


Abstract
Most patients with BRAFV600-mutant metastatic melanoma develop resistance to selective RAF kinase inhibitors. The spectrum of clinical genetic resistance mechanisms to RAF inhibitors and options for salvage therapy are incompletely understood. We performed whole-exome sequencing on formalin-fixed, paraffin-embedded tumors from 45 patients with BRAFV600-mutant metastatic melanoma who received vemurafenib or dabrafenib monotherapy. Genetic alterations in known or putative RAF inhibitor resistance genes were observed in 23 of 45 patients (51%). Besides previously characterized alterations, we discovered a “long tail” of new mitogen-activated protein kinase (MAPK) pathway alterations (MAP2K2, MITF) that confer RAF inhibitor resistance. In three cases, multiple resistance gene alterations were observed within the same tumor biopsy. Overall, RAF inhibitor therapy leads to diverse clinical genetic resistance mechanisms, mostly involving MAPK pathway reactivation. Novel therapeutic combinations may be needed to achieve durable clinical control of BRAFV600-mutant melanoma. Integrating clinical genomics with preclinical screens may model subsequent resistance studies.
Significance: The use of RAF inhibitors for BRAFV600-mutant metastatic melanoma improves patient outcomes, but most patients demonstrate early or acquired resistance to this targeted therapy. We reveal the genetic landscape of clinical resistance mechanisms to RAF inhibitors from patients using whole-exome sequencing, and experimentally assess new observed mechanisms to define potential subsequent treatment strategies. Cancer Discov; 4(1); 94–109. ©2013 AACR.
See related commentary by Solit and Rosen,
This article is highlighted in the In This Issue feature,
Introduction
RAF inhibitors (vemurafenib and dabrafenib) administered alone or in combination with MAP–ERK kinase (MEK) inhibitors have improved progression-free and overall survival in patients with BRAFV600-mutated metastatic melanoma (1–4). Although the vast majority of patients experience clinical benefit, almost all develop resistance to these agents (5). Furthermore, some of these patients show either intrinsic resistance or short-lived responses (e.g., disease progression in less than 12 weeks).
Several mechanisms of RAF inhibitor resistance have been identified. Genetic causes of resistance identified thus far include NRAS (6) and MEK1 (5, 7) mutations, BRAF amplification (8), and NF1 loss (9). Nongenetic causes of RAF inhibitor resistance include activation of COT (a kinase encoded by the MAP3K8 gene; ref. 10) or EGF receptor (EGFR; ref. 11), suppression of BIM expression via PTEN loss (12), alternative splicing of BRAF RNA transcripts (13), disrupted feedback regulation (14), receptor tyrosine kinase dysregulation (6, 15), and stromal secretion of growth factors like hepatocyte growth factor (HGF; refs. 16, 17).
Most previously described resistance mechanisms were identified using preclinical models and confirmed in limited numbers of clinical specimens. A detailed understanding of the somatic genetic causes of RAF inhibitor resistance may have longstanding clinical impact, as these mechanisms may inform future clinical development priorities. However, our understanding of the spectrum of genetic resistance mechanisms is incomplete. Therefore, we performed whole-exome sequencing of BRAFV600-mutant melanoma tissue obtained before treatment and after the development of resistance to RAF inhibitors to characterize the clinical spectrum of genetic resistance for this patient population.
Results
A Spectrum of Genetic Alterations Is Associated with Clinical Resistance to RAF Inhibition
Among the 45 patients in our cohort (Fig. 1A), 14 (31%) had early resistance (on therapy for less than 12 weeks) and 31 (68.9%) developed acquired resistance (Table 1). Among the early-resistance patients, 7 (50%) had progressive disease as best response, 6 (43%) had short-lived stable disease, and 1 (7%) had a brief partial response. The mean target coverage for tumor samples was 200×, and 92× for germline DNA (Supplementary Table S1). BRAF mutations were detected in all pretreatment biopsy specimens by whole-exome sequencing, of which 44 of 45 were missense mutations affecting codon V600. Patient 11 had an in-frame deletion event predicted to generate a functional effect similar to V600E (Val600_Lys601delinsGlu).
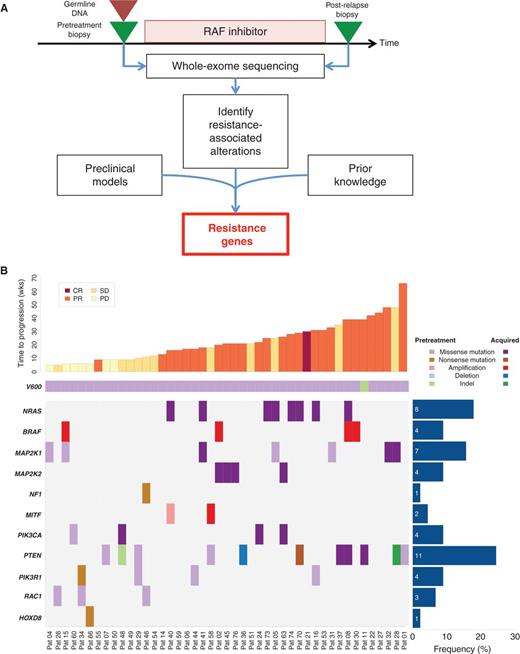
Genetic alterations in the context of RAF inhibitor therapy. A, schematic overview of tumor biopsy collection in the context of RAF inhibitor therapy, followed by whole-exome sequencing and analysis. B, spectrum of putative resistance genes, including known genes (NRAS, BRAF, and MEK1) and new genes (MEK2 and MITF). Additional recurrently altered pathways (PI3K pathway) or genomic correlates of early resistance (HOXD8 and RAC1) are also shown. Results are sorted by duration of therapy (weeks). CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; Pat, patient.
Genetic alterations in the context of RAF inhibitor therapy. A, schematic overview of tumor biopsy collection in the context of RAF inhibitor therapy, followed by whole-exome sequencing and analysis. B, spectrum of putative resistance genes, including known genes (NRAS, BRAF, and MEK1) and new genes (MEK2 and MITF). Additional recurrently altered pathways (PI3K pathway) or genomic correlates of early resistance (HOXD8 and RAC1) are also shown. Results are sorted by duration of therapy (weeks). CR, complete response; PR, partial response; SD, stable disease; PD, progressive disease; Pat, patient.
Clinical characteristics
| Total | Early | Acquired | |
|---|---|---|---|
| Number of patients | 45 | 14 | 31 |
| Age, y | 51.0 | 48.8 | 52.0 |
| Gender (% female) | 48.9 | 50 | 48.3 |
| Medication | |||
| Vemurafenib (%) | 66.6 | 64.3 | 67.7 |
| Dabrafenib (%) | 33.3 | 35.7 | 32.3 |
| Median duration of therapy, wks | 20 (SD 14.1) | 9 (SD 2.3) | 25 (SD 12.4) |
| Best response (%) | |||
| CR | 2.2 | 0 | 3.2 |
| PR | 55.6 | 7.1 | 80.6 |
| SD | 24.4 | 42.9 | 16.1 |
| PD | 17.8 | 50 | 0 |
| Total | Early | Acquired | |
|---|---|---|---|
| Number of patients | 45 | 14 | 31 |
| Age, y | 51.0 | 48.8 | 52.0 |
| Gender (% female) | 48.9 | 50 | 48.3 |
| Medication | |||
| Vemurafenib (%) | 66.6 | 64.3 | 67.7 |
| Dabrafenib (%) | 33.3 | 35.7 | 32.3 |
| Median duration of therapy, wks | 20 (SD 14.1) | 9 (SD 2.3) | 25 (SD 12.4) |
| Best response (%) | |||
| CR | 2.2 | 0 | 3.2 |
| PR | 55.6 | 7.1 | 80.6 |
| SD | 24.4 | 42.9 | 16.1 |
| PD | 17.8 | 50 | 0 |
NOTE: The RAF inhibitor used, duration of response, and best clinical response data for 45 patients are detailed here, further stratified by whether the patient exhibited early or acquired resistance to therapy.
Age is defined as the age at time of starting therapy. All values are medians across the cohort.
Abbreviations: CR, complete response; PD, progressive disease; PR, partial response; SD, stable disease.
Mutational analysis of resistant tumors revealed several genes shown previously to confer resistance to RAF inhibition (Supplementary Tables S2–S4). These include somatic mutations in NRAS (17.8%; seven involving Q61 and one involving T58), amplifications of BRAF (8.9%), and mutations in MAP2K1 (15.6%), although MAP2K1 mutations did not universally preclude clinical response (Fig. 1B). As expected, acquired NRAS mutations occurred exclusively in patients on therapy for more than 12 weeks (P = 0.04). We also observed multiple additional putative resistance drivers that occurred at low frequencies across the cohort (Fig. 1B).
Globally, these events could be aggregated on the basis of the cellular pathways or mechanisms implicated by the resistance-associated genes. Resistance alterations predominantly involved the mitogen-activated protein kinase (MAPK) pathway or downstream effectors (NRAS, BRAF, MAP2K1, MAP2K2, MITF, and NF1), and occurred in 44.4% (20 of 45) of the patients. Additional alterations with less clear resistance relationships were observed in the phosphoinositide 3-kinase (PI3K) pathway (PIK3CA, PTEN, and PIK3R1), and in HOXD8 or RAC1 (Fig. 1B).
MEK2 Mutations Confer Resistance to RAF and MEK Inhibitors
We identified four mutations involving the MAP2K2 gene (which encodes the MEK2 kinase) in drug-resistant melanoma specimens (Fig. 2A and B). Like its homolog MEK1, MEK2 is situated immediately downstream of RAF proteins in the MAPK pathway. MEK2 forms a heterodimer with MEK1 that promotes extracellular signal–regulated kinase (ERK) phosphorylation (18). One of these mutations (MEK2C125S) is homologous to a previously described MEK1C121S mutation that confers cross-resistance to RAF and MEK inhibitors in vitro (7).

MEK2 mutations confer resistance to RAF/MEK but not ERK inhibition. A, a stick plot of MAP2K2 (which encodes the MEK2 kinase); the location of putative resistance-associated mutations observed in the patient cohort are indicated. B, the crystal structure for MEK2. The locations of somatically mutated bases are denoted in yellow; the first stretch of amino acids are missing from the MEK2 structure in PDB, so the V35M and L46F mutations cannot be shown on the structure. C–E, growth inhibition curves are shown for MEK2 mutants in the context of RAF (C), MEK (D), or ERK (E) inhibitors. F, the effect of dabrafenib or trametinib on ERK1/2 phosphorylation (pERK1/2) in wild-type A375 cells (BRAFV600E) and those expressing wild-type MEK2 (WT) or mutant constructs for MEK2. The levels of pERK1/2, total ERK1/2, pMEK1/2, MEK1/2, and vinculin are shown for A375 cells expressing novel MEK2 mutations after a 16-hour incubation at various drug concentrations as indicated. p, phosphorylated.
MEK2 mutations confer resistance to RAF/MEK but not ERK inhibition. A, a stick plot of MAP2K2 (which encodes the MEK2 kinase); the location of putative resistance-associated mutations observed in the patient cohort are indicated. B, the crystal structure for MEK2. The locations of somatically mutated bases are denoted in yellow; the first stretch of amino acids are missing from the MEK2 structure in PDB, so the V35M and L46F mutations cannot be shown on the structure. C–E, growth inhibition curves are shown for MEK2 mutants in the context of RAF (C), MEK (D), or ERK (E) inhibitors. F, the effect of dabrafenib or trametinib on ERK1/2 phosphorylation (pERK1/2) in wild-type A375 cells (BRAFV600E) and those expressing wild-type MEK2 (WT) or mutant constructs for MEK2. The levels of pERK1/2, total ERK1/2, pMEK1/2, MEK1/2, and vinculin are shown for A375 cells expressing novel MEK2 mutations after a 16-hour incubation at various drug concentrations as indicated. p, phosphorylated.
To verify the predicted resistance phenotypes conferred by MEK2 mutations, MEK2-mutant constructs were cloned into a doxycycline-inducible vector and expressed in A375 melanoma cells that harbor the BRAFV600E mutation and are sensitive to RAF inhibition, which were then treated with increasing concentrations of MAPK pathway inhibitors. Compared with the effects of wild-type MEK2, cells expressing resistance-associated MEK2 mutations were less sensitive to both RAF (dabrafenib; Fig. 2C) and MEK (trametinib; Fig. 2D) inhibition. As with the homologous and previously reported MEK1C121S resistance mutation (7), MEK2C125S conferred profound resistance to both RAF and MEK inhibition, with fold changes in GI50 (half-maximal inhibitor concentration) greater than 100. The MEK2V35M, MEK2L46F, and MEK2N126D mutants also engendered resistance to RAF and MEK inhibition, although their effect was not as pronounced as those of MEK2C125S. In contrast, all resistance-associated MEK2 mutations remained sensitive to ERK inhibition using the tool compound VRT11E (Fig. 2E). All MEK2-mutant alleles examined conferred sustained MEK and ERK phosphorylation in the context of RAF inhibitor treatment (Fig. 2F).
MEK1 Mutations Confer Resistance to RAF and MEK Inhibitors When Expressed Inducibly
Five MAP2K1 gene mutations (encodes the MEK1 kinase) were detected in either drug-resistant specimens (three mutations; MEK1V60E, MEK1G128V, and MEK1V154I in patients 41, 32, and 28, respectively) or pretreatment tumors that progressed rapidly in the face of clinical RAF inhibition (two mutations; MEK1P124S and MEK1P124L in patients 4 and 15, respectively; Fig. 1B). Two additional MEK1 mutations (MEK1G276W and MEK1F53Y) occurred in pretreatment tumor samples from patients (patient 5 and 31) who experienced a clinical benefit from RAF inhibitors based on time on therapy (25–33 weeks; Fig. 1B). This finding was consistent with prior studies indicating that the presence of MEK1 mutations does not necessarily preclude a clinical benefit from RAF inhibitors (5, 19). Nonetheless, several MEK1 mutations involved residues predicted to cause RAF inhibitor resistance based on mutagenesis screens and experimental studies reported previously (ref. 7; Fig. 3A and B), including MEK1V60E and MEK1G128V. These results raised the possibility that somatic MEK1 mutations might promote resistance to RAF inhibition in some contexts but not others.

MEK1 mutations confer varying degrees of resistance to RAF/MEK but not ERK inhibition. A, a stick plot of MAP2K1 (which encodes the MEK1 kinase); the location of putative resistance-associated mutations observed in the patient cohort are indicated. B, the crystal structure for MEK1. The locations of somatically mutated bases are denoted in yellow. C–E, growth inhibition curves are shown for MEK1 mutants in the context of RAF (C), MEK (D), or ERK (E) inhibitors. F, the effect of dabrafenib or trametinib on ERK1/2 phosphorylation (pERK1/2) in wild-type A375 cells (BRAFV600E) and those expressing wild-type MEK1 (WT) or mutant constructs for MEK1. The levels of pERK1/2, total ERK1/2, pMEK1/2, MEK1/2, and vinculin are shown for A375 cells expressing novel MEK1 mutations after a 16-hour incubation at various drug concentrations as indicated.
MEK1 mutations confer varying degrees of resistance to RAF/MEK but not ERK inhibition. A, a stick plot of MAP2K1 (which encodes the MEK1 kinase); the location of putative resistance-associated mutations observed in the patient cohort are indicated. B, the crystal structure for MEK1. The locations of somatically mutated bases are denoted in yellow. C–E, growth inhibition curves are shown for MEK1 mutants in the context of RAF (C), MEK (D), or ERK (E) inhibitors. F, the effect of dabrafenib or trametinib on ERK1/2 phosphorylation (pERK1/2) in wild-type A375 cells (BRAFV600E) and those expressing wild-type MEK1 (WT) or mutant constructs for MEK1. The levels of pERK1/2, total ERK1/2, pMEK1/2, MEK1/2, and vinculin are shown for A375 cells expressing novel MEK1 mutations after a 16-hour incubation at various drug concentrations as indicated.
To explore this possibility, we cloned several MEK1-mutant cDNAs identified herein into a doxycycline-inducible expression vector, and expressed the mutant proteins alongside wild-type MEK1 in A375 melanoma cells. Cell growth inhibition curves were generated in the presence of dabrafenib (RAF inhibitor), trametinib (MEK inhibitor), or VRT11E (ERK inhibitor), as shown in Fig. 3 (see Methods for additional details). All MEK1 mutations examined conferred robust resistance to both RAF and MEK inhibition following doxycycline induction, with fold changes in GI50 of 10- to 80-fold for dabrafenib (Fig. 3C) and 3- to 20-fold for trametinib (Fig. 3D) as compared with wild-type MEK1. Of note, doxycycline induction of wild-type MEK1 produced modest (∼3-fold) resistance to MEK inhibition but had no effect on the dabrafenib GI50 value (Fig. 3C and D). In contrast, doxycycline induction of these MEK1 mutants had no effect on BRAFV600E-mutant melanoma cell sensitivity to ERK inhibition using VRT11E (Fig. 3E). As seen with the somatic MEK2 mutations described above, doxycycline-inducible expression of MEK1 mutations resulted in elevated MEK and ERK phosphyorylation, which was sustained in the presence of drug concentrations that inhibited MAPK signaling in A375 melanoma cells that overexpressed wild-type MEK1 (Fig. 3F). Together, these results indicated that inducible expression of somatic MEK1 mutations can confer resistance to RAF/MEK inhibition, and suggested that dynamic regulation of mutant MEK1 protein may conceivably modulate clinical sensitivity to these agents in melanoma tumors.
MITF Amplification Is Associated with Resistance to MAPK Inhibition
In one patient whose melanoma tumor lacked a previously described genetic resistance mechanism, we identified a relapse-associated focal amplification of MITF (Fig. 4A). This gene encodes a master lineage transcription factor that governs melanocyte development, and is also an amplified oncogene within the melanocyte lineage (20). Although paired RNA or immunohistochemistry analysis was not possible with this clinical sample, the highly focal nature of the amplification and its specificity for the relapsed tumor suggested that this amplicon in general, and MITF in particular, might contribute to the resistance phenotype. To test this hypothesis, we overexpressed wild-type MITF alongside a DNA-binding impaired MITF mutant (MITFR217Δ), or negative control (LacZ) in WM266.4 (BRAFV600E) melanoma cells. The resulting cells were cultured in the presence of RAF (PLX4720), MEK (AZD6244), or ERK (VRT11E) inhibitors at fixed concentrations. Indeed, forced MITF overexpression rendered these BRAFV600E melanoma cells resistant to RAF, MEK, and ERK inhibition (Fig. 4B and C).

Acquired MITF amplification confers resistance to MAPK pathway inhibition. A, focal genomic amplification of MITF occurs in a post-relapse sample in the absence of additional, known resistance mechanisms. B, relative viability of BRAFV600E-mutant WM266.4 cells following overexpression of wild-type MITF, a DNA-binding impaired MITF-mutant (MITF-mR217Δ), or control (LacZ) in the presence of RAF, MEK, combined RAF/MEK, or ERK inhibitors. C, Western blot analysis of WM266.4 cells expressing the constructs used in B. Half-maximal drug response curves in BRAFV600E-mutant melanoma cell lines SKMEL19 (D) and UACC62 (E) expressing either LacZ or the melanocyte-specific isoform of MITF (MITFm) in the presence of increasing concentrations of PLX4720. CR, copy ratio. DMSO, dimethyl sulfoxide.
Acquired MITF amplification confers resistance to MAPK pathway inhibition. A, focal genomic amplification of MITF occurs in a post-relapse sample in the absence of additional, known resistance mechanisms. B, relative viability of BRAFV600E-mutant WM266.4 cells following overexpression of wild-type MITF, a DNA-binding impaired MITF-mutant (MITF-mR217Δ), or control (LacZ) in the presence of RAF, MEK, combined RAF/MEK, or ERK inhibitors. C, Western blot analysis of WM266.4 cells expressing the constructs used in B. Half-maximal drug response curves in BRAFV600E-mutant melanoma cell lines SKMEL19 (D) and UACC62 (E) expressing either LacZ or the melanocyte-specific isoform of MITF (MITFm) in the presence of increasing concentrations of PLX4720. CR, copy ratio. DMSO, dimethyl sulfoxide.
Next, we overexpressed MITF in two additional BRAFV600E-mutant melanoma cell lines (SKMEL19 and UACC62) and performed cell growth inhibition studies using the RAF inhibitor tool compound (PLX4720). In both cell lines, MITF overexpression conferred a 30- to 80-fold increase in the PLX4720 GI50 values relative to control (LacZ) gene expression (Fig. 4D and E). Because MITF activity is regulated by MAPK signaling in melanocytes and melanoma, these results suggest that restoration of a MITF-driven transcriptional output (by genomic amplification or other means) comprises a newly recognized clinical resistance mechanism (21).
Intratumor Heterogeneity of Resistance Mechanisms
In three patients, multiple independent resistance mechanisms were evident within the same resistant tumor biopsy. For example, one tumor biopsied at progression after 18 weeks on treatment contained resistance-associated mutations in both NRAS and MEK1 (patient 41; Fig. 5A). Moreover, two distinct somatic NRAS mutations were observed exclusively in the resistant tumor: a validated resistance alteration (Q61R), and a second alteration (T58I) that is homologous to germline KRAS alterations that cause Noonan syndrome (22). These alterations occurred on mutually exclusive reads that spanned both loci, suggesting that either the two alterations occur in trans in the same resistant tumor cells or they represent separate subclonal resistant populations harboring different NRAS mutations (Fig. 5A).
![Figure 5. Intratumoral resistance heterogeneity. A, co-occuring NRAS [Integrated Genomics Viewer (IGV) compressed window] andMEK1V60E (IGV regular window) missense mutations in a relapsed tumor sample (patient 41). NRASQ61R occurs in mutually exclusive reads compared with a neighboring NRAST58I somatic missense mutation in this patient. B, acquired NRASQ61K missense mutation together with BRAF amplification in the same tumor (patient 8). C, a MEK2L46F missense mutation coincident with BRAF amplification in a resistant tumor specimen (patient 02—no pretreatment tumor sample was available in this case). CR, copy ratio.](https://aacr.silverchair-cdn.com/aacr/content_public/journal/cancerdiscovery/4/1/10.1158_2159-8290.cd-13-0617/4/m_94fig5.jpeg?Expires=1716299458&Signature=OP6d~7Q0HgqUVavBjrAznjZ6LcYJxVOcic7~o2oLHsmS9UnD9pGwXWuQeuZeJKyHhIRrnYhvlsy1Mlz58DOWbsc8T3b6ROPF1Lt4qRiBHe45c9O~cMwus4m46~QPhkAZl2Q3hKRVwIGRr7FSbGRXIDogvlaFJNPe0w3mqA0uLxTJw0A8hgPImS9mYIK3jUkH4791Hvqqo9wk8rNukYrfwFNneNqcYyJ6AqeVGaqjdNkA6yQ9RRtdW~fDidlwBhKMF-AKR9o-Be9HXlYURxx36bTeP7CwmnL2DoAIupwwr6tKR24~UBp62KxiOQAZu7R-EixUJjUXITTTwHCyb6RGNg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Intratumoral resistance heterogeneity. A, co-occuring NRAS [Integrated Genomics Viewer (IGV) compressed window] andMEK1V60E (IGV regular window) missense mutations in a relapsed tumor sample (patient 41). NRASQ61R occurs in mutually exclusive reads compared with a neighboring NRAST58I somatic missense mutation in this patient. B, acquired NRASQ61K missense mutation together with BRAF amplification in the same tumor (patient 8). C, a MEK2L46F missense mutation coincident with BRAF amplification in a resistant tumor specimen (patient 02—no pretreatment tumor sample was available in this case). CR, copy ratio.
Intratumoral resistance heterogeneity. A, co-occuring NRAS [Integrated Genomics Viewer (IGV) compressed window] andMEK1V60E (IGV regular window) missense mutations in a relapsed tumor sample (patient 41). NRASQ61R occurs in mutually exclusive reads compared with a neighboring NRAST58I somatic missense mutation in this patient. B, acquired NRASQ61K missense mutation together with BRAF amplification in the same tumor (patient 8). C, a MEK2L46F missense mutation coincident with BRAF amplification in a resistant tumor specimen (patient 02—no pretreatment tumor sample was available in this case). CR, copy ratio.
Another post-relapse tumor (patient 08) harbored an acquired NRASQ61K missense mutation together with focal BRAF amplification (Fig. 5B). The resistant tumor from a third patient harbored both a MEK2 mutation and BRAF amplification (patient 02; Fig. 5C). Although each of these events has been shown to confer a RAF inhibitor resistance phenotype, the pretreatment tumor was unavailable for this case. Thus, we cannot formally exclude the possibility that these mutations were present before treatment with RAF inhibition. Nonetheless, these results further indicate that BRAF-mutant melanomas may elaborate multiple resistance mechanisms simultaneously, as suggested by an earlier study involving a single case (23). Given that “nongenetic” resistance mechanisms such as BRAF alternative splicing or COT overexpression could not be assessed in this cohort, these findings may vastly undersample the actual clinical incidence of intratumoral resistance heterogeneity.
PI3K Pathway Activation May Contribute to Clinical RAF Inhibitor Resistance
In contrast to MAPK pathway activation, the clinical importance of PI3K pathway dysregulation as a RAF inhibitor resistance mechanism has been less clear (12, 24, 25). Prior studies have demonstrated that PI3K pathway activation through multiple mechanisms (e.g., PTEN loss, AKT activation) is associated with RAF inhibitor resistance in some preclinical models (12, 26), and that patients with PTEN loss trend toward shorter progression-free survival to dabrafenib (27). However, as is the case with MEK1 (above), PI3K pathway alterations did not necessarily preclude clinical response in tumors characterized herein. For example, patient 58 harbored a pretreatment PTENK128T missense mutation but was on therapy for 18 weeks with stable disease before developing an acquired MITF amplification. Patient 1 had a PTENH93D missense mutation in the pretreatment tumor but was on therapy for 66 weeks and achieved a partial response.
We noted that several resistant tumors contained PI3K pathway gene alterations in the absence of known MAPK resistance mechanisms (Fig. 1B and Supplementary Table S5). For example, one patient (patient 48) whose pretreatment tumor harbored a PTENY86fs frameshift exhibited a new PIK3CAH1047R mutation in the resistant tumor biopsy (PIK3CAH1047R is a well-characterized oncogenic mutation in the catalytic PI3Kα subunit; ref. 28). The resistant tumor from patient 36 harbored a PTEN deletion (Fig. 6A), although the pretreatment tumor was unavailable for this case. In 9 of 11 patients with PTEN missense mutations, the mutation occurred in the phosphatase domain of the protein (Supplementary Table S5). Such mutations have been shown previously to confer a loss-of-function phenotype (29, 30). Other patients with known resistance mechanisms also harbored post-relapse somatic variants of uncertain significance in the PI3K pathway (predominantly PTEN missense mutations, but also PIK3R1 alterations) that lack experimental validation. For instance, patient 70 was on therapy for 29 weeks and achieved a partial response before acquiring an NRASQ60H mutation and a PTENR233* nonsense mutation that may be inactivating but requires experimental confirmation.

Combined inhibition of BRAF and PI3K can confer synergistic activity. A, copy number profile from a resistant tumor sample (patient 36—no pretreatment tumor sample was available in this case) demonstrates a focal homozygous deletion in chromosome 10 that includes PTEN. B, A375 (BRAFV600E, PTENWT) and A2058 (BRAFV600E, PTENnull) cells were treated with multiples of their respective GI50s for PLX4720 or GDC0941 alone and in combination for 4 days. Cell proliferation was assessed using the CellTiter-Glo reagent. Combined BRAF and PI3K inhibition demonstrated synergistic activity in A2058 cells but was less synergistic in A375 cells. The combination index (CI) was determined at the GI50 concentration using the Chou and Talalay method and CalcuSyn (Biosoft; ref. 52). C, A375 and A2058 cells were treated with 4× GI50 concentrations of PLX4720, GDC0941, or a combination of both inhibitors for 20 hours. Cell lysates were analyzed by Western blotting for the indicated proteins. CR, copy ratio; DMSO, dimethyl sulfoxide.
Combined inhibition of BRAF and PI3K can confer synergistic activity. A, copy number profile from a resistant tumor sample (patient 36—no pretreatment tumor sample was available in this case) demonstrates a focal homozygous deletion in chromosome 10 that includes PTEN. B, A375 (BRAFV600E, PTENWT) and A2058 (BRAFV600E, PTENnull) cells were treated with multiples of their respective GI50s for PLX4720 or GDC0941 alone and in combination for 4 days. Cell proliferation was assessed using the CellTiter-Glo reagent. Combined BRAF and PI3K inhibition demonstrated synergistic activity in A2058 cells but was less synergistic in A375 cells. The combination index (CI) was determined at the GI50 concentration using the Chou and Talalay method and CalcuSyn (Biosoft; ref. 52). C, A375 and A2058 cells were treated with 4× GI50 concentrations of PLX4720, GDC0941, or a combination of both inhibitors for 20 hours. Cell lysates were analyzed by Western blotting for the indicated proteins. CR, copy ratio; DMSO, dimethyl sulfoxide.
In light of these observations, we sought to probe the possible therapeutic implications of PI3K pathway alterations in BRAF-mutant melanoma. Accordingly, we treated BRAF-mutant melanoma cells that were either wild-type or mutant for PTEN with both a RAF inhibitor and a PI3K inhibitor in vitro. Combined MAPK (PLX4720) and PI3K (GDC0941) inhibition had a modest but discernible impact on cell proliferation in BRAFV600E/PTENWT cells (A375), but in the BRAFV600E/PTENnull cell line (A2058) the combination has a marked effect on cell proliferation and apoptosis (Fig. 6B and C), consistent with prior reports (24, 31–33). Despite these results (which are limited to only one cell line per condition), the cellular contexts in which PI3K pathway activation may promote cell viability in the setting of RAF inhibition remains unclear (5, 9, 10). Some in vitro studies argue against a strong proliferative effect (21), therefore implying that PI3K pathway dysregulation may augment cell survival in some contexts and allow such tumor cells the opportunity to elaborate additional (nongenetic) adaptive resistance mechanisms. This model is consistent with the observation that many, though not all, tumors with resistance-associated PI3K pathway mutations lack genetic resistance mechanisms linked to MAPK pathway activation (Fig. 1B).
Genetic Correlates of Early Resistance
Among the 14 patients exhibiting early disease progression in the setting of RAF inhibition (disease progression within 12 weeks of initial RAF inhibitor therapy), three pretreatment tumor biopsies harbored RAC1P29S mutations. Patients 26 and 46 had transient stable disease, and patient 34 had immediate progressive disease. RAC1P29S was previously identified as a gain-of-function oncogene mutation in melanoma (34, 35). No patients who demonstrated a sustained response to therapy (≥12 weeks on therapy) exhibited this mutation (P = 0.026; Fig. 1B). Conceivably, the RAC1P29S mutations may mark a tumor cell population that is partially resistant to single-agent RAF inhibition, although experimental evidence in support of this notion is necessary.
Finally, a nonsense mutation in the HOXD8 gene was observed in a single resistant tumor from a patient with early resistance (Fig. 1B). HOXD8 is a homeobox transcription factor (36) that has been shown to be dysregulated in multiple cancers (37, 38), although it has not previously been associated with melanoma, nor is there a known functional relationship between this gene and RAF signaling. Interestingly, HOXD8 suppression was implicated in RAF inhibitor resistance following a genome-scale RNA interference (RNAi) suppressor screen reported previously (9). The presence of a HOXD8 nonsense mutation in the absence of other known resistance-associated alterations raises the possibility that inactivation of this homeobox transcription factor may represent another new resistance mechanism. However, this gene has not previously been interrogated in drug-resistant clinical specimens.
Discussion
Whole-exome sequencing of BRAFV600-mutant melanoma tumors obtained before treatment with selective class I RAF inhibitors and following resistance has revealed a landscape of putative clinical resistance gene mutations linked to this therapy. This study used clinical formalin-fixed, paraffin-embedded (FFPE) tumor samples rather than research-grade frozen tissue. The regular use of FFPE material for massively parallel sequencing studies should greatly expand the specimens available for systematic studies of response and resistance to anticancer agents. In addition to known alterations, we found multiple new gene mutations that may implicate novel resistance mechanisms. Consequently, this work has extended knowledge of potential genetic avenues through which BRAFV600-mutant melanomas can achieve resistance to RAF inhibition.
This study was performed with patient samples longitudinally obtained from a nationwide clinical consortium [the Dermatologic Cooperative Oncology Group of Germany (DeCOG)]. Whole-exome sequencing was completed using genomic DNA obtained from archival (e.g., FFPE) tumor material. This study therefore demonstrates the feasibility of comprehensive clinical resistance studies that unite the potentially expansive clinical access of consortium groups with the production capabilities of a large-scale genome center. Such efforts may guide future cancer genomics studies that incorporate multiple biopsies obtained from patients for clinical purposes to effectively study therapeutic resistance and other salient clinical questions. The use of FFPE tumor samples for whole-exome sequencing did not limit this study; indeed, sequencing metrics for this cohort met or exceeded standards that have been used for The Cancer Genome Atlas and other cancer genome projects of similar scale. This technical success may therefore provide a framework for many new clinical studies that allow insights into the molecular basis of therapeutic response and resistance in cancer.
Computational analysis of the tumor genomic data in this study was enhanced by integration with available systematic functional screening data to prioritize candidate RAF inhibitor resistance-associated genes. Because the pattern or resistance genes may approximate a “long tail” distribution (similar to the distribution of cancer genes; ref. 39), many resistance-associated alterations may not reach statistical significance without much larger clinical cohorts than the one employed here. By analyzing exome sequencing results in the context of experimental data obtained from large-scale resistance screens, rare resistance-associated events that arise in functionally credentialed genes may be prioritized for future studies. Integration of in vitro genome-wide resistance screens with in vivo comprehensive genomic profiling may enrich for robust (albeit less common) resistance mechanisms, and may articulate a generalizable approach for resistance studies that is relevant to many tumor types.
The majority of RAF inhibitor resistance alterations involve reactivation of the MAPK pathway. Experimental assessment of a subset of resistance mutations further stratified these events into discrete categories that may inform future therapeutic or clinical trial options. Several MEK1 and MEK2 resistance mutations confer cross-resistance to MEK inhibitor therapy and raise the possibility that combined RAF/MEK inhibition may also have limited efficacy in this patient subpopulation (40). In contrast, NRAS mutations and BRAF amplifications may still prove responsive to subsequent MEK inhibitor-based regimens, although the existing clinical data suggest that patients who progress following single-agent RAF inhibition are less likely to benefit from MEK inhibitors (41). Most of these mutations remain sensitive to ERK inhibitors in vitro, suggesting that such agents (42) may represent a high-priority avenue for new targeted therapeutic development.
Alterations in genes encoding transcription factors may also implicate a rare category of clinical resistance mechanisms that involves restoration (by MITF amplification) of an oncogenic transcriptional output downstream of the MAPK pathway. Unlike signaling-based resistance, transcriptional effectors may confer cross-resistance to all MAPK pathway inhibitors (including ERK inhibitors) and therefore require alternative therapeutic approaches. HOXD8, another transcriptional effector that emerged in an RNAi screen and was subsequently observed to be mutated in a patient with early resistance, may also contribute to RAF inhibitor resistance, although further functional studies are necessary to confirm this model. Moreover, the identification of alterations that activate PI3K signaling suggests that novel combinations of MAPK and PI3K pathway inhibitors may merit clinical evaluation in BRAF-mutant melanoma.
Comprehensive cancer genome characterization studies have revealed a “long tail” of mutated genes that drive carcinogenesis (39, 43). The present study suggests that a similar long tail of genetic effectors may mediate resistance to RAF inhibitors. In addition, multiple resistance mechanisms may arise within the same tumor, and the mechanisms observed in this study differ from those reported in the case report form (23). As noted earlier, given we could not interrogate nonsomatic mechanisms of resistance, such as BRAF alternative splicing, this result likely underestimates the true incidence of intratumor resistance heterogeneity observed clinically. Therapeutically, this finding implies that multiple pathways may need to be targeted either in parallel if not limited by toxicity, or in series as part of an intermittent dosing schedule (44).
Although informative, this study is limited in several aspects. For example, multiple known resistance mechanisms cannot be identified by whole-exome sequencing (including BRAF alternative splicing, COT upregulation, and ligand/receptor tyrosine kinase overexpression); thus, this study undoubtedly underestimates the true prevalence of molecular changes that drive clinical resistance to RAF inhibition. Also, because serial biopsies cannot always be performed on the same tumor focus, it may be difficult to discern resistance-associated changes between pretreatment and resistant tumor samples given preexisting tumor heterogeneity. The advent of systematic approaches to functional evaluation of candidate resistance effectors may help address such challenges, which are inherent to many genomics studies using clinical samples.
Moreover, some genetic alterations observed in this cohort (e.g., RAC1, HOXD8) require further experimental validation and assessment in large clinical cohorts to determine their specific impact on mediating RAF inhibitor resistance. It also seems possible that, in certain settings, dysregulation of the PI3K pathway may help improve tumor cell survival during treatment with these agents until a proliferative resistance allele emerges, although experimental validation of many PI3K variants observed in this cohort is necessary.
In conclusion, this work demonstrates the promise of serial tumor biopsies coupled with systematic genetic characterization to reveal a wide spectrum of clinical resistance mechanisms. Widespread clinical application of similar approaches may illuminate new biologic and therapeutic insights across many cancer types and inform clinical trial design. Furthermore, these findings underscore the future importance of novel therapeutic combinations (including both targeted agents and immunotherapy) to achieve more durable control of metastatic melanoma and other advanced cancers.
Methods
Patients and Tumor Specimens
Biopsy samples were secured from 45 patients with metastatic melanoma under an Institutional Review Board (IRB)–approved protocol to obtain research biopsies before and after resistance developed to systemic therapy (University Hospital Essen, Essen, Germany, 12-4961-BO). All patients provided written informed consent. The clinical characteristics of these patients are described in Table 1. Clinical responses to therapy were determined using Response Evaluation Criteria in Solid Tumors (RECIST) criteria. Pretreatment biopsies were obtained before starting therapy, and resistant biopsy samples were obtained upon discontinuation of vemurafenib or dabrafenib at disease progression (Fig. 1A). For patients where complete “trios” (germline sample, pretreatment tumor, postprogression tumor) were not available (n = 13), pretreatment biopsy results were included if the patient experienced rapid disease progression, and resistant biopsy results were included if an objective clinical response was achieved. Results from one responding patient (patient 65) were included to inform analyses related to predictors of early resistance, although this patient did not have a matched resistant tumor sample for analysis. Additional details about the patient cohort can be found in Supplementary Table S1.
FFPE DNA Extraction.
After fixation and mounting, 5 to 10 10-μm slices from an FFPE tumor block were obtained, and tumor-enriched tissue was macrodissected. Germline DNA was obtained from adjacent normal tissue or peripheral blood mononuclear cells. Paraffin was removed from FFPE sections and cores using CitriSolv (Fisher Scientific) followed by ethanol washes, then tissue was lysed overnight at 56°C. Samples were then incubated at 90°C to remove DNA cross-links, and extraction was performed using the Qiagen QIAamp DNA Mini Kit (#51306).
Whole-Exome Sequencing
We performed whole-exome sequencing on the extracted DNA using the Illumina HiSeq platform (ref. 45; Van Allen and colleagues, in press).
Library Construction.
DNA libraries for massively parallel sequencing were generated as previously described (45) with the following modifications: the initial genomic DNA input into the shearing step was reduced from 3 μg to 10–100 ng in 50 μL of solution. For adapter ligation, Illumina paired-end adapters were replaced with palindromic forked adapters (purchased from Integrated DNA Technologies) with unique 8-base index molecular barcode sequences included in the adapter sequence to facilitate downstream pooling. With the exception of the palindromic forked adapters, all reagents used for end repair, A-base addition, adapter ligation, and library enrichment PCR were purchased from KAPA Biosciences in 96-reaction kits. In addition, during the postenrichment solid phase reversible immobilization (SPRI) bead cleanup, elution volume was reduced to 20 μL to maximize library concentration, and a vortexing step was added to maximize the amount of template eluted from the beads. Libraries with concentrations above 40 ng/μL, as measured by a PicoGreen assay automated on an Agilent Bravo instrument, were considered acceptable for hybrid selection and sequencing.
Solution-Phase Hybrid Selection.
The exon capture procedure was performed as previously described (45) with the following modifications: before hybridization, any libraries with concentrations more than 60 ng/μL (as determined by PicoGreen) were brought to 60 ng/μL, and 8.3 μL of library was combined with blocking agent, bait, and hybridization buffer. Libraries with concentrations between 50 and 60 ng/μL were normalized to 50 ng/μL, and 10.3 μL of library was combined with blocking agent, bait, and hybridization buffer. Libraries with concentrations between 40 and 50 ng/μL were normalized to 40 ng/μL, and 12.3 μL of library was combined with blocking agent, bait, and hybridization buffer. Finally, the hybridization reaction was reduced to 17 hours, with no changes to the downstream capture protocol.
Preparation of Libraries for Cluster Amplification and Sequencing.
After postcapture enrichment, libraries were quantified using PicoGreen, normalized to equal concentration using a PerkinElmer MiniJanus instrument, and pooled by equal volume on the Agilent Bravo platform. Library pools were then quantified using quantitative PCR (qPCR; KAPA Biosystems) with probes specific to the ends of the adapters; this assay was automated using Agilent's Bravo liquid handling platform. On the basis of qPCR quantification, libraries were brought to 2 nmol/L and denatured using 0.2 N NaOH on the PerkinElmer MiniJanus. After denaturation, libraries were diluted to 20 pmol/L using hybridization buffer purchased from Illumina.
Cluster Amplification and Sequencing.
Cluster amplification of denatured templates was performed according to the manufacturer's protocol (Illumina) HiSeq v3 cluster chemistry and flow cells, as well as Illumina's Multiplexing Sequencing Primer Kit. DNA was added to flow cells and sequenced using the HiSeq 2000 v3 Sequencing-by-Synthesis method, then analyzed using RTA v.1.12.4.2 or later. Each pool of whole-exome libraries was subjected to paired 76-bp runs. An 8-base index sequencing read was performed to read molecular indices across the number of lanes needed to meet coverage for all libraries in the pool.
Sequencing Analysis and Interpretation
Sequence Data Processing.
Exome sequence data processing was performed using established analytic pipelines at the Broad Institute of MIT and Harvard (Cambridge, MA). A BAM file was produced with the Picard pipeline (http://picard.sourceforge.net/), which aligns the tumor and normal sequences to the hg19 human genome build using Illumina sequencing reads. The BAM was uploaded into the Firehose pipeline (http://www.broadinstitute.org/cancer/cga/Firehose), which manages input and output files to be executed by GenePattern (46).
Sequencing Quality Control.
Quality control modules within Firehose were applied to all sequencing data for comparison of the origin for tumor and normal genotypes and to assess fingerprinting concordance. Cross-contamination of samples was estimated using ContEst (47). Where 5% to 15% contamination was observed (n = 2), single-nucleotide polymorphism (SNP) fingerprints from each lane of a tumor/normal pair were cross-checked to confirm concordance; those without global concordance were excluded from subsequent analysis.
Somatic Alterations.
MuTect (48) was applied to identify somatic single-nucleotide variants. Indelocator (http://www.broadinstitute.org/cancer/cga/indelocator) was applied to identify small insertions or deletions. Artifacts introduced by DNA oxidation during sequencing were computationally removed using a filter-based method (49). Annotation of identified variants was done using Oncotator (http://www.broadinstitute.org/cancer/cga/oncotator). Copy ratios were calculated for each captured target by dividing the tumor coverage by the median coverage obtained in a set of reference normal samples. The resulting copy ratios were segmented using the circular binary segmentation algorithm (50). Genes in copy ratio regions with segment means of greater than log2(4) were evaluated for focal amplifications, and genes in regions with segment means of less than log2(0.5) were evaluated for deletions. For cases where we had pretreatment and post-resistance tumors (n = 32), alterations that were present in the post-resistance tumor only were selected; in other cases, we included all nonsynonymous alterations (Supplementary Tables S2–S3). We then integrated these data with preclinical functional models of resistance and existing literature on melanoma oncogenesis (7, 9, 34, 35, 51). The analyses were performed using the R statistical software. All somatic mutations and short insertion/deletions are provided in Supplementary Table S4.
Experimental Analysis
Physical and biologic containment procedures for recombinant DNA followed institutional protocols in accordance with the NIH (Bethesda, MD) Guidelines for Research Involving Recombinant DNA Molecules.
Plasmids and Site-Directed Mutagenesis.
The tet-inducible construct, pCW57.1, was a generous gift from Dr. David Root and Dr. John Doench (The Broad Institute of Harvard and MIT). The V5-tagged tet-inducible construct, pLIX_403, was obtained from Addgene (41395). MEK1 cDNA was described previously (7), and MEK2 cDNA was obtained from Addgene (23555). The m-isoform of MITF was previously described (20). Site-directed mutagenesis was performed in the pDONR (Invitrogen) construct using QuickChange II (Stratagene) according to the manufacturer's instructions. MEK1/2 cDNA was then transferred from pDONR to pCW57.1 or pLIX_403 using LR Clonase II (Invitrogen). Arginine 217 of MITF-m (37) was deleted using the QuikChange Lightning Mutagenesis Kit (Agilent), performed in pDonor223 (Invitrogen). MITF-mR217Δ was transferred into pLX304 using LR Clonase (Invitrogen) as per the manufacturer's recommendation.
Viral Infections.
293T cells (70% confluent) were transfected with expression vectors pCW57.1 MEK1 (tet-inducible), pLIX_403MEK2 (V5-tagged, tet-inducible), or pLX_304 MITF-m (V5 tagged), together with packaging vectors Δ8.91 and VSVG, using X-treme Gene 9 (Roche). Viral supernatants were passed through a 0.45-μm syringe. A375 cells were infected for 16 hours with virus in the presence of polybrene (4 μg/mL; Sigma); puromycin was introduced 48 hours postinfection to create stable cell lines. For MITF expression in WM266.4 cells, cells were infected at a 1:10 to 1:20 dilution of virus in 6-well plates (2.0 × 105 cells/well for immunoblot assays) or 96-well plates (3.0 × 103 for cell growth assays) in the presence of 5.5 μg/mL polybrene and centrifuged at 2,250 rpm for 60 minutes at 37°C, followed immediately by removal of media and replacement with complete growth media.
Cell Lines.
A375, SKMEL28, A2058, and WM266.4 cells were purchased from American Type Culture Collection (ATCC) and were maintained in Dulbecco's Modified Eagle Medium (DMEM) or RPMI-1640 with 10% heat-inactivated FBS. A375 cells stably expressing tet-inducible MEK1 (pCW57.1 MEK1) or tet-inducible MEK2-V5 (pLIX_403 MEK2) were maintained in 10% tet-approved FBS (Clontech) and 2 μg/mL puromycin. Cell lines obtained from ATCC, which verify identity by short-tandem repeat profiling, were passaged less than 6 months following receipt.
In Vitro Pharmacologic Growth Inhibition Assays.
Dabrafenib (RAF inhibitor), trametinib (MEK inhibitor), and VRT11E (ERK inhibitor) were purchased from ChemieTek. PLX4720 (RAF inhibitor), GDC0941 (PI3K inhibitor), and selumetinib (MEK inhibitor) were purchased from Selleck Chemicals. For growth inhibition analysis, A375 cells expressing WT or mutant MEK1 or MEK2 were seeded in 96-well plates (2,000 cells/well for A375 cells) and allowed to adhere for 16 hours. Afterward, media containing serial dilutions of inhibitor were added, ensuring that the final volume of dimethyl sulfoxide (DMSO) did not exceed 0.1%. Doxycycline (1 μg/mL) was added to the media at the time of drug treatment to induce the protein expression. Cells were incubated for 72 hours in the presence of drug, and viability was measured by the CellTiter96 AQueous Assay (Promega). For short-term culture growth inhibition analysis, cells were seeded in 96-well plates and treated the following day with serial dilutions of inhibitor. Cells were incubated for 72 hours in the presence of drug, and viability was measured by the CellTiter96 Aqueous Assay (Promega). WM266.4 cells engineered to express MITF were seeded into 96-well, white-walled, clear bottom plates. Seventy-two hours after viral infection, dilutions of the relevant compound were prepared in DMSO to 1,000× stocks. Drug stocks were then diluted 1:100 into appropriate growth media and added to cells at a dilution of 1:10 (1× final). Cell viability was measured using CellTiter-Glo Viability Assay (Promega) at a dilution of 1:6. Viability was calculated as a percentage of the control (untreated cells) after background subtraction. Six replicates were performed in each cell line and drug combination experiment, and the entire experiment was also repeated three times. Data from the pharmacologic growth-inhibition assays were modeled using a nonlinear regression curve fit with a sigmoidal dose response. These curves were displayed using GraphPad Prism 5 (GraphPad Software).
Western Blot Analysis.
Immunoblot studies were performed using standard procedures. Briefly, melanoma cells were lysed with radioimmunoprecipitation assay (RIPA) or 1% NP-40 lysis buffer containing both protease and phosphatase inhibitor cocktails (Roche). Lysates were quantified (Bradford assay), denatured (95°C), and resolved by SDS gel electrophoresis. Protein was transferred to polyvinylidene difluoride (PVDF) or nitrocellulose membranes and probed with primary antibodies recognizing pERK1/2, pMEK1/2 (Ser217/221), MEK1/2, total AKT, pAKT Ser473, PARP, actin, and α-tubulin (Cell Signaling Technology; 1:1,000 dilution) or MITF (NeoMarkers, C5), Silver, pERK (Sigma), V5 (Invitrogen), ERK2 and Melan-A (Santa Cruz Biotechnology). After incubation with the appropriate secondary antibody [anti-rabbit or anti-mouse immunoglobulin G (IgG), horseradish peroxidase (HRP)-linked; 1:1,000 dilution; Cell Signaling Technology], proteins were detected using chemiluminescence (Pierce).
Statistical Analysis
Analyses of clinical parameters were performed using the R statistical package. Significance between two means was calculated with the two-tailed Mann–Whitney test. The two-tailed Fisher exact test was used to test the statistical significance of the contingency table represented by NRAS or RAC1 mutation status in early resistance and responding cohorts. P < 0.05 was considered significant.
Disclosure of Potential Conflicts of Interest
N. Wagle has ownership interest (including patents) in Foundation Medicine and is a consultant/advisory board member of the same. L. Zimmer has received honoraria from the Speakers Bureaus of Roche and Bristol-Myers Squibb, is a consultant/advisory board member of Roche and Bristol-Myers Squibb, and has given expert testimony for the same. R. Gutzmer has received honoraria from the Speakers Bureaus of Bristol-Myers Squibb, Roche Pharma, Merck, Sharp & Dome, GlaxoSmithKline, Novartis, Amgen, Janssen, and Merck Serono and is a consultant/advisory board member of Bristol-Myers Squibb, Roche Pharma, Merck, Sharp & Dome, GlaxoSmithKline, Novartis, and Almirall Hermal. S.M. Goldinger is a consultant/advisory board member of Bristol-Myers Squibb and has given expert testimony for MSD. S. Ugurel has received a commercial research grant from Medac and is a consultant/advisory board member of Roche. C. Berking has received honoraria from the Speakers Bureaus of Roche and GlaxoSmithKline and is a consultant/advisory board member of Roche. U. Trefzer has received honoraria from the Speakers Bureaus of Merck, Sharp & Dome, Bristol-Myers Squibb, Roche, and GlaxoSmithKline and is a consultant/advisory board member of Roche, GlaxoSmithKline, and Merck, Sharp & Dome. C. Loquai has received honoraria from the Speakers Bureau of Roche and is a consultant/advisory board member of the same. L.A. Garraway has received a commercial research grant from Novartis, has ownership interest (including patents) in Foundation Medicine, and is a consultant/advisory board member of Novartis, Foundation Medicine, Boehringer–Ingelheim, and Millennium. D. Schadendorf has received honoraria from the Speakers Bureaus of Merck, GlaxoSmithKline, Roche, Novartis, Bristol-Myers Squibb, and Amgen and is a consultant/advisory board member of Merck, GlaxoSmithKline, Roche, Novartis, and Bristol-Myers Squibb. No potential conflicts of interest were disclosed by the other authors.
Authors' Contributions
Conception and design: E.M. Van Allen, N. Wagle, C.M. Johannessen, L.A. Garraway, D. Schadendorf
Development of methodology: E.M. Van Allen, N. Wagle, S.L. Carter, G. Getz, L.A. Garraway
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): E.M. Van Allen, A. Sucker, D.J. Treacy, C.M. Johannessen, E.M. Goetz, C.S. Place, S. Whittaker, L. Zimmer, U. Hillen, S.M. Goldinger, S. Ugurel, H.J. Gogas, F. Egberts, C. Berking, U. Trefzer, C. Loquai, B. Weide, S.B. Gabriel, L.A. Garraway, D. Schadendorf
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): E.M. Van Allen, N. Wagle, D. J. Treacy, A. Taylor-Weiner, S. Whittaker, G. V. Kryukov, E. Hodis, M. Rosenberg, A. McKenna, K. Cibulskis, U. Hillen, S. Ugurel, C. Loquai, S.L. Carter, L.A. Garraway, D. Schadendorf
Writing, review, and/or revision of the manuscript: E.M. Van Allen, N. Wagle, C.M. Johannessen, C.S. Place, S. Whittaker, L. Zimmer, R. Gutzmer, S.M. Goldinger, S. Ugurel, H.J. Gogas, F. Egberts, C. Berking, U. Trefzer, C. Loquai, B. Weide, L.A. Garraway, D. Schadendorf
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): E.M. Van Allen, N. Wagle, A. Taylor-Weiner, D. Farlow, L. Zimmer, R. Gutzmer, S.M. Goldinger, S. Ugurel, F. Egberts, D. Schadendorf
Study supervision: E.M. Van Allen, N. Wagle, D. Farlow, S.B. Gabriel, L.A. Garraway, D. Schadendorf
Acknowledgments
The authors thank the patients who were enrolled in this study.
Grant Support
This work was supported by the National Cancer Institute (Award #P01 CA163222 01A1, P50 CA 93683; to L.A. Garraway), National Human Genome Research Institute (Award #5U54HG003067-11; to L.A. Garraway, G. Getz, and S.B. Gabriel), the Melanoma Research Alliance (to L.A. Garraway), the Dr. Miriam and Sheldon G. Adelson Medical Research Foundation (to L.A. Garraway), the Harvard Clinical and Translational Science Center NIH Training Grant Award (Award #UL1 RR 025758; to C.S. Place), the National Institute of General Medical Sciences Grant T32GM07753 (to E. Hodis), the Conquer Cancer Foundation (to E.M. Van Allen and N. Wagle), the Dana-Farber Leadership Council (to E.M. Van Allen), and the American Cancer Society (to E.M. Van Allen).