBC tumors (even though with fluctuating levels) correlating with poorer OS (219). Dinutuximab treatment meaningfully reduced adhesion and migration of MDA-MB-231 and SUM159 TNBC cells and suppressed GD2-upregulated mTOR signaling in BCSCs (219). This mAb also exhibited tumor outgrowth suppression capability in MDA-MB-231-established TNBC xenograft models suggesting the applicability of dinutuximab for TNBC control in the preclinical stage (219).
There are not many studies investigating CAR-Ts targeting GD2 in TNBC. In 2020, Seitz et al. used the scFv derived from ch14.18 to generate GD2-redirected CAR-Ts (221). In detail, TNBC cell lines including MDA-MB-468, MDA-MB-231, Hs 578T, and BT-549 were screened for GD2 expression and it was demonstrated that MDA-MB-231 expressed GD2, even though at very low levels, and Hs 578T and BT-549 exhibited uniform GD2 expression (221). In vitro cytotoxicity assay demonstrated these CAR-Ts did not mediate any specific tumoricidal activity towards MDA-MB-231 (221). However, these CAR-Ts induced specific cytotoxicity and cytokine secretion upon co-cultivation with the Hs 578T and BT-549 cell lines (221). Moreover, GD2-redirected CAR-Ts mediated tumor growth suppression in the preclinical orthotopic model of TNBC (established using MDA-MB-231) and inhibited lung metastasis (221). Conclusively, these researchers suggested CAR-T-mediated GD2 targeting as an approach to eradicate disseminated malignant cells and inhibit metastasis (221). However, more preclinical and clinical findings may be required.
Mucin 1 (MUC1)
MUC1 is a heavily glycosylated membrane-spanning mucin protein expressed on glandular epithelial cells (222–224). The extracellular domain of MUC1 has a variable number tandem repeats (VNTR) region which is rich in serine and threonine residues acting as a platform for the attachment of O-glycans (222–224). O-glycosylation of MUC1 leads to the generation of a tumor-associated aberrantly glycosylated form of MUC1 known as tMUC1 (51, 222–224). tMUC1 is overexpressed in all subtypes of breast cancer, including in 95% of TNBCs (225). Moreover, this antigen has no detectable expression on the cells of normal breast tissues (225). Such characteristics render tMUC1 a great CAR-T therapy target antigen in various types of malignancies including TNBC. To this day, various studies have demonstrated that CAR-T-mediated targeting of the aberrantly glycosylated tumor forms of surface antigens is a safe and feasible approach (51). In this regard, Zhou et al. generated second-generation CAR-Ts using a mAb named TAB004, which is capable of tMUC1 recognition (225). These tMUC1-redirected CAR-Ts demonstrated target antigen-dependent cytotoxicity against nine different TNBC cell lines and released cytokines, chemokines, and granzyme B (225). Moreover, these CAR-Ts inhibited HCC70 tumor outgrowth and maintained their prolonged antitumor activity for HCC70 tumor burden reduction in vivo (225). In a nutshell, this study indicated that tMUC1-redirected CAR-Ts harbor significant therapeutic value against tMUC1-positive TNBC with minimal on-target off-tumor toxicity toward normal breast epithelial cells (225).
In addition to t-MUC1, TnMUC1 is another aberrant glycoform of MUC1 that is also highly expressed by TNBC cells (226). Posey et al. generated TnMUC1-redirected CAR-Ts using a Tn-MUC1-specific antibody named 5E5, and reported target antigen-specific antitumor activity of these CAR-Ts against a panel of human primary cells and several cell lines (226). Moreover, these researchers reported that these CAR-Ts managed to suppress tumor growth in T-cell leukemia and pancreatic cancer xenograft in vivo models (226). Recently, Zhai et al. reported that their TnMUC1-redirected CAR-Ts generated using Vγ9Vδ2 T cells exhibited “similar or stronger” antitumor effects, than CAR-expressing αβ T cells, against breast cancer cell lines with various levels of Tn-MUC1 expression (including T47D, MDA-MB-231, and MDA-MB-468) (227). Similar results were observed upon the co-cultivation of these CAR-Ts with gastric cancer cells (227). However, these researchers indicated that Vγ9Vδ2 TnMUC1-redirected CAR-Ts demonstrated persistence deficiencies for addressing which IL-2 is critical (227). Moreover, Zhai also reported that Vγ9Vδ2 TnMUC1-redirected CAR-Ts had significant tumoricidal activity in gastric cancer preclinical mouse models (227).
It is worth mentioning that clinical trials (NCT04020575 and NCT04025216) are currently evaluating second generation CAR-Ts redirected against cancer glycoforms of MUC1. In an early report from one of these trials (NCT04025216), the researchers indicated that since this study is still in the dose-escalation step and it has completed only 2 of 6 planned dose levels, safety issues or on-target off-tumor toxicity of this platform are not yet up for debate (228). Moreover, another clinical trial (NCT02587689) investigating MUC1-redirected CAR-Ts in patients with advanced refractory solid tumors, including TNBC, has been finished. However, no official report of the results of this trial has been published yet.
Trophoblast cell-surface antigen 2 (TROP2)
TROP2 is a transmembrane protein expressed on the surface of human trophoblast cells (229). The expression of this antigen has been often detected in various epithelium tissue malignancies mediating their tumor-associated behavior and correlating with poor prognosis (229, 230). Such characteristics have rendered TROP2 an attractive target antigen for various types of cancer immunotherapy, especially in TNBC (229). In this regard, a recent study by Liu et al. has reported that TRBAs targeting TROP2 and CD3 suppress tumor growth in both TNBC cell lines and primary tumor cells (231). In detail, the TROP2-CD3 TRBAs developed by these researchers were capable of recruiting T lymphocytes to TROP2-positive tumor cells in vitro and into tumor tissues in xenograft TNBC preclinical models (231). The data presented by this study supports the potential of TROP2 for the immunotherapy of TNBC in patients with advanced/metastatic neoplasms (231). Moreover, in 2021, the US FDA approved sacituzumab govitecan, a TROP2-redirected mAb conjugated to a topoisomerase I inhibitor drug, for the treatment of patients with R/R unresectable locally advanced or metastatic TNBC who have received two or more prior systemic therapies (232). FDA granted medical use permit to this ADC based on a clinical trial report (NCT01631552) by Bardia et al. which demonstrated that sacituzumab govitecan mediated a response rate of 33% (3 complete and 33 partial responses) and a median duration of response of 7.7 months in patients with TNBC who had received a range of 2 to 10 previous lines of therapies (232). In the case of CAR-T-mediated TROP2 targeting, Zhao et al. developed bi-specific TROP2- and PD-L1-redirected CAR-Ts and evaluated their antitumor activity in vitro (using TROP2-positive and PD-L1-positive gastric cancer cell lines) and in vivo (233). According to the in vitro results obtained by these researchers, the tumoricidal activity of bi-specific CAR-Ts was higher than that of mono-specific CAR-Ts (233). These bi-specific CAR-Ts also managed to secrete pro-inflammatory cytokines upon encountering TROP2-positive and PD-L1-positive gastric cancer cells (233). Additionally, in vivo results demonstrated that these CAR-Ts remarkably suppressed tumor growth via intratumoral injection in preclinical mouse models established using human gastric tumors (233). However, these results only support the suitability of TROP2 as a CAR-T target but not specifically in TNBC CAR-T therapy. In the case of TROP2-redirected CAR-Ts for targeting TNBC, Bedoya et al. generated TROP2-redirected CAR-Ts and reported that these cells demonstrated antitumor activity against TROP2-negative cells in the presence of TROP2-positive cells (234). As claimed by these researchers, this phenomenon has also been observed in the case of CD19-redirected CAR-Ts but it was resolved by the blockade of death receptor ligands; however, Bedoya et al. indicated that this approach only resulted in the partial blockade of TROP2-redirected CAR-T cytotoxicity in this case (234). Moreover, these researchers transferred TROP2-positive tumor exosomes to TROP2-negative tumor cells to elevate the percentage of tumor cells targetable by TROP2-redirected CAR-Ts and to tackle the limitation of antigen heterogeneity in solid tumors (234). In a nutshell, even though CAR-T-mediated TROP2 targeting in TNBC has not been comprehensively investigated, the data discussed here might highlight the potential applicability of this target antigen for the treatment of TNBC.
The potential toxicities of CAR-T therapy in TNBC
Identifying suitable target antigens is one of the most important steps of CAR-T cell therapy in solid tumors. “On-target off-tumor” effect is a CAR-T cell-mediated adverse event that takes place when a CAR-T-redirected target antigen is present (even at very low levels) on normal tissues, especially vital organs. The best strategy to prevent this adverse event is to select target antigens that are completely absent from normal tissues or have undetectable expression levels. However, this strategy is quite unpractical since every target antigen, which is overexpressed on malignant tissue cells, can also be detected in normal tissues. Therefore, researchers have focused on strategies that can be beneficial for preventing or reducing the occurrence of such events. For instance, “safety switches” which include suicide genes have been utilized in the construct of CAR-Ts for targeting TNBC (173). In detail, Wallstabe et al. have studied the applicability of αvβ3-redirected CAR-Ts equipped with EGFRt which allows the eradication of the infused CAR-Ts using the anti-EGFR mAb cetuximab (173). Furthermore, other novel CAR designs such as dual CARs and synNotch CARs have also been investigated in solid tumor CAR-T therapy for preventing off-tumor toxicities (182). Researchers have claimed that ROR1-expressing stromal cells are simultaneously targeted by ROR1-redirected CAR-Ts in the context of targeting ROR1-expressing malignant cells (182). This “on-target off-tumor” toxicity can lead to lethal bone marrow failure; therefore, an efficient strategy is required for preventing this adverse event. Srivastava et al. generated ROR1-redirected CAR-Ts with synNotch receptors specific for EpCAM or B7-H3 (182). Conclusively, these researchers added that such CAR-Ts mediated efficient antitumor responses without causing toxicity (182). In addition to these strategies, trans-signaling CARs are also considered innovative in terms of preventing on-target off-tumor toxicities (198). In the context of TNBC, Lanitis et al. generated trans-signaling FRα-redirected CAR-Ts in which the CAR activation domain was physically detached from the CAR co-stimulatory domain and they were incorporated into two separate CARs one targeting mesothelin and the other targeting FRα (198). These CAR-Ts were evaluated in preclinical assessments, and it was proposed that this strategy can decrease the risk of on-target off-tumor toxicity against normal tissues (198).
In the context of CAR-T therapy of hematologic malignancies, such as those targeting CD19, due to the expression of CD19 by both normal cells and malignant blasts, CAR-T therapy results in the elimination of both of these cells leading to a phenomenon known as “B-cell aplasia” (235). This occurrence is a marker of an efficient CAR-T therapy of hematologic malignancies, even though it makes the relative patients susceptible to opportunistic infections to tackle which the patients should undergo immunoglobulin replacement (235).
As mentioned earlier, the application and efficiency of CAR-Ts whose scFv targeting domains have been subjected to affinity-tuning have also been studied (52). Such CAR-Ts can be favorable in distinguishing between tumor cells overexpressing the target antigen and normal cells expressing the antigen at physiologic levels (52). In this regard, Park et al. demonstrated that affinity-tuned ICAM-1-redirected CAR-Ts are capable of targeting malignant cells with ICAM-1 overexpression while sparing normal cells expressing ICAM-1 at basal physiologic levels (52). Moreover, mRNA-based CAR-Ts have also been proposed as a platform for preventing on-target off-tumor toxicities. In this regard, Tchou et al. developed mRNA-based c-Met-redirected CAR-Ts to avoid CAR-T-mediated targeting of c-Met-expressing normal cells (189). They reported that these cells demonstrated efficient antitumor activity against TNBC both in vitro and in vivo (189). These researchers also evaluated the safety and efficacy of these CAR-Ts in a Phase I clinical trial (NCT01837602) (189).
CRS is well-known CAR-T cell therapy adverse event which is more likely to occur in CAR-T therapy of hematologic malignancies (235, 236). CRS results from the rapid activation of various immunological pathways. Its critical damages can include cardiac-related toxicities and hyponatremia. CRS has also been detected in patients with solid tumors undergoing CAR-T therapy (236). As discussed throughout the article, there are not many clinical trials investigating CAR-T therapy for the treatment of patients with TNBC. Some of these trials have been completed and some are still ongoing. Among the trials that have been completed, only a few of them have reported their findings. In detail, in a Phase I clinical trial (NCT02706392) studying ROR1-redirected CAR-Ts in patients with various solid tumors including metastatic TNBC, it was reported that out of 4 TNBC patients, half of them experienced grade 1 CRS (184). According to a report by Tchou et al. from a Phase I clinical trial (NCT01837602) assessing the safety and feasibility of intratumoral delivery of c-Met-redirected CAR-Ts in patients with metastatic breast cancer, it was reported that the CAR-Ts were well-tolerated and no CAR-T-associated toxicities (> grade 1) were documented (189). Moreover, in another a Phase I clinical trial (NCT03060356) investigating the same target antigen, it was reported that 5 patients experienced grade 1 or 2 CAR-T delivery-associated adverse events (no grade 3 or CRS were reported) (190).
CRS is a medical condition that demands rapid clinical intervention to prevent the condition from worsening. Generally, antihistamines or corticosteroids are recommended for low-grade CRS management (236). However, in the case of CAR-T therapy-mediated CRS, more efficient strategies are required, especially in the case of hematologic malignancies such as B-cell acute lymphoblastic leukemia (B-ALL) (235). IL-1 and IL-6 blockade, GM-CSF blockade, antibody-based immunotherapy pretreatment, therapeutic plasma exchange, hemofiltration, and fractionated CAR-T infusion are among strategies proven to be efficient in the management of severe lethal CRS after CAR-T therapy (235, 237–245). Such strategies can also be applied in the case of solid tumor CAR-T therapy including TNBC CAR-T therapy.
Conclusion
CAR-T products have been available on the market as a treatment option for R/R hematologic malignancies in the past recent years. However, CAR-T-mediated targeting of solid tumors or some hematologic malignancies, such as T-cell neoplasms, face additional unexpected challenges that remarkably restrain the antitumor potential of these therapeutics. TNBC is known as a heterogeneous breast cancer subtype that is mainly resistant to conventional therapies. The immunogenic nature of this neoplasm has proven that immunotherapy-based therapeutics can lead to favorable clinical benefits. For instance, the promising clinical outcomes of the checkpoint inhibitor atezolizumab in combination with nab-paclitaxel led to its approval by the US FDA for the treatment of locally or metastatic advanced unresectable TNBC (18). However, CAR-T therapy of TNBC is in an emerging field as it is focused on discovering the suitable and targetable TAAs, mostly in preclinical and early clinical stages. Of note, to overcome the hurdles of CAR-T therapy in TNBC, various challenges of solid tumor CAR-T therapy should be tackled beforehand. Various critical stratagems should be employed to ensure the safety and effectiveness of CAR-T therapy in solid tumors. Combinatorial CAR-T therapy with other types of therapies can result in improved clinical outcomes of CAR-T therapy in solid tumors (70). For instance, ECM- or CAF-targeting agents can be applied for maximizing CAR-T antitumor effects (70). Also, macrophage- or monocyte-eliminating agents are beneficial for amplifying the tumoricidal impact of CAR-Ts, or anti-angiogenic agents can be leveraged for enhancing the tumor site trafficking of CAR-Ts (70). Moreover, other types of effector cells have also been investigated for the expression of CARs. In this regard, γδ-CAR-Ts and CAR-expressing NK cells (CAR-NKs) have been investigated for the treatment of both hematologic and solid malignancies, including TNBC (68, 246–248). Such alternative CAR-expressing effector cells might be beneficial for tackling various types of CAR-T therapy challenges (68, 246–248).
Throughout this article, we reviewed various novel CAR-T target antigens for the selective targeting of TNBC. A number of these target antigens have only been evaluated in in vitro and in vivo studies while some of them have found their way to in-human clinical investigations, as summarized in Table 3. Moreover, in addition to the discussed target antigens, new targets can also be discovered. For instance, stage-specific embryonic antigen-4 (SSEA-4) is among more novel antigens detected in a proportion of TNBCs, and CAR-Ts against this target antigen have been investigated in vitro and in vivo (249). However, such antigens are not comprehensively studied as TNBC CAR-T-targetable antigens and require more in-depth assessments.
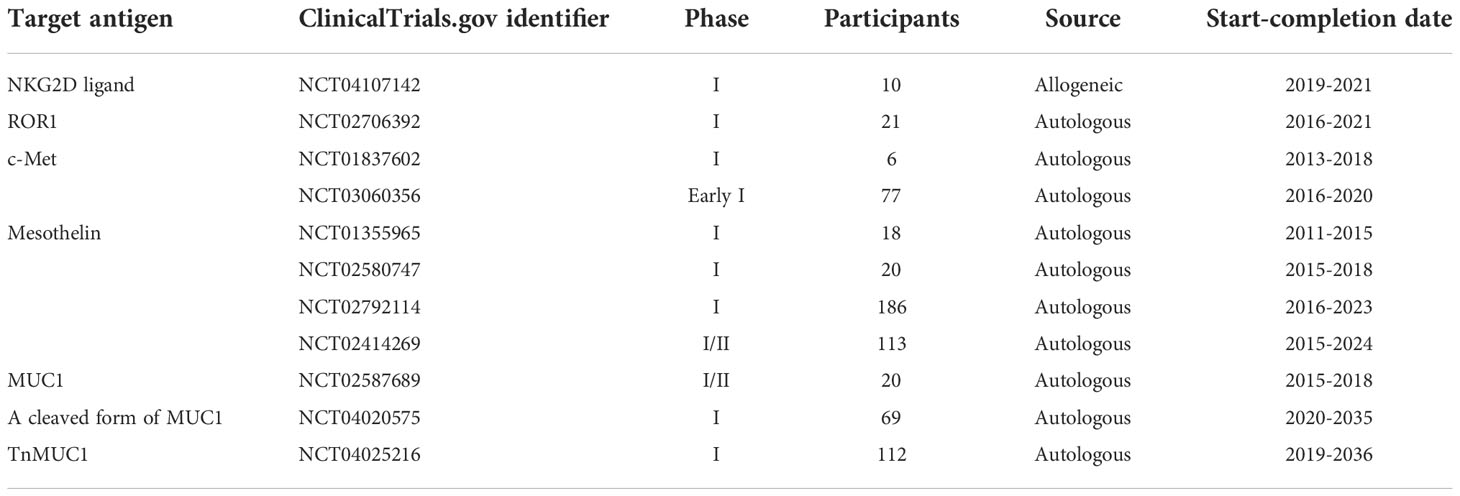
Table 3 A summary of different clinical trials investigating CAR-Ts against different target antigens for the treatment of TNBC and other solid tumors.
In a nutshell, it is safe to mention that considerable efforts have been made in the field of TNBC CAR-T therapy as demonstrated by several ongoing preclinical and clinical studies. However, there is still a long way to go for some of the proposed strategies (for enhancing the efficacy of CAR-T therapy in solid tumors) and several TNBC target antigens discussed in this article. Successful CAR-T therapy in TNBC is substantially reliant on improving the specificity, safety, and efficacy of CAR-T therapy in solid tumors by both choosing the most appropriate target antigens and addressing the unmet restricting challenges. Until then, fingers are crossed for CAR-T therapy to be able to act as an efficient treatment approach alongside the conventional TNBC treatment modalities.
Author contributions
FN: Investigation, writing - original draft, validation. MK: Writing - original draft. SMJM: Writing - original draft. MMK: Writing - original draft. MAN: Conceptualization, writing - review & editing. FS: Conceptualization, writing - review & editing. SDS: Writing - original draft. SEB: Writing - original draft. PouSK: Conceptualization, investigation, writing - original draft, writing - review & editing, validation, supervision. PooSK: Conceptualization, investigation, writing - original draft, writing - review & editing, validation, supervision. All authors contributed to the article and approved the submitted version.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: A Cancer J Clin (2022) 72:7–33. doi: 10.3322/caac.21708
2. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers (2019) 5:66. doi: 10.1038/s41572-019-0111-2
3. Feng Y, Spezia M, Huang S, Yuan C, Zeng Z, Zhang L, et al. Breast cancer development and progression: Risk factors, cancer stem cells, signaling pathways, genomics, and molecular pathogenesis. Genes Dis (2018) 5:77–106. doi: 10.1016/j.gendis.2018.05.001
4. Makki J. Diversity of breast carcinoma: Histological subtypes and clinical relevance. Clin Med Insights Pathol (2015) 8:23–31. doi: 10.4137/CPath.S31563
5. Malhotra GK, Zhao X, Band H, Band V. Histological, molecular and functional subtypes of breast cancers. Cancer Biol Ther (2010) 10:955–60. doi: 10.4161/cbt.10.10.13879
6. Rakha EA, Reis-Filho JS, Baehner F, Dabbs DJ, Decker T, Eusebi V, et al. Breast cancer prognostic classification in the molecular era: The role of histological grade. Breast Cancer Res (2010) 12:207. doi: 10.1186/bcr2607
7. Russnes HG, Lingjærde OC, Børresen-Dale AL, Caldas C. Breast cancer molecular stratification: From intrinsic subtypes to integrative clusters. Am J Pathol (2017) 187:2152–62. doi: 10.1016/j.ajpath.2017.04.022
8. Lumachi F, Santeufemia DA, Basso SM. Current medical treatment of estrogen receptor-positive breast cancer. World J Biol Chem (2015) 6:231–9. doi: 10.4331/wjbc.v6.i3.231
9. Pernas S, Tolaney SM. HER2-positive breast cancer: new therapeutic frontiers and overcoming resistance. Ther Adv Med Oncol (2019) 11:1758835919833519. doi: 10.1177/1758835919833519
10. Waks AG, Winer EP. Breast cancer treatment: A review. JAMA (2019) 321:288–300. doi: 10.1001/jama.2018.19323
11. Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. New Engl J Med (2010) 363:1938–48. doi: 10.1056/NEJMra1001389
12. Wahba HA, El-Hadaad HA. Current approaches in treatment of triple-negative breast cancer. Cancer Biol Med (2015) 12:106–16. doi: 10.7497/j.issn.2095-3941.2015.0030
13. Zeichner SB, Terawaki H, Gogineni K. A review of systemic treatment in metastatic triple-negative breast cancer. Breast Cancer (Auckl) (2016) 10:25–36. doi: 10.4137/bcbcr.S32783
14. Rose M, Burgess JT, O’Byrne K, Richard DJ, Bolderson E. PARP inhibitors: Clinical relevance, mechanisms of action and tumor resistance. Front Cell Dev Biol (2020) 8:564601. doi: 10.3389/fcell.2020.564601
15. Luo C, Wang P, He S, Zhu J, Shi Y, Wang J. Progress and prospect of immunotherapy for triple-negative breast cancer. Front Oncol (2022) 12:919072. doi: 10.3389/fonc.2022.919072
16. Marra A, Viale G, Curigliano G. Recent advances in triple negative breast cancer: the immunotherapy era. BMC Med (2019) 17:90. doi: 10.1186/s12916-019-1326-5
17. Yu L, Wang J. T Cell-redirecting bispecific antibodies in cancer immunotherapy: recent advances. J Cancer Res Clin Oncol (2019) 145:941–56. doi: 10.1007/s00432-019-02867-6
18. Schmid P, Rugo HS, Adams S, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2020) 21:44–59. doi: 10.1016/s1470-2045(19)30689-8
19. Li Z, Qiu Y, Lu W, Jiang Y, Wang J. Immunotherapeutic interventions of triple negative breast cancer. J Transl Med (2018) 16:147. doi: 10.1186/s12967-018-1514-7
20. Jia H, Truica CI, Wang B, Wang Y, Ren X, Harvey HA, et al. Immunotherapy for triple-negative breast cancer: Existing challenges and exciting prospects. Drug Resist Update (2017) 32:1–15. doi: 10.1016/j.drup.2017.07.002
21. Dieci MV, Criscitiello C, Goubar A, Viale G, Conte P, Guarneri V, et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann Oncol (2014) 25:611–8. doi: 10.1093/annonc/mdt556
22. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Rivoltini L, Topalian SL, et al. Cloning of the gene coding for a shared human melanoma antigen recognized by autologous T cells infiltrating into tumor. Proc Natl Acad Sci United States America (1994) 91:3515–9. doi: 10.1073/pnas.91.9.3515
23. Kawakami Y, Eliyahu S, Delgado CH, Robbins PF, Sakaguchi K, Appella E, et al. Identification of a human melanoma antigen recognized by tumor-infiltrating lymphocytes associated with in vivo tumor rejection. Proc Natl Acad Sci United States America (1994) 91:6458–62. doi: 10.1073/pnas.91.14.6458
24. Magalhaes I, Carvalho-Queiroz C, Hartana CA, Kaiser A, Lukic A, Mints M, et al. Facing the future: challenges and opportunities in adoptive T cell therapy in cancer. Expert Opin Biol Ther (2019) 19:811–27. doi: 10.1080/14712598.2019.1608179
25. Mayor P, Starbuck K, Zsiros E. Adoptive cell transfer using autologous tumor infiltrating lymphocytes in gynecologic malignancies. Gynecol Oncol (2018) 150:361–9. doi: 10.1016/j.ygyno.2018.05.024
26. Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: Recognition and management. Blood (2016) 127:3321–30. doi: 10.1182/blood-2016-04-703751
27. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med (2014) 371:1507–17. doi: 10.1056/NEJMoa1407222
28. Paul Symonds R, Foweraker K. Principles of chemotherapy and radiotherapy. Curr Obstetrics Gynaecol (2006) 16:100–6. doi: 10.1016/j.curobgyn.2006.01.006
29. Monsuez JJ, Charniot JC, Vignat N, Artigou JY. Cardiac side-effects of cancer chemotherapy. Int J Cardiol (2010) 144:3–15. doi: 10.1016/j.ijcard.2010.03.003
30. Barazzuol L, Coppes RP, van Luijk P. Prevention and treatment of radiotherapy-induced side effects. Mol Oncol (2020) 14:1538–54. doi: 10.1002/1878-0261.12750
31. DiNofia AM, Grupp SA. Will allogeneic CAR T cells for CD19+ malignancies take autologous CAR T cells ‘off the shelf’? Nat Rev Clin Oncol (2021) 18:195–6. doi: 10.1038/s41571-021-00485-1
32. June CH, O'Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science (2018) 359:1361–5. doi: 10.1126/science.aar6711
33. Safarzadeh Kozani P, Naseri A, Mirarefin SMJ, Salem F, Nikbakht M, Evazi Bakhshi S, et al. Nanobody-based CAR-T cells for cancer immunotherapy. biomark Res (2022) 10:24. doi: 10.1186/s40364-022-00371-7
34. Fujiwara K, Tsunei A, Kusabuka H, Ogaki E, Tachibana M, Okada N. Hinge and transmembrane domains of chimeric antigen receptor regulate receptor expression and signaling threshold. Cells (2020) 9:1182. doi: 10.3390/cells9051182
35. Muller YD, Nguyen DP, Ferreira LMR, Ho P, Raffin C, Valencia RVB, et al. The CD28-transmembrane domain mediates chimeric antigen receptor heterodimerization with CD28. Front Immunol (2021) 12:639818. doi: 10.3389/fimmu.2021.639818
36. Ferreira LMR, Muller YD. CAR T-cell therapy: Is CD28-CAR heterodimerization its achilles’ heel? Front Immunol (2021) 12:766220. doi: 10.3389/fimmu.2021.766220
37. Frigault MJ, Lee J, Basil MC, Carpenito C, Motohashi S, Scholler J, et al. Identification of chimeric antigen receptors that mediate constitutive or inducible proliferation of T cells. Cancer Immunol Res (2015) 3:356–67. doi: 10.1158/2326-6066.CIR-14-0186
38. Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol Ther (2017) 25:2452–65. doi: 10.1016/j.ymthe.2017.07.013
39. Majzner RG, Rietberg SP, Sotillo E, Dong R, Vachharajani VT, Labanieh L, et al. Tuning the antigen density requirement for CAR T-cell activity. Cancer Discovery (2020) 10:702–23. doi: 10.1158/2159-8290.Cd-19-0945
40. van der Stegen SJ, Hamieh M, Sadelain M. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discovery (2015) 14:499–509. doi: 10.1038/nrd4597
41. Zhong XS, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther (2010) 18:413–20. doi: 10.1038/mt.2009.210
42. Duell J, Lurati S, Dittrich M, Bedke T, Pule M, Einsele H, et al. First generation chimeric antigen receptor display functional defects in key signal pathways upon antigen stimulation. Blood (2010) 116:2088. doi: 10.1182/blood.V116.21.2088.2088
43. Kershaw MH, Westwood JA, Parker LL, Wang G, Eshhar Z, Mavroukakis SA, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res (2006) 12:6106–15. doi: 10.1158/1078-0432.ccr-06-1183
44. Chmielewski M, Abken H. TRUCKs: The fourth generation of CARs. Expert Opin Biol Ther (2015) 15:1145–54. doi: 10.1517/14712598.2015.1046430
45. Tokarew N, Ogonek J, Endres S, von Bergwelt-Baildon M, Kobold S. Teaching an old dog new tricks: next-generation CAR T cells. Br J Cancer (2019) 120:26–37. doi: 10.1038/s41416-018-0325-1
46. Yeku OO, Brentjens RJ. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem Soc Trans (2016) 44:412–8. doi: 10.1042/bst20150291
47. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res (2011) 71:5697–706. doi: 10.1158/0008-5472.Can-11-0103
48. Kagoya Y, Tanaka S, Guo T, Anczurowski M, Wang CH, Saso K, et al. A novel chimeric antigen receptor containing a JAK-STAT signaling domain mediates superior antitumor effects. Nat Med (2018) 24:352–9. doi: 10.1038/nm.4478
49. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med (2015) 21:581–90. doi: 10.1038/nm.3838
50. Landoni E, Fucá G, Wang J, Chirasani VR, Yao Z, Dukhovlinova E, et al. Modifications to the framework regions eliminate chimeric antigen receptor tonic signaling. Cancer Immunol Res (2021) 9:441–53. doi: 10.1158/2326-6066.Cir-20-0451
51. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. CAR T cells redirected against tumor-specific antigen glycoforms: can low-sugar antigens guarantee a sweet success? Front Med (2022) 16:322–38. doi: 10.1007/s11684-021-0901-2
52. Park S, Shevlin E, Vedvyas Y, Zaman M, Park S, Hsu Y-MS, et al. Micromolar affinity CAR T cells to ICAM-1 achieves rapid tumor elimination while avoiding systemic toxicity. Sci Rep (2017) 7:14366. doi: 10.1038/s41598-017-14749-3
53. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365:725–33. doi: 10.1056/NEJMoa1103849
54. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368:1509–18. doi: 10.1056/NEJMoa1215134
55. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with b-cell lymphoblastic leukemia. N Engl J Med (2018) 378:439–48. doi: 10.1056/NEJMoa1709866
56. Bouchkouj N, Kasamon YL, de Claro RA, George B, Lin X, Lee S, et al. FDA Approval summary: Axicabtagene ciloleucel for relapsed or refractory large b-cell lymphoma. Clin Cancer Res (2019) 25:1702–8. doi: 10.1158/1078-0432.CCR-18-2743
57. Mullard A. FDA Approves first BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discovery (2021) 20:332. doi: 10.1038/d41573-021-00063-1
58. Voelker R. CAR-T therapy is approved for mantle cell lymphoma. Jama (2020) 324:832. doi: 10.1001/jama.2020.15456
59. Mullard A. FDA Approves first CAR T therapy. Nat Rev Drug Discovery (2017) 16:669. doi: 10.1038/nrd.2017.196
60. Mullard A. FDA Approves fourth CAR-T cell therapy. Nat Rev Drug Discovery (2021) 20:166. doi: 10.1038/d41573-021-00031-9
61. FDA Approves second CAR T-cell therapy. Cancer Discovery (2018) 8:5–6. doi: 10.1158/2159-8290.Cd-nb2017-155
62. Prasad V. Immunotherapy: Tisagenlecleucel - the first approved CAR-t-cell therapy: implications for payers and policy makers. Nat Rev Clin Oncol (2018) 15:11–2. doi: 10.1038/nrclinonc.2017.156
63. Mullard A. FDA Approves second BCMA-targeted CAR-T cell therapy. Nat Rev Drug Discovery (2022) 21:249. doi: 10.1038/d41573-022-00048-8
64. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in adult relapsed or refractory diffuse Large b-cell lymphoma. New Engl J Med (2018) 380:45–56. doi: 10.1056/NEJMoa1804980
65. Fowler NH, Dickinson M, Dreyling M, Martinez-Lopez J, Kolstad A, Butler J, et al. Tisagenlecleucel in adult relapsed or refractory follicular lymphoma: the phase 2 ELARA trial. Nat Med (2022) 28:325–32. doi: 10.1038/s41591-021-01622-0
66. Jacobson CA, Chavez JC, Sehgal AR, William BM, Munoz J, Salles G, et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): a single-arm, multicentre, phase 2 trial. Lancet Oncol (2022) 23:91–103. doi: 10.1016/s1470-2045(21)00591-x
67. Frey NV. Approval of brexucabtagene autoleucel for adults with relapsed and refractory acute lymphocytic leukemia. Blood (2022) 140:11–5. doi: 10.1182/blood.2021014892
68. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. CAR-T cell therapy in T-cell malignancies: Is success a low-hanging fruit? Stem Cell Res Ther (2021) 12:527. doi: 10.1186/s13287-021-02595-0
69. Safarzadeh Kozani P, Safarzadeh Kozani P, Ahmadi Najafabadi M, Yousefi F, Mirarefin SMJ, Rahbarizadeh F. Recent advances in solid tumor CAR-T cell therapy: Driving tumor cells from hero to zero? Front Immunol (2022) 13:795164. doi: 10.3389/fimmu.2022.795164
70. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. Addressing the obstacles of CAR T cell migration in solid tumors: wishing a heavy traffic. Crit Rev Biotechnol (2022) 42:1079–98. doi: 10.1080/07388551.2021.1988509
71. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. Novel antigens of CAR T cell therapy: New roads; old destination. Transl Oncol (2021) 14:101079. doi: 10.1016/j.tranon.2021.101079
72. Kailayangiri S, Altvater B, Wiebel M, Jamitzky S, Rossig C. Overcoming heterogeneity of antigen expression for effective CAR T cell targeting of cancers. Cancers (2020) 12:1075. doi: 10.3390/cancers12051075
73. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med (2017) 9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984
74. Anurathapan U, Chan RC, Hindi HF, Mucharla R, Bajgain P, Hayes BC, et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol Ther (2014) 22:623–33. doi: 10.1038/mt.2013.262
75. Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ. The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology (2013) 138:105–15. doi: 10.1111/imm.12036
76. Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA, Kang CY. Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: An alternative cell-based antitumor vaccine. J Immunol (2009) 182:1818–28. doi: 10.4049/jimmunol.0802430
77. Davis RJ, Van Waes C, Allen CT. Overcoming barriers to effective immunotherapy: MDSCs, TAMs, and tregs as mediators of the immunosuppressive microenvironment in head and neck cancer. Oral Oncol (2016) 58:59–70. doi: 10.1016/j.oraloncology.2016.05.002
78. Che Y, Yang Y, Suo J, An Y, Wang X. Induction of systemic immune responses and reversion of immunosuppression in the tumor microenvironment by a therapeutic vaccine for cervical cancer. Cancer Immunol Immunother (2020) 69:2651–64. doi: 10.1007/s00262-020-02651-3
79. Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol (2005) 25:9543–53. doi: 10.1128/mcb.25.21.9543-9553.2005
80. Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med (2013) 5:215ra172. doi: 10.1126/scitranslmed.3006597
81. Sharma P, Wagner K, Wolchok JD, Allison JP. Novel cancer immunotherapy agents with survival benefit: recent successes and next steps. Nat Rev Cancer (2011) 11:805–12. doi: 10.1038/nrc3153
82. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer (2012) 12:252–64. doi: 10.1038/nrc3239
83. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523:231–5. doi: 10.1038/nature14404
84. Li X, Xiang Y, Li F, Yin C, Li B, Ke X. WNT/β-catenin signaling pathway regulating T cell-inflammation in the tumor microenvironment. Front Immunol (2019) 10:2293. doi: 10.3389/fimmu.2019.02293
85. Xue J, Yu X, Xue L, Ge X, Zhao W, Peng W. Intrinsic β-catenin signaling suppresses CD8(+) T-cell infiltration in colorectal cancer. BioMed Pharmacother (2019) 115:108921. doi: 10.1016/j.biopha.2019.108921
86. Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest (2012) 122:899–910. doi: 10.1172/jci45817
87. Poznansky MC, Olszak IT, Evans RH, Wang Z, Foxall RB, Olson DP, et al. Thymocyte emigration is mediated by active movement away from stroma-derived factors. J Clin Invest (2002) 109:1101–10. doi: 10.1172/jci13853
88. Li J, Li W, Huang K, Zhang Y, Kupfer G, Zhao Q. Chimeric antigen receptor T cell (CAR-T) immunotherapy for solid tumors: Lessons learned and strategies for moving forward. J Hematol Oncol (2018) 11:22. doi: 10.1186/s13045-018-0568-6
89. Beyer I, Li Z, Persson J, Liu Y, van Rensburg R, Yumul R, et al. Controlled extracellular matrix degradation in breast cancer tumors improves therapy by trastuzumab. Mol Ther (2011) 19:479–89. doi: 10.1038/mt.2010.256
90. Choi BD, Yu X, Castano AP, Bouffard AA, Schmidts A, Larson RC, et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat Biotechnol (2019) 37:1049–58. doi: 10.1038/s41587-019-0192-1
91. Choi BD, Cai M, Bigner DD, Mehta AI, Kuan CT, Sampson JH. Bispecific antibodies engage T cells for antitumor immunotherapy. Expert Opin Biol Ther (2011) 11:843–53. doi: 10.1517/14712598.2011.572874
92. Wing A, Fajardo CA, Posey AD Jr., Shaw C, Da T, Young RM, et al. Improving CART-cell therapy of solid tumors with oncolytic virus-driven production of a bispecific T-cell engager. Cancer Immunol Res (2018) 6:605–16. doi: 10.1158/2326-6066.Cir-17-0314
93. Porter CE, Rosewell Shaw A, Jung Y, Yip T, Castro PD, Sandulache VC, et al. Oncolytic adenovirus armed with BiTE, cytokine, and checkpoint inhibitor enables CAR T cells to control the growth of heterogeneous tumors. Mol Ther (2020) 28:1251–62. doi: 10.1016/j.ymthe.2020.02.016
94. Aalipour A, Le Boeuf F, Tang M, Murty S, Simonetta F, Lozano AX, et al. Viral delivery of CAR targets to solid tumors enables effective cell therapy. Mol Ther Oncol (2020) 17:232–40. doi: 10.1016/j.omto.2020.03.018
95. Tang X, Li Y, Ma J, Wang X, Zhao W, Hossain MA, et al. Adenovirus-mediated specific tumor tagging facilitates CAR-T therapy against antigen-mismatched solid tumors. Cancer Lett (2020) 487:1–9. doi: 10.1016/j.canlet.2020.05.013
96. Park AK, Fong Y, Kim SI, Yang J, Murad JP, Lu J, et al. Effective combination immunotherapy using oncolytic viruses to deliver CAR targets to solid tumors. Sci Transl Med (2020) 12:eaaz1863. doi: 10.1126/scitranslmed.aaz1863
97. Mei H, Li C, Jiang H, Zhao X, Huang Z, Jin D, et al. A bispecific CAR-T cell therapy targeting BCMA and CD38 in relapsed or refractory multiple myeloma. J Hematol Oncol (2021) 14:161. doi: 10.1186/s13045-021-01170-7
98. Fousek K, Watanabe J, George A, An X, Samaha H, Navai SA, et al. Targeting primary pre-b cell acute lymphoblastic leukemia and CD19-negative relapses using trivalent CAR T cells. Blood (2017) 130:4614. doi: 10.1182/blood.V130.Suppl_1.4614.4614
99. Fousek K, Watanabe J, Joseph SK, George A, An X, Byrd TT, et al. CAR T-cells that target acute b-lineage leukemia irrespective of CD19 expression. Leukemia (2021) 35:75–89. doi: 10.1038/s41375-020-0792-2
100. Wang N, Hu X, Cao W, Li C, Xiao Y, Cao Y, et al. Efficacy and safety of CAR19/22 T-cell cocktail therapy in patients with refractory/relapsed b-cell malignancies. Blood (2020) 135:17–27. doi: 10.1182/blood.2019000017
101. Halpin-Veszeleiova K, Hatfield SM. Oxygenation and A2AR blockade to eliminate hypoxia/HIF-1α-adenosinergic immunosuppressive axis and improve cancer immunotherapy. Curr Opin Pharmacol (2020) 53:84–90. doi: 10.1016/j.coph.2020.07.005
102. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co-Inhibition of CD73 and A2AR adenosine signaling improves anti-tumor immune responses. Cancer Cell (2016) 30:391–403. doi: 10.1016/j.ccell.2016.06.025
103. Hatfield SM, Kjaergaard J, Lukashev D, Belikoff B, Schreiber TH, Sethumadhavan S, et al. Systemic oxygenation weakens the hypoxia and hypoxia inducible factor 1α-dependent and extracellular adenosine-mediated tumor protection. J Mol Med (Berl) (2014) 92:1283–92. doi: 10.1007/s00109-014-1189-3
104. Juillerat A, Marechal A, Filhol JM, Valogne Y, Valton J, Duclert A, et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci Rep (2017) 7:39833. doi: 10.1038/srep39833
105. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
106. Grosser R, Cherkassky L, Chintala N, Adusumilli PS. Combination immunotherapy with CAR T cells and checkpoint blockade for the treatment of solid tumors. Cancer Cell (2019) 36:471–82. doi: 10.1016/j.ccell.2019.09.006
107. Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, et al. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother (2019) 68:365–77. doi: 10.1007/s00262-018-2281-2
108. Rafiq S, Yeku OO, Jackson HJ, Purdon TJ, van Leeuwen DG, Drakes DJ, et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat Biotechnol (2018) 36:847–56. doi: 10.1038/nbt.4195
109. Zhou JT, Liu JH, Song TT, Ma B, Amidula N, Bai C. EGLIF-CAR-T cells secreting PD-1 blocking antibodies significantly mediate the elimination of gastric cancer. Cancer Manag Res (2020) 12:8893–902. doi: 10.2147/cmar.S260915
110. Theruvath J, Sotillo E, Mount CW, Graef CM, Delaidelli A, Heitzeneder S, et al. Locoregionally administered B7-H3-targeted CAR T cells for treatment of atypical teratoid/rhabdoid tumors. Nat Med (2020) 26:712–9. doi: 10.1038/s41591-020-0821-8
111. Nellan A, Rota C, Majzner R, Lester-McCully CM, Griesinger AM, Mulcahy Levy JM, et al. Durable regression of medulloblastoma after regional and intravenous delivery of anti-HER2 chimeric antigen receptor T cells. J Immunother Cancer (2018) 6:30. doi: 10.1186/s40425-018-0340-z
112. Katz SC, Point GR, Cunetta M, Thorn M, Guha P, Espat NJ, et al. Regional CAR-T cell infusions for peritoneal carcinomatosis are superior to systemic delivery. Cancer Gene Ther (2016) 23:142–8. doi: 10.1038/cgt.2016.14
113. Jin L, Tao H, Karachi A, Long Y, Hou AY, Na M, et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat Commun (2019) 10:4016. doi: 10.1038/s41467-019-11869-4
114. Whilding LM, Halim L, Draper B, Parente-Pereira AC, Zabinski T, Davies DM, et al. CAR T-cells targeting the integrin αvβ6 and Co-expressing the chemokine receptor CXCR2 demonstrate enhanced homing and efficacy against several solid malignancies. Cancers (2019) 11:674. doi: 10.3390/cancers11050674
115. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE, et al. T Lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood (2009) 113:6392–402. doi: 10.1182/blood-2009-03-209650
116. Newick K, O'Brien S, Sun J, Kapoor V, Maceyko S, Lo A, et al. Augmentation of CAR T-cell trafficking and antitumor efficacy by blocking protein kinase a localization. Cancer Immunol Res (2016) 4:541–51. doi: 10.1158/2326-6066.Cir-15-0263
117. Chen Q, Hu Q, Dukhovlinova E, Chen G, Ahn S, Wang C, et al. Photothermal therapy promotes tumor infiltration and antitumor activity of CAR T cells. Adv Mater (2019) 31:e1900192. doi: 10.1002/adma.201900192
118. Miller IC, Zamat A, Sun LK, Phuengkham H, Harris AM, Gamboa L, et al. Enhanced intratumoural activity of CAR T cells engineered to produce immunomodulators under photothermal control. Nat BioMed Eng (2021) 5:1348–59. doi: 10.1038/s41551-021-00781-2
119. Bocca P, Di Carlo E, Caruana I, Emionite L, Cilli M, De Angelis B, et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology (2017) 7:e1378843. doi: 10.1080/2162402x.2017.1378843
120. Deng C, Zhao J, Zhou S, Dong J, Cao J, Gao J, et al. The vascular disrupting agent CA4P improves the antitumor efficacy of CAR-T cells in preclinical models of solid human tumors. Mol Ther (2020) 28:75–88. doi: 10.1016/j.ymthe.2019.10.010
121. Ferrara N, Adamis AP. Ten years of anti-vascular endothelial growth factor therapy. Nat Rev Drug Discovery (2016) 15:385–403. doi: 10.1038/nrd.2015.17
122. Wang W, Marín-Ramos NI, He H, Zeng S, Cho HY, Swenson SD, et al. NEO100 enables brain delivery of blood‒brain barrier impermeable therapeutics. Neuro Oncol (2021) 23:63–75. doi: 10.1093/neuonc/noaa206
123. Ghassemi S, Martinez-Becerra F, Master A, Richman SA, Heo D, Leferovich J, et al. Novel media formulations to enhance chimeric antigen receptor (CAR) T-cell potency and anti-tumor cell function for adoptive immunotherapy. Cytotherapy (2020) 22:S133. doi: 10.1016/j.jcyt.2020.03.260
124. Ghassemi S, Martinez-Becerra FJ, Master AM, Richman SA, Heo D, Leferovich J, et al. Enhancing chimeric antigen receptor T cell anti-tumor function through advanced media design. Mol Ther - Methods Clin Dev (2020) 18:595–606. doi: 10.1016/j.omtm.2020.07.008
125. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell (2016) 167:829–42:e13. doi: 10.1016/j.cell.2016.09.031
126. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood (2014) 123:3750–9. doi: 10.1182/blood-2014-01-552174
127. Giuffrida L, Sek K, Henderson MA, House IG, Lai J, Chen AXY, et al. IL-15 preconditioning augments CAR T cell responses to checkpoint blockade for improved treatment of solid tumors. Mol Ther (2020) 28:2379–93. doi: 10.1016/j.ymthe.2020.07.018
128. Alizadeh D, Wong RA, Yang X, Wang D, Pecoraro JR, Kuo CF, et al. IL15 enhances CAR-T cell antitumor activity by reducing mTORC1 activity and preserving their stem cell memory phenotype. Cancer Immunol Res (2019) 7:759–72. doi: 10.1158/2326-6066.Cir-18-0466
129. Canestrari E, Steidinger HR, McSwain B, Charlebois SJ, Dann CT. Human platelet lysate media supplement supports lentiviral transduction and expansion of human T lymphocytes while maintaining memory phenotype. J Immunol Res (2019) 2019:3616120. doi: 10.1155/2019/3616120
130. Torres Chavez A, McKenna MK, Canestrari E, Dann CT, Ramos CA, Lulla P, et al. Expanding CAR T cells in human platelet lysate renders T cells with in vivo longevity. J Immunother Cancer (2019) 7:330. doi: 10.1186/s40425-019-0804-9
131. Slaney CY, von Scheidt B, Davenport AJ, Beavis PA, Westwood JA, Mardiana S, et al. Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of Large solid tumors in an immunocompetent, self-antigen setting. Clin Cancer Res (2017) 23:2478–90. doi: 10.1158/1078-0432.Ccr-16-1860
132. Tanaka M, Tashiro H, Omer B, Lapteva N, Ando J, Ngo M, et al. Vaccination targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T cells. Clin Cancer Res (2017) 23:3499–509. doi: 10.1158/1078-0432.Ccr-16-2138
133. Wu M, Zhang L, Zhang H, Ning J, Tu S, He Y, et al. CD19 chimeric antigen receptor-redirected T cells combined with epidermal growth factor receptor pathway substrate 8 peptide-derived dendritic cell vaccine in leukemia. Cytotherapy (2019) 21:659–70. doi: 10.1016/j.jcyt.2019.03.313
134. Chan JD, von Scheidt B, Zeng B, Oliver AJ, Davey AS, Ali AI, et al. Enhancing chimeric antigen receptor T-cell immunotherapy against cancer using a nanoemulsion-based vaccine targeting cross-presenting dendritic cells. Clin Transl Immunol (2020) 9:e1157. doi: 10.1002/cti2.1157
135. Ma L, Dichwalkar T, Chang JYH, Cossette B, Garafola D, Zhang AQ, et al. Enhanced CAR-T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science (2019) 365:162–8. doi: 10.1126/science.aav8692
136. Reinhard K, Rengstl B, Oehm P, Michel K, Billmeier A, Hayduk N, et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science (2020) 367:446–53. doi: 10.1126/science.aay5967
137. Ilieva KM, Cheung A, Mele S, Chiaruttini G, Crescioli S, Griffin M, et al. Chondroitin sulfate proteoglycan 4 and its potential as an antibody immunotherapy target across different tumor types. Front Immunol (2018) 8:1911. doi: 10.3389/fimmu.2017.01911
138. Amoury M, Mladenov R, Nachreiner T, Pham AT, Hristodorov D, Di Fiore S, et al. A novel approach for targeted elimination of CSPG4-positive triple-negative breast cancer cells using a MAP tau-based fusion protein. Int J Cancer (2016) 139:916–27. doi: 10.1002/ijc.30119
139. Geldres C, Savoldo B, Hoyos V, Caruana I, Zhang M, Yvon E, et al. T Lymphocytes redirected against the chondroitin sulfate proteoglycan-4 control the growth of multiple solid tumors both in vitro and in vivo. Clin Cancer Res (2014) 20:962–71. doi: 10.1158/1078-0432.ccr-13-2218
140. Beard RE, Zheng Z, Lagisetty KH, Burns WR, Tran E, Hewitt SM, et al. Multiple chimeric antigen receptors successfully target chondroitin sulfate proteoglycan 4 in several different cancer histologies and cancer stem cells. J immunother Cancer (2014) 2:25. doi: 10.1186/2051-1426-2-25
141. Müller N. The role of intercellular adhesion molecule-1 in the pathogenesis of psychiatric disorders. Front Pharmacol (2019) 10:1251. doi: 10.3389/fphar.2019.01251
142. Guo P, Huang J, Wang L, Jia D, Yang J, Dillon DA, et al. ICAM-1 as a molecular target for triple negative breast cancer. Proc Natl Acad Sci United States America (2014) 111:14710–5. doi: 10.1073/pnas.1408556111
143. Vedvyas Y, McCloskey JE, Yang Y, Min IM, Fahey TJ, Zarnegar R, et al. Manufacturing and preclinical validation of CAR T cells targeting ICAM-1 for advanced thyroid cancer therapy. Sci Rep (2019) 9:10634. doi: 10.1038/s41598-019-46938-7
144. Yang Y, Vedvyas Y, McCloskey JE, Min IM, Jin MM. Abstract 2322: ICAM-1 targeting CAR T cell therapy for triple negative breast cancer. Cancer Res (2019) 79:2322. doi: 10.1158/1538-7445.AM2019-2322
145. Morisaki T, Onishi H, Katano M. Cancer immunotherapy using NKG2D and DNAM-1 systems. Anticancer Res (2012) 32:2241–7.
146. Zhang T, Lemoi BA, Sentman CL. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood (2005) 106:1544–51. doi: 10.1182/blood-2004-11-4365
147. Sallman DA, Brayer J, Sagatys EM, Lonez C, Breman E, Agaugué S, et al. NKG2D-based chimeric antigen receptor therapy induced remission in a relapsed/refractory acute myeloid leukemia patient. Haematologica (2018) 103:e424–e6. doi: 10.3324/haematol.2017.186742
148. Murad JM, Baumeister SH, Werner L, Daley H, Trébéden-Negre H, Reder J, et al. Manufacturing development and clinical production of NKG2D chimeric antigen receptor-expressing T cells for autologous adoptive cell therapy. Cytotherapy (2018) 20:952–63. doi: 10.1016/j.jcyt.2018.05.001
149. Han Y, Xie W, Song DG, Powell DJ Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J Hematol Oncol (2018) 11:92. doi: 10.1186/s13045-018-0635-z
150. Feneyrolles C, Spenlinhauer A, Guiet L, Fauvel B, Daydé-Cazals B, Warnault P, et al. Axl kinase as a key target for oncology: Focus on small molecule inhibitors. Mol Cancer Ther (2014) 13:2141–8. doi: 10.1158/1535-7163.mct-13-1083
151. Barata PC, Rini BI. Treatment of renal cell carcinoma: Current status and future directions. CA: Cancer J Clin (2017) 67:507–24. doi: 10.3322/caac.21411
152. Holland SJ, Pan A, Franci C, Hu Y, Chang B, Li W, et al. R428, a selective small molecule inhibitor of axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res (2010) 70:1544–54. doi: 10.1158/0008-5472.can-09-2997
153. Ye X, Li Y, Stawicki S, Couto S, Eastham-Anderson J, Kallop D, et al. An anti-axl monoclonal antibody attenuates xenograft tumor growth and enhances the effect of multiple anticancer therapies. Oncogene (2010) 29:5254–64. doi: 10.1038/onc.2010.268
154. Wei J, Sun H, Zhang A, Wu X, Li Y, Liu J, et al. A novel AXL chimeric antigen receptor endows T cells with anti-tumor effects against triple negative breast cancers. Cell Immunol (2018) 331:49–58. doi: 10.1016/j.cellimm.2018.05.004
155. Zhao Z, Li Y, Liu W, Li X. Engineered IL-7 receptor enhances the therapeutic effect of AXL-CAR-T cells on triple-negative breast cancer. BioMed Res Int (2020) 2020:4795171. doi: 10.1155/2020/4795171
156. Fu S, Tong X, Cai C, Zhao Y, Wu Y, Li Y, et al. The structure of tumor endothelial marker 8 (TEM8) extracellular domain and implications for its receptor function for recognizing anthrax toxin. PloS One (2010) 5:e11203. doi: 10.1371/journal.pone.0011203
157. Davies G, Rmali KA, Watkins G, Mansel RE, Mason MD, Jiang WG. Elevated levels of tumour endothelial marker-8 in human breast cancer and its clinical significance. Int J Oncol (2006) 29:1311–7. doi: 10.3892/ijo.29.5.1311
158. Gutwein LG, Al-Quran SZ, Fernando S, Fletcher BS, Copeland EM, Grobmyer SR. Tumor endothelial marker 8 expression in triple-negative breast cancer. Anticancer Res (2011) 31:3417–22.
159. Opoku-Darko M, Yuen C, Gratton K, Sampson E, Bathe OF. Tumor endothelial marker 8 overexpression in breast cancer cells enhances tumor growth and metastasis. Cancer Invest (2011) 29:676–82. doi: 10.3109/07357907.2011.626474
160. Chaudhary A, Hilton MB, Seaman S, Haines DC, Stevenson S, Lemotte PK, et al. TEM8/ANTXR1 blockade inhibits pathological angiogenesis and potentiates tumoricidal responses against multiple cancer types. Cancer Cell (2012) 21:212–26. doi: 10.1016/j.ccr.2012.01.004
161. Fernando S, Fletcher BS. Targeting tumor endothelial marker 8 in the tumor vasculature of colorectal carcinomas in mice. Cancer Res (2009) 69:5126–32. doi: 10.1158/0008-5472.can-09-0725
162. Byrd TT, Fousek K, Pignata A, Szot C, Samaha H, Seaman S, et al. TEM8/ANTXR1-specific CAR T cells as a targeted therapy for triple-negative breast cancer. Cancer Res (2018) 78:489–500. doi: 10.1158/0008-5472.can-16-1911
163. Petrovic K, Robinson J, Whitworth K, Jinks E, Shaaban A, Lee SP. TEM8/ANTXR1-specific CAR T cells mediate toxicity in vivo. PloS One (2019) 14:e0224015. doi: 10.1371/journal.pone.0224015
164. Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer (2010) 10:9–22. doi: 10.1038/nrc2748
165. Brooks PC, Clark RA, Cheresh DA. Requirement of vascular integrin alpha v beta 3 for angiogenesis. Science (1994) 264:569–71. doi: 10.1126/science.7512751
166. Kumar CC. Integrin alpha v beta 3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr Drug Targets (2003) 4:123–31. doi: 10.2174/1389450033346830
167. Lim M, Guccione S, Haddix T, Sims L, Cheshier S, Chu P, et al. alpha(v)beta(3) integrin in central nervous system tumors. Hum Pathol (2005) 36:665–9. doi: 10.1016/j.humpath.2005.03.014
168. Schnell O, Krebs B, Wagner E, Romagna A, Beer AJ, Grau SJ, et al. Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol (2008) 18:378–86. doi: 10.1111/j.1750-3639.2008.00137.x
169. Wick W, Platten M, Wick A, Hertenstein A, Radbruch A, Bendszus M, et al. Current status and future directions of anti-angiogenic therapy for gliomas. Neuro Oncol (2016) 18:315–28. doi: 10.1093/neuonc/nov180
170. Gutheil JC, Campbell TN, Pierce PR, Watkins JD, Huse WD, Bodkin DJ, et al. Targeted antiangiogenic therapy for cancer using vitaxin: a humanized monoclonal antibody to the integrin alphavbeta3. Clin Cancer Res (2000) 6:3056–61.
171. Patel SR, Jenkins J, Papadopolous N, Burgess MA, Plager C, Gutterman J, et al. Pilot study of vitaxin–an angiogenesis inhibitor-in patients with advanced leiomyosarcomas. Cancer (2001) 92:1347–8. doi: 10.1002/1097-0142(20010901)92:5<1347::aid-cncr1456>3.0.co;2-0
172. Posey JA, Khazaeli MB, DelGrosso A, Saleh MN, Lin CY, Huse W, et al. A pilot trial of vitaxin, a humanized anti-vitronectin receptor (anti alpha v beta 3) antibody in patients with metastatic cancer. Cancer Biother Radiopharm (2001) 16:125–32. doi: 10.1089/108497801300189218
173. Wallstabe L, Mades A, Frenz S, Einsele H, Rader C, Hudecek M. CAR T cells targeting α(v)β(3) integrin are effective against advanced cancer in preclinical models. Adv Cell Gene Ther (2018) 1:e11. doi: 10.1002/acg2.11
174. Rader C, Cheresh DA, Barbas C. A phage display approach for rapid antibody humanization: designed combinatorial V gene libraries. Proc Natl Acad Sci U.S.A. (1998) 95:8910–5. doi: 10.1073/pnas.95.15.8910
175. Cobb DA, de Rossi J, Liu L, An E, Lee DW. Targeting of the alpha(v) beta(3) integrin complex by CAR-T cells leads to rapid regression of diffuse intrinsic pontine glioma and glioblastoma. J Immunother Cancer (2022) 10:e003816. doi: 10.1136/jitc-2021-003816
176. Matsuda T, Nomi M, Ikeya M, Kani S, Oishi I, Terashima T, et al. Expression of the receptor tyrosine kinase genes, Ror1 and Ror2, during mouse development. Mech Dev (2001) 105:153–6. doi: 10.1016/s0925-4773(01)00383-5
177. Hudecek M, Schmitt TM, Baskar S, Lupo-Stanghellini MT, Nishida T, Yamamoto TN, et al. The b-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood (2010) 116:4532–41. doi: 10.1182/blood-2010-05-283309
178. Hudecek M, Lupo-Stanghellini M-T, Kosasih PL, Sommermeyer D, Jensen MC, Rader C, et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin Cancer Res (2013) 19:3153. doi: 10.1158/1078-0432.CCR-13-0330
179. Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS, et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell (2021) 39:193–208:e10. doi: 10.1016/j.ccell.2020.11.005
180. Gohil SH, Della Peruta M, Paredes-Moscosso SR, Harrasser M, Cheung GW-K, Davies DM, et al. Novel humanised ROR1 chimeric antigen receptors for the treatment of haematological malignancies. Blood (2016) 128:3361. doi: 10.1182/blood.V128.22.3361.3361
181. Gohil S, Paredes-Moscosso S, Harrasser M, Davidoff A, Pule M, Della Peruta M, et al. Preclinical development of novel humanised ROR1 targeting chimeric antigen receptor T cells and bispecific T-cell engagers. Lancet (2017) 389:S40. doi: 10.1016/S0140-6736(17)30436-1
182. Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, et al. Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell (2019) 35:489–503:e8. doi: 10.1016/j.ccell.2019.02.003
183. Wallstabe L, Göttlich C, Nelke LC, Kühnemundt J, Schwarz T, Nerreter T, et al. ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight (2019) 4:e126345. doi: 10.1172/jci.insight.126345
184. Specht JM, Lee SM, Turtle C, Berger C, Balakrishnan A, Srivastava S, et al. Abstract P2-09-13: A phase I study of adoptive immunotherapy for ROR1+ advanced triple negative breast cancer (TNBC) with defined subsets of autologous T cells expressing a ROR1-specific chimeric antigen receptor (ROR1-CAR). Cancer Res (2019) 79:P2-09-13. doi: 10.1158/1538-7445.SABCS18-P2-09-13
185. Zhao X, Qu J, Hui Y, Zhang H, Sun Y, Liu X, et al. Clinicopathological and prognostic significance of c-met overexpression in breast cancer. Oncotarget (2017) 8:56758–67. doi: 10.18632/oncotarget.18142
186. Kim YJ, Choi JS, Seo J, Song JY, Lee SE, Kwon MJ, et al. MET is a potential target for use in combination therapy with EGFR inhibition in triple-negative/basal-like breast cancer. Int J Cancer (2014) 134:2424–36. doi: 10.1002/ijc.28566
187. Han K, Chanu P, Jonsson F, Winter H, Bruno R, Jin J, et al. Exposure-response and tumor growth inhibition analyses of the monovalent anti-c-MET antibody onartuzumab (MetMAb) in the second- and third-line non-small cell lung cancer. AAPS J (2017) 19:527–33. doi: 10.1208/s12248-016-0029-5
188. Wang J, Cheng JX. C-met inhibition enhances chemosensitivity of human ovarian cancer cells. Clin Exp Pharmacol Physiol (2017) 44:79–87. doi: 10.1111/1440-1681.12672
189. Tchou J, Zhao Y, Levine BL, Zhang PJ, Davis MM, Melenhorst JJ, et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol Res (2017) 5:1152–61. doi: 10.1158/2326-6066.Cir-17-0189
190. Shah PD, Huang ACC, Xu X, Zhang PJ, Orlowski R, Matlawski T, et al. Phase I trial of autologous cMET-directed CAR-t cells administered intravenously in patients with melanoma & breast carcinoma. J Clin Oncol (2020) 38:10035. doi: 10.1200/JCO.2020.38.15_suppl.10035
191. Xia W, Low PS. Folate-targeted therapies for cancer. J medicinal Chem (2010) 53:6811–24. doi: 10.1021/jm100509v
192. Salazar MD, Ratnam M. The folate receptor: what does it promise in tissue-targeted therapeutics? Cancer metastasis Rev (2007) 26:141–52. doi: 10.1007/s10555-007-9048-0
193. Boogerd LS, Boonstra MC, Beck AJ, Charehbili A, Hoogstins CE, Prevoo HA, et al. Concordance of folate receptor-α expression between biopsy, primary tumor and metastasis in breast cancer and lung cancer patients. Oncotarget (2016) 7:17442–54. doi: 10.18632/oncotarget.7856
194. Toffoli G, Cernigoi C, Russo A, Gallo A, Bagnoli M, Boiocchi M. Overexpression of folate binding protein in ovarian cancers. Int J Cancer (1997) 74:193–8. doi: 10.1002/(sici)1097-0215(19970422)74:2<193::aid-ijc10>3.0.co;2-f
195. O'Shannessy DJ, Somers EB, Maltzman J, Smale R, Fu YS. Folate receptor alpha (FRA) expression in breast cancer: identification of a new molecular subtype and association with triple negative disease. SpringerPlus (2012) 1:22. doi: 10.1186/2193-1801-1-22
196. Song DG, Ye Q, Carpenito C, Poussin M, Wang LP, Ji C, et al. In vivo persistence, tumor localization, and antitumor activity of CAR-engineered T cells is enhanced by costimulatory signaling through CD137 (4-1BB). Cancer Res (2011) 71:4617–27. doi: 10.1158/0008-5472.can-11-0422
197. Song DG, Ye Q, Poussin M, Chacon JA, Figini M, Powell DJ Jr. Effective adoptive immunotherapy of triple-negative breast cancer by folate receptor-alpha redirected CAR T cells is influenced by surface antigen expression level. J Hematol Oncol (2016) 9:56. doi: 10.1186/s13045-016-0285-y
198. Lanitis E, Poussin M, Klattenhoff AW, Song D, Sandaltzopoulos R, June CH, et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res (2013) 1:43–53. doi: 10.1158/2326-6066.cir-13-0008
199. Kim MS, Ma JS, Yun H, Cao Y, Kim JY, Chi V, et al. Redirection of genetically engineered CAR-T cells using bifunctional small molecules. J Am Chem Soc (2015) 137:2832–5. doi: 10.1021/jacs.5b00106
200. Nakai K, Hung MC, Yamaguchi H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am J Cancer Res (2016) 6:1609–23.
201. Xia L, Z-z Z, Liu J-y, Chen Y-j, Ding J-c, Xia N-s, et al. EGFR-targeted CAR-T cells are potent and specific in suppressing triple-negative breast cancer both in vitro and in vivo. Clin Trans Immunol (2020) 9:e1135. doi: 10.1002/cti2.1135
202. Zhang Y, Zhang Z, Ding Y, Fang Y, Wang P, Chu W, et al. Phase I clinical trial of EGFR-specific CAR-T cells generated by the piggyBac transposon system in advanced relapsed/refractory non-small cell lung cancer patients. J Cancer Res Clin Oncol (2021) 147:3725–34. doi: 10.1007/s00432-021-03613-7
203. Li H, Huang Y, Jiang D-Q, Cui L-Z, He Z, Wang C, et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis (2018) 9:177. doi: 10.1038/s41419-017-0238-6
204. Li G, Guo J, Zheng Y, Ding W, Han Z, Qin L, et al. CXCR5 guides migration and tumor eradication of anti-EGFR chimeric antigen receptor T cells. Mol Ther - Oncol (2021) 22:507–17. doi: 10.1016/j.omto.2021.07.003
205. Liu Y, Zhou Y, Huang KH, Li Y, Fang X, An L, et al. EGFR-specific CAR-T cells trigger cell lysis in EGFR-positive TNBC. Aging (2019) 11:11054–72. doi: 10.18632/aging.102510
206. Caruso HG, Tanaka R, Liang J, Ling X, Sabbagh A, Henry VK, et al. Shortened ex vivo manufacturing time of EGFRvIII-specific chimeric antigen receptor (CAR) T cells reduces immune exhaustion and enhances antiglioma therapeutic function. J neuro-oncol (2019) 145:429–39. doi: 10.1007/s11060-019-03311-y
207. Yoon DH, Osborn MJ, Tolar J, Kim CJ. Incorporation of immune checkpoint blockade into chimeric antigen receptor T cells (CAR-ts): Combination or built-in CAR-T. Int J Mol Sci (2018) 19:340. doi: 10.3390/ijms19020340
208. Tozbikian G, Brogi E, Kadota K, Catalano J, Akram M, Patil S, et al. Mesothelin expression in triple negative breast carcinomas correlates significantly with basal-like phenotype, distant metastases and decreased survival. PloS One (2014) 9:e114900. doi: 10.1371/journal.pone.0114900
209. Parinyanitikul N, Blumenschein GR, Wu Y, Lei X, Chavez-Macgregor M, Smart M, et al. Mesothelin expression and survival outcomes in triple receptor negative breast cancer. Clin Breast Cancer (2013) 13:378–84. doi: 10.1016/j.clbc.2013.05.001
210. Klampatsa A, Dimou V, Albelda SM. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin Biol Ther (2021) 21:473–86. doi: 10.1080/14712598.2021.1843628
211. Tchou J, Wang LC, Selven B, Zhang H, Conejo-Garcia J, Borghaei H, et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res Treat (2012) 133:799–804. doi: 10.1007/s10549-012-2018-4
212. Del Bano J, Florès-Florès R, Josselin E, Goubard A, Ganier L, Castellano R, et al. A bispecific antibody-based approach for targeting mesothelin in triple negative breast cancer. Front Immunol (2019) 10:1593. doi: 10.3389/fimmu.2019.01593
213. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res (2014) 2:361–70. doi: 10.1158/2326-6066.Cir-13-0127
214. Voeller J, Sondel PM. Advances in anti-GD2 immunotherapy for treatment of high-risk neuroblastoma. J Pediatr hematology/oncol (2019) 41:163–9. doi: 10.1097/mph.0000000000001369
215. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD, et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood (2011) 118:6050–6. doi: 10.1182/blood-2011-05-354449
216. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14:1264–70. doi: 10.1038/nm.1882
217. Dhillon S. Dinutuximab: First global approval. Drugs (2015) 75:923–7. doi: 10.1007/s40265-015-0399-5
218. Battula VL, Nguyen K, Somanchi A, Mu H, Ueno NT, Lee D, et al. Abstract 1766: Dinutuximab targets GD2<sup<+</sup< breast cancer stem cells and inhibits TNBC tumor growth. Cancer Res (2018) 78:1766. doi: 10.1158/1538-7445.AM2018-1766
219. Ly S, Anand V, El-Dana F, Nguyen K, Cai Y, Cai S, et al. Anti-GD2 antibody dinutuximab inhibits triple-negative breast tumor growth by targeting GD2(+) breast cancer stem-like cells. J immunother Cancer (2021) 9:e001197. doi: 10.1136/jitc-2020-001197
220. Battula VL, Shi Y, Evans KW, Wang RY, Spaeth EL, Jacamo RO, et al. Ganglioside GD2 identifies breast cancer stem cells and promotes tumorigenesis. J Clin Invest (2012) 122:2066–78. doi: 10.1172/jci59735
221. Seitz CM, Schroeder S, Knopf P, Krahl AC, Hau J, Schleicher S, et al. GD2-targeted chimeric antigen receptor T cells prevent metastasis formation by elimination of breast cancer stem-like cells. Oncoimmunology (2020) 9:1683345. doi: 10.1080/2162402x.2019.1683345
222. Gendler S, Taylor-Papadimitriou J, Duhig T, Rothbard J, Burchell J. A highly immunogenic region of a human polymorphic epithelial mucin expressed by carcinomas is made up of tandem repeats. J Biol Chem (1988) 263:12820–3. doi: 10.1016/S0021-9258(18)37632-4
223. Müller S, Alving K, Peter-Katalinic J, Zachara N, Gooley AA, Hanisch FG. High density O-glycosylation on tandem repeat peptide from secretory MUC1 of T47D breast cancer cells. J Biol Chem (1999) 274:18165–72. doi: 10.1074/jbc.274.26.18165
224. Gendler SJ, Lancaster CA, Taylor-Papadimitriou J, Duhig T, Peat N, Burchell J, et al. Molecular cloning and expression of human tumor-associated polymorphic epithelial mucin. J Biol Chem (1990) 265:15286–93. doi: 10.1016/S0021-9258(18)77254-2
225. Zhou R, Yazdanifar M, Roy LD, Whilding LM, Gavrill A, Maher J, et al. CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front Immunol (2019) 10:1149. doi: 10.3389/fimmu.2019.01149
226. Posey AD Jr., Schwab RD, Boesteanu AC, Steentoft C, Mandel U, Engels B, et al. Engineered CAR T cells targeting the cancer-associated tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity (2016) 44:1444–54. doi: 10.1016/j.immuni.2016.05.014
227. Zhai X, You F, Xiang S, Jiang L, Chen D, Li Y, et al. MUC1-tn-targeting chimeric antigen receptor-modified Vγ9Vδ2 T cells with enhanced antigen-specific anti-tumor activity. Am J Cancer Res (2021) 11:79–91.
228. Gutierrez R, Shah PD, Hamid O, Garfall AL, Posey A, Bishop MR, et al. Phase I experience with first in class TnMUC1 targeted chimeric antigen receptor T-cells in patients with advanced TnMUC1 positive solid tumors. J Clin Oncol (2021) 39:e14513. doi: 10.1200/JCO.2021.39.15_suppl.e14513
229. Goldenberg DM, Stein R, Sharkey RM. The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget (2018) 9:28989–9006. doi: 10.18632/oncotarget.25615
230. Shvartsur A, Bonavida B. Trop2 and its overexpression in cancers: regulation and clinical/therapeutic implications. Genes Cancer (2015) 6:84–105. doi: 10.18632/genesandcancer.40
231. Liu H, Bai L, Huang L, Ning N, Li L, Li Y, et al. Bispecific antibody targeting TROP2xCD3 suppresses tumor growth of triple negative breast cancer. J ImmunoTher Cancer (2021) 9:e003468. doi: 10.1136/jitc-2021-003468
232. Bardia A, Mayer IA, Vahdat LT, Tolaney SM, Isakoff SJ, Diamond JR, et al. Sacituzumab govitecan-hziy in refractory metastatic triple-negative breast cancer. N Engl J Med (2019) 380:741–51. doi: 10.1056/NEJMoa1814213
233. Zhao W, Jia L, Zhang M, Huang X, Qian P, Tang Q, et al. The killing effect of novel bi-specific Trop2/PD-L1 CAR-T cell targeted gastric cancer. Am J Cancer Res (2019) 9:1846–56.
234. Bedoya DM, King T, Posey AD. Generation of CART cells targeting oncogenic TROP2 for the elimination of epithelial malignancies. Cytotherapy (2019) 21:S11–S2. doi: 10.1016/j.jcyt.2019.03.570
235. Safarzadeh Kozani P, Safarzadeh Kozani P, Rahbarizadeh F. Optimizing the clinical impact of CAR-T cell therapy in b-cell acute lymphoblastic leukemia: Looking back while moving forward. Front Immunol (2021) 12:765097. doi: 10.3389/fimmu.2021.765097
236. Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood (2014) 124:188–95. doi: 10.1182/blood-2014-05-552729
237. Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric antigen receptor T-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol (2020) 38:415–22. doi: 10.1200/jco.19.01892
238. Constantinescu C, Pasca S, Tat T, Teodorescu P, Vlad C, Iluta S, et al. Continuous renal replacement therapy in cytokine release syndrome following immunotherapy or cellular therapies? J immunother Cancer (2020) 8:e000742. doi: 10.1136/jitc-2020-000742
239. Liu Y, Chen X, Wang D, Li H, Huang J, Zhang Z, et al. Hemofiltration successfully eliminates severe cytokine release syndrome following CD19 CAR-T-Cell therapy. J Immunother (2018) 41:406–10. doi: 10.1097/cji.0000000000000243
240. Heng G, Jia J, Li S, Fu G, Wang M, Qin D, et al. Sustained therapeutic efficacy of humanized anti-CD19 chimeric antigen receptor T cells in Relapsed/Refractory acute lymphoblastic leukemia. Clin Cancer Res (2020) 26:1606–15. doi: 10.1158/1078-0432.Ccr-19-1339
241. Stoner A, Miller JO, Flatt T, Wallisch JS. Extracorporeal membrane oxygenation as a bridge to chimeric antigen receptor T-cell therapy for severe refractory sepsis in the setting of relapsed refractory pediatric acute lymphoblastic leukemia: A case report. Crit Care Explor (2020) 2:e0093. doi: 10.1097/cce.0000000000000093
242. Danylesko I, Chowers G, Shouval R, Besser MJ, Jacoby E, Shimoni A, et al. Treatment with anti CD19 chimeric antigen receptor T cells after antibody-based immunotherapy in adults with acute lymphoblastic leukemia. Curr Res Trans Med (2020) 68:17–22. doi: 10.1016/j.retram.2019.12.001
243. Sterner RM, Sakemura R, Cox MJ, Yang N, Khadka RH, Forsman CL, et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood (2019) 133:697–709. doi: 10.1182/blood-2018-10-881722
244. Giavridis T, van der Stegen SJC, Eyquem J, Hamieh M, Piersigilli A, Sadelain M. CAR T cell-induced cytokine release syndrome is mediated by macrophages and abated by IL-1 blockade. Nat Med (2018) 24:731–8. doi: 10.1038/s41591-018-0041-7
245. Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med (2018) 24:739–48. doi: 10.1038/s41591-018-0036-4
246. Patel S, Burga RA, Powell AB, Chorvinsky EA, Hoq N, McCormack SE, et al. Beyond CAR T cells: Other cell-based immunotherapeutic strategies against cancer. Front Oncol (2019) 9:196. doi: 10.3389/fonc.2019.00196
247. Hu Z. Tissue factor as a new target for CAR-NK cell immunotherapy of triple-negative breast cancer. Sci Rep (2020) 10:2815. doi: 10.1038/s41598-020-59736-3
248. Wu Y, Kyle-Cezar F, Woolf RT, Naceur-Lombardelli C, Owen J, Biswas D, et al. An innate-like Vδ1(+) γδ T cell compartment in the human breast is associated with remission in triple-negative breast cancer. Sci Transl Med (2019) 11:eaax9364. doi: 10.1126/scitranslmed.aax9364
Keywords: triple-negative breast cancer, chimeric antigen receptor, solid tumors, cancer immunotherapy, adoptive cell therapy, T-cell therapy
Citation: Nasiri F, Kazemi M, Mirarefin SMJ, Mahboubi Kancha M, Ahmadi Najafabadi M, Salem F, Dashti Shokoohi S, Evazi Bakhshi S, Safarzadeh Kozani P and Safarzadeh Kozani P (2022) CAR-T cell therapy in triple-negative breast cancer: Hunting the invisible devil. Front. Immunol. 13:1018786. doi: 10.3389/fimmu.2022.1018786
Received: 13 August 2022; Accepted: 24 October 2022;
Published: 22 November 2022.
Edited by:
Cristina Maccalli, Sidra Medicine, QatarReviewed by:
Hany E. Marei, Mansoura University, EgyptLeonardo M. R. Ferreira, Medical University of South Carolina, United States
Copyright © 2022 Nasiri, Kazemi, Mirarefin, Mahboubi Kancha, Ahmadi Najafabadi, Salem, Dashti Shokoohi, Evazi Bakhshi, Safarzadeh Kozani and Safarzadeh Kozani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pooria Safarzadeh Kozani, pooriasafarzadeh@modares.ac.ir
†These authors have contributed equally to this work