





Aims Patients with acute coronary syndrome (ACS) in the Myocardial Ischaemia Reduction with Aggressive Cholesterol Lowering (MIRACL) study had diminished cardiovascular events after 16 weeks of treatment of atorvastatin 80 mg daily. We determined whether plasma lipoproteins at baseline and then at 6 weeks after randomization predicted clinical outcome.
Methods and results Cox proportional hazards models were constructed to determine relations between lipoproteins and clinical endpoint events. Baseline LDL cholesterol (LDL-C) did not predict outcome. In contrast, baseline HDL-C predicted outcome with a hazard ratio of 0.986 per mg/dL increment in HDL-C, P<0.001, indicating 1.4% reduction in risk for each 1 mg/dL increase in HDL-C. Atorvastatin treatment profoundly lowered LDL-C, but had minimal effect on HDL-C. Neither Week 6 LDL-C nor absolute change of LDL-C from baseline by Week 6 had any significant impact on clinical endpoints occurring between Week 6 and Week 16 after randomization.
Conclusion Plasma HDL-C, but not LDL-C, measured in the initial stage of ACS predicts the risk of recurrent cardiovascular events over the ensuing 16 weeks. LDL-C reduction does not account for the clinical risk reduction with atorvastatin treatment after ACS. This finding may suggest that the clinical benefit of atorvastatin after ACS is mediated by qualitative changes in the LDL particle and/or by non-lipid (pleiotropic) effects of the drug.
for the editorial comment on this article (doi:10.1093/eurheartj/ehi218)
Introduction
Epidemiological studies have shown that long-term morbidity and mortality in coronary heart disease (CHD), manifest over years, is directly related to circulating levels of atherogenic lipoproteins, in particular LDL-cholesterol (LDL-C).1 In addition, long-term prospective randomized trials of statins in chronic CHD indicate a direct relation between the LDL-C concentration achieved during treatment and the risk of a new ischaemic cardiovascular event.2 These data provide cogent evidence for a relationship between spontaneous or pharmacologically modulated LDL-C levels and the long-term risk of CHD events.
Acute coronary syndrome (ACS), including acute myocardial infarction and unstable angina pectoris, is associated with a high short-term risk for recurrent ischaemic events.3,4 There has been no prior study concerning the relationship between the serum lipoproteins at the time of ACS and the risk of early, recurrent ischaemic events following ACS. Nor has there been any analysis to determine whether pharmacological modulation of lipoproteins affects short-term risk after ACS. Because of different physiology of unstable vs. stable atherosclerotic plaques, it is possible that the relationship between serum lipoproteins and events may differ in acute and chronic CHD.
Statins may act rapidly to improve vascular endothelial function, attenuate vascular inflammation, and correct prothrombotic tendencies.5 Such effects have been observed within a few weeks of initiating treatment with statins in experimental animals or human subjects. These effects of statins may be related to their lipid-lowering actions and/or mechanisms unrelated to circulating lipoproteins.
The Myocardial Ischaemia Reduction with Aggressive Cholesterol Lowering (MIRACL) trial demonstrated that treatment with atorvastatin 80 mg daily, initiated early after presentation with ACS and maintained for 16 weeks, resulted in a significant reduction of early recurrent ischaemic events, including both cardiac events and strokes when compared with treatment with placebo.6
The present analysis of data from the MIRACL trial had two objectives: first, we sought to determine the relationship between baseline serum lipoproteins (i.e. prior to randomized treatment assignment) and 16 week outcomes following ACS. Secondly, we sought to determine the relationship between the changes in serum lipoproteins during randomized treatment and the short-term risk of recurrent cardiovascular events.
Methods
Study population
The design7 and the results of the MIRACL study have been reported.6 MIRACL was a worldwide randomized double-blind study comprising 3086 patients. All were admitted to hospital with unstable angina or non-Q-wave acute myocardial infarction (index events). These diagnoses required chest discomfort lasting at least 15 min within the 24 h preceding hospitalization and representing a change in the usual pattern of angina. The diagnosis of unstable angina required: new or dynamic ST-wave or T-wave changes in at least two electrocardiographic leads, a new wall motion abnormality by echocardiography, a new and reversible myocardial perfusion defect by radionuclide scintigraphy, or elevation of cardiac troponin to a level not exceeding twice the upper limit of normal. The diagnosis of non-Q-wave myocardial infarction required elevation of serum creatine kinase or its MB fraction or troponin to a level exceeding twice the upper limit of normal.
Most important exclusion criteria were serum cholesterol >7.0 mmol/L (270 mg/dL), anticipated coronary revascularization, Q-wave myocardial infarction within the previous month, coronary artery bypass grafting within 3 months, percutaneous coronary intervention within 6 months of enrolment, or treatment with other lipid-lowering drugs. There was no lower limit for total or LDL-C or upper limit for HDL-cholesterol (HDL-C) at entry.
All patients provided informed consent. The protocol was approved by local institutional review boards.
Study design
Between 24 and 96 h after hospital admission, patients were randomly assigned to double-blind treatment with atorvastatin 80 mg/day or matching placebo for 16 weeks. The dose was chosen to allow for maximal effects on LDL-C and possible pleiotropism in this proof-of-concept trial. Follow-up visits were scheduled at 2, 6, and 16 weeks. Plasma lipid and lipoprotein concentrations were measured at baseline, 6, and 16 weeks. Apolipoprotein B (apoB) and A-1 (apoA-1) were determined at baseline and at 16 weeks.
The primary efficacy measure was the time to first occurrence of death, non-fatal acute myocardial infarction, cardiac arrest with resuscitation, or worsening angina with new objective evidence of ischaemia and requiring emergency rehospitalization.7
Lipid and lipoprotein analysis
Venous blood was collected after subjects had fasted for 12–14 h. For analysis of changes in lipids, frozen sera obtained immediately before the start of active treatment and at the Week 6 and Week 16 visits were assayed. The central laboratory (Medical Research Laboratories, Cincinnati, OH, USA, and Brussels, Belgium) utilized assay methods standardized by the Centers for Disease Control, USA. In all samples where serum triglyceride levels did not exceed 4.40 mmol/L (400 mg/dL), LDL-C values were calculated on the basis of the Friedewald formula.8 If the serum triglyceride level exceeded 4.40 mmol/L, plasma LDL-C was measured directly.
Statistical analysis
Pre-specified Cox proportional hazards models stratified by country and inclusion event (unstable angina or non-Q-wave myocardial infarction) were constructed to determine relationships between lipoprotein fractions (total cholesterol, LDL-C, HDL-C, triglycerides, apoA-1, and apoB), randomized treatment assignment, and time to occurrence of clinical endpoint events (death, non-fatal acute myocardial infarction, cardiac arrest, or urgent rehospitalization for recurrent myocardial ischaemia). Two analysis populations were considered (Figure 1).
The first population consisted of all MIRACL subjects with baseline lipid and lipoprotein measurements. Analysis of this population related lipid and lipoprotein measurements at baseline (before randomized treatment) to clinical events occurring from randomization to Week 16 (end of study). The second population consisted of all MIRACL subjects who had measurements of lipids and lipoproteins both at baseline and at Week 6 of randomized treatment, excluding those who had experienced a primary endpoint event before Week 6. Analysis of this population related lipid and lipoprotein measurements at Week 6 (or the absolute change in these measurements from baseline to Week 6) to clinical events occurring from Week 6 to Week 16.
Hazard ratios were expressed as the relative hazard for each 1 mg/dL increase in a specific lipid parameter, using a linear model. Absolute changes in concentrations were used in the models to maintain the same continuous scale as the baseline and Week 6 analyses. The validity of the proportional hazard assumption was tested by adding a time-dependent variable to each model to confirm that the hazard ratio for each lipid parameter did not increase or decrease over time. To determine whether non-linear relationships existed between lipid and lipoprotein concentrations and events, baseline, Week 6, and absolute change from baseline to Week 6 concentrations of total cholesterol, LDL-C, HDL-C, and triglycerides were also analysed in quartiles. In each model, the relative hazard of an event in quartiles 2, 3, and 4 was expressed with quartile 1 serving as the reference category (hazard ratio=1.000). All hypothesis testing was two-sided, and statistical significance was declared if P<0.05. Owing to the exploratory nature of the analyses, there was no adjustment to maintain an overall Type I error rate of 5%.
Results
Characteristics of the study population
Among the 3086 randomized patients in the MIRACL trial, 3038 had baseline and 2739 had baseline and Week 6 lipid and lipoprotein measurements. Characteristics of both analysis populations were similar to those of the entire MIRACL cohort.6 Mean age was 65 years. About one-third of the patients were women, with the vast majority of them post-menopausal. There were slightly more patients included with the diagnosis non-Q-wave myocardial infarction (54%) than with unstable angina (46%). Approximately one-fourth of the patients had diabetes mellitus. The primary reason for and likelihood of missing lipoprotein data at Week 6 was related to the occurrence of endpoints: subjects who had an endpoint during the first 6 weeks of the trial were less likely to have post-baseline lipid measurements than subjects who did not experience a recurrent event.
Lipid and lipoprotein concentrations at baseline and on randomized treatment
At the time of randomization, mean (SD) baseline LDL-C was 3.22 (0.86) mmol/L. There was no difference in baseline LDL-C between placebo and atorvastatin groups (Table 1). Mean baseline LDL-C in the non-Q-wave myocardial infarction group and in the unstable angina group were 3.15 and 3.33 mmol/L, respectively. This difference is probably related to greater tissue damage, more pronounced acute phase reaction, and thereby lower LDL-C among patients with non-Q-wave myocardial infarction than among patients with unstable angina. Six weeks after randomization, LDL-C had increased by ∼10% to a mean concentration of 3.46 mmol/L in the placebo group. Six weeks after randomization, LDL-C decreased by 46% to a mean concentration of 1.69 mmol/L in the atorvastatin group.
Changes in apoB during randomized treatment followed essentially the pattern of LDL-C, with a slight increase in the placebo group and a substantial decrease in the atorvastatin group, resulting in a net concentration difference of 51 mg/dL between the two groups at Week 6. In both groups, apoA-1 increased slightly from baseline to Week 16 of randomized treatment.
Mean (SD) baseline HDL-C was 1.22 (0.32) mmol/L for the whole cohort and showed only minute changes in both groups 6 weeks after randomization.
Mean (SD) baseline triglyceride concentration for this cohort was moderately elevated at 2.01 (1.02) mmol/L. At Week 6, triglycerides remained essentially unchanged in the placebo group, but decreased by 21% in the atorvastatin group.
At Week 16, mean plasma lipid measurements were similar to those at Week 6; in both groups, total cholesterol, LDL-C, HDL-C, and triglycerides increased by 2–6% between the Week 6 and the Week 16 measurement (data not shown).
Relation of baseline lipids to 16 week risk of ischaemic events
Relationships among baseline lipids, lipoproteins, and apolipoproteins, and endpoints are shown in Figure 2. Between Week 0 and Week 16, a primary endpoint event occurred in 491 patients or 16.2% of patients (Figure 1). In a model incorporating treatment assignment and baseline LDL-C, assignment to treatment with atorvastatin was associated with a decreased risk of a primary endpoint event [hazard ratio 0.835, 95% confidence interval (CI) 0.699–0.999, P=0.049].
Surprisingly, baseline LDL-C bore no relation to 16 week outcomes, with a univariate hazard ratio of 1.000 (95% CI 0.997–1.003) per mg/dL increment in baseline LDL-C. Time from admission to randomization did not confound the relationship (or lack of relationship) between any lipid parameter at baseline and recurrent cardiac events. There was no significant interaction between treatment assignment and baseline LDL-C. In an analysis by quartiles of baseline LDL-C, the hazard ratios [(95% CI), P-value] for recurrent ischaemic events in quartiles 2–4 were 0.890 [(0.693, 1.144), 0.364], 0.934 [(0.728, 1.200), 0.594], and 0.924 [(0.718, 1.188), 0.535], respectively, when quartile 1 was set to 1.000. In an analysis confined to the placebo group (i.e. an analysis of ‘natural history’ without lipid intervention), there was also no significant effect of baseline LDL-C on risk of a primary endpoint event (hazard ratio 1.004, 95% CI 0.998–1.010). Similar to baseline LDL-C, baseline total cholesterol, triglycerides, and apoB were not significantly related to the 16 week risk of clinical events.
In contrast to LDL-C, baseline HDL-C was significantly related to outcome at 16 weeks. In a model incorporating treatment assignment, baseline HDL-C, and their interaction, for each mg/dL increment in baseline HDL-C, the hazard ratio for an event was 0.986 (95% CI 0.979–0.994; P<0.001). Expressed another way, the risk of an event diminished by 1.4% for each 1 mg/dL increment in baseline HDL-C. An analysis by quartiles of baseline HDL-C suggests a linear relationship; hazard ratios [(95% CI), P-value] for events in quartiles 2–4 (relative to quartile 1) were 0.896 [(0.706, 1.136), 0.364], 0.822 [(0.642, 1.053), 0.121], and 0.620 [(0.477, 0.806), <0.001]. Owing to the unexpected non-significant relationship between baseline LDL-C and outcome, the relationship of baseline HDL-C and atorvastatin treatment to outcome was further explored in a post hoc analysis by level of LDL-C (below or above median of 3.17 mmol/L). The protective effect of HDL-C was more pronounced for those patients with baseline LDL-C below median, indicating that each mg/dL increase in baseline HDL-C was associated with a 2.2% reduction in risk (95% CI 1.2–3.3%; P<0.001). In contrast, among those with baseline LDL-C above median, there was no significant relationship between baseline HDL-C and risk of an ischaemic event between Week 0 and Week 16 [hazard ratio (95% CI)=0.998 (0.987, 1.010); P=0.744]. Baseline apoA-1 was inversely related to 16 week risk (P<0.001).
There was no evidence against the proportional hazards assumption in all of the models involving baseline lipid parameters.
Relationship between on-treatment lipids and outcome
The relationship between modification of lipids by randomized treatment and outcomes was studied using lipid values at baseline and Week 6 and clinical events occurring from Week 6 to Week 16 (n=121). Although this analysis excluded events occurring from baseline to Week 6, the alternative approach (relating lipid changes between baseline and Week 16 to events during the same period) would be logically invalid as it would use future lipid values (at Week 6) to ‘predict’ past clinical events (prior to Week 6). The proportion of fatal events was greater in the period after 6 weeks than in the period prior to 6 weeks, even though the overall event rate was lower after 6 weeks. Therefore, we do not believe that by excluding events between 0 and 6 weeks, this resulted in a population at lower risk.
Assignment to treatment with atorvastatin tended to lower the risk of an ischaemic event between Week 6 and Week 16, with hazard ratio of 0.721 (95% CI 0.501–1.037; P=0.077 and model including Week 6 HDL-C). However, neither Week 6 LDL-C or HDL-C nor absolute changes of these lipoproteins from baseline to Week 6 had any significant relationship to the risk of a clinical endpoint between Week 6 and Week 16, Figure 2. In addition, interactions between Week 6 lipoprotein levels and treatment assignment were not statistically significant, indicating that atorvastatin treatment did not modify the relationships between on-treatment LDL-C, HDL-C, or absolute change in these parameters and the incidence of recurrent events.
We also computed the univariate hazard ratio for the effect of LDL-C at 6 weeks (or change from Week 0 to Week 6) on the risk of an event between Week 6 and Week 16. This univariate hazard ratio was 1.003 (95% CI 0.999–1.007) when compared with the multivariate hazard ratio for LDL-C of 1.001 (95% CI 0.996–1.007), i.e. in a model also including treatment assignment.
If the Week 6 plasma samples were divided into quartiles of LDL-C with the lowest quartile serving as a reference, there was no discernible relationship between quartile of LDL-C at Week 6 or absolute change to Week 6 and events between Week 6 and Week 16. This was true both for quartiles defined across both treatment groups and for quartiles defined within treatment groups. Thus, there was no linear relationship between Week 6 or absolute change in LDL-C and events between Week 6 and Week 16, nor did analysis by quartiles suggest a threshold or quadratic relationship.
In contrast, patients in the highest quartile of HDL-C at Week 6 had a substantially reduced risk of an event between Week 6 and Week 16 with a hazard ratio of 0.520 (95% CI 0.305–0.886; P<0.02 vs. the lowest quartile). Neither Week 6 values of total cholesterol or triglycerides nor changes in these values from baseline to Week 6 bore a relation to outcomes between Week 6 and Week 16.
In all of the models involving on-treatment lipid parameters, the proportional hazards assumption appeared to be valid.
Discussion
The present analysis of the MIRACL trial provides three key observations concerning the relationship between lipids and short-term risk following ACS. First, we demonstrate that no relationship exists between LDL-C levels at the time of ACS and the risk of recurrent cardiovascular events over the ensuing 16 weeks. Secondly, we demonstrate a significant relationship between HDL-C at the time of ACS and 16 week risk of recurrent events, with a 1.4% reduction in risk for each 1 mg/dL increment in baseline HDL-C. Thirdly, we demonstrate that the pronounced LDL-C lowering induced by a high dose of atorvastatin bears no relation to the short-term risk of recurrent ischaemic events after ACS and does not account for the benefit of atorvastatin treatment after ACS.
Interpretation of a relationship between baseline LDL-C and risk of early, recurrent ischaemic events after ACS is complicated by acute phase effects on this lipoprotein.9 In the present study, this phenomenon is demonstrated in two ways. First, after 6 weeks on placebo, LDL-C increased significantly by 10% from baseline, suggesting recovery from an initial acute phase depression of plasma lipid levels. Secondly, those patients with more severe initial myocardial injury—whose index event was non-Q-wave myocardial infarction—had lower baseline LDL-C and higher levels of inflammatory markers, such as C-reactive protein,10 than those with less severe injury—index event unstable angina. Thus, as a result of the acute phase response, baseline LDL-C may be lower among patients with a poorer prognosis owing to greater initial myocardial injury. This relationship is directionally opposite to the relationship of LDL-C to long-term risk for CHD events. Operating concurrently, these two relationships may negate one another, resulting in no net relationship between LDL-C close to the time of ACS and short-term risk of recurrent events after ACS.
In contrast to LDL-C, acute phase effects on HDL-C were minimal in MIRACL. The cause of this, which is at variance with some other observations indicating that HDL-C and apoA-1 decrease in ACSs, was probably owing to the early blood sampling during the index event and to the relatively minor myocardial damage sustained. Therefore, levels of HDL-C measured shortly after ACS may more accurately reflect the physiological role of this lipoprotein in promoting plaque stability through mechanisms including reverse cholesterol transport and protection of LDL-C from oxidative stress.
Our second key finding was a highly significant relationship between baseline levels of HDL-C and 16 week risk of recurrent events after ACS. Because atorvastatin treatment had minimal effect on HDL-C in MIRACL, it is not possible to determine whether the relationship between HDL-C and risk would apply if HDL-C were modulated by drug therapy. Of particular interest is that atorvastatin, with unchanged apoA-1 concentrations, has been demonstrated to shift the HDL subpopulation profile of CHD patients towards that observed in subjects without CHD.11 The observed relationship raises the possibility that HDL might be an attractive target for modifying the high-risk state following ACS. The striking effect of direct apolipoprotein A-I infusions on coronary atherosclerosis in patients with ACS supports this view.12
The third finding, the absence of a discernible relation between changes in lipoproteins and the reduction in ischaemic events resulting from statin treatment is unexpected. It should be borne in mind that estimation of LDL-C with the Friedewald formula is a crude measure of the lipoprotein. Despite similar LDL-C concentrations, LDL particles may differ in their susceptibility to oxidation and thereby their ability to elicit an inflammatory response. It is possible that changes in the physicochemical properties of LDL-C (e.g. size, chemical composition, or oxidation state) by atorvastatin,13 rather than changes in the concentration of LDL-C, could account for the beneficial effect on ischaemic events observed in MIRACL. This explanation for the positive outcome on MIRACL is supported by our suggestion that atorvastatin promotes mobilization and clearance of proinflammatory oxidized phospholipids, which may contribute to a reduction in ischaemic events after ACS.14
Alternatively, our data are also consistent with the hypothesis that atorvastatin produces beneficial clinical effects after ACS entirely independent of effects on LDL-C or other lipoproteins, acting instead through non-lipid, or pleiotropic mechanisms. In experimental studies, many of the potentially beneficial effects of statins, such as enhanced expression of endothelial nitric oxide synthase, reduced expression of matrix metalloproteinases, and attenuation of ischaemia/reperfusion injury, may be demonstrated independently of any changes in the lipid milieu.15 The lipid-independent mechanisms of action may include modulation of the rho/rho-kinase pathway. By inhibiting 3-hydroxy-3-methyl glutaryl CoA reductase, statins reduce the intracellular pool of isoprenoid compounds. Reduced prenylation of the rho protein may attenuate vascular inflammation through a combination of different actions.16 We have previously demonstrated that atorvastatin treatment potentiates the decline in inflammation as estimated by CRP in MIRACL, a finding supporting the concept of an anti-inflammatory effect mediating the beneficial clinical effect in ACS.10 We demonstrated significant effects by atorvastatin on non-fatal and fatal strokes in the MIRACL trial.17 When the outcome measure included non-fatal stroke in addition to primary endpoint events in the present analysis, assignment to atorvastatin was associated with a hazard ratio of 0.824 (95% CI 0.693–0.979; P<0.028). As the risk of stroke bears little relation to LDL-C in epidemiological studies, this finding favours the view of a pleiotropic effect to explain the clinical benefit in MIRACL.
Several limitations of the present analysis should be considered. Our analysis of relationships between changes in lipoproteins on randomized treatment and events utilized lipid values at Week 6 (or changes from Week 0 to Week 6) and events occurring between Week 6 and Week 16. Events occurring between Week 0 and Week 6 were not considered. This resulted in considerably fewer endpoints and diminished statistical power when compared with an analysis including all events between Week 0 and Week 16. However, the latter analysis would be logically flawed, as it would use future lipid values (at Week 6) to predict past events (prior to Week 6).
This analysis of the MIRACL trial may provide the first evidence of a clinically significant effect induced by non-lipid or pleiotropic effect(s) of statins. Regardless of the underlying mechanism, the practical message from this analysis is that, within the lipoprotein ranges included in MIRACL, the benefit of statin treatment after ACS is not influenced by LDL-C levels either at baseline or on treatment, and measurements of serum lipoproteins should not guide the use of this therapeutic intervention in the high-risk period immediately following ACS.
Acknowledgement
Supported by a grant from Pfizer Inc. Pfizer provided the atorvastatin and matching placebo used in this study.
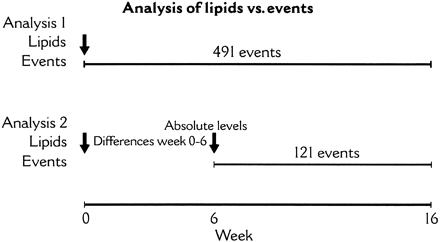
Figure 1 Analysis of lipids vs. events in the MIRACL study.
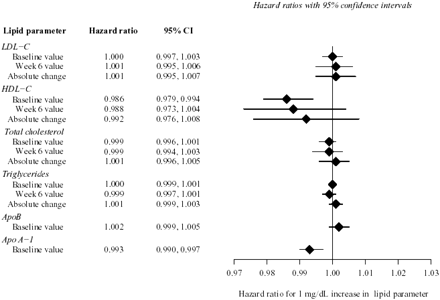
Figure 2 Relationship between time to first occurrence of a primary clinical endpoint up to 16 weeks after randomization and baseline, Week 6, and Week 0 to Week 6 absolute change values.
Baseline and 6 weeks on-treatment lipid, lipoprotein, and apolipoprotein concentrations
| Lipid parameter and treatment group | Baseline value | Six week value | Percentage change from baseline at 6 weeks | P-value for percentage change treatment comparison |
|---|---|---|---|---|
| Total cholesterol (mmol/L) | ||||
| Atorvastatin | 5.33 (5.29, 5.38) | 3.54 (3.48, 3.59) | −33 (−34, −32) | <0.001 |
| Placebo | 5.38 (5.33, 5.43) | 5.54 (5.49, 5.62) | 4 (4, 5) | |
| LDL-C (mmol/L) | ||||
| Atorvastatin | 3.20 (3.15, 3.25) | 1.69 (1.64, 1.72) | −46 (−48, −45) | <0.001 |
| Placebo | 3.25 (3.20, 3.28) | 3.46 (3.41, 3.51) | 10 (8, 11) | |
| HDL-C (mmol/L) | ||||
| Atorvastatin | 1.22 (1.20, 1.22) | 1.20 (1.17, 1.20) | −1 (−2, 0) | 0.431 |
| Placebo | 1.20 (1.17, 1.22) | 1.17 (1.17, 1.20) | 0 (−1, 1) | |
| Triglycerides (mmol/L) | ||||
| Atorvastatin | 1.99 (1.94, 2.05) | 1.43 (1.39, 1.47) | −23 (−25, −21) | <0.001 |
| Placebo | 2.05 (1.98, 2.09) | 2.02 (1.96, 2.09) | 5 (3, 7) | |
| ApoB (mg/dL) | ||||
| Atorvastatin | 132 (131, 134) | 87 (86, 89)a | −33 (−34, −32) | <0.001 |
| Placebo | 133 (131, 135) | 138 (136, 140)a | 6 (5, 7) | |
| ApoA-1 (mg/dL) | ||||
| Atorvastatin | 133 (131, 134) | 137 (136, 139)a | 6 (4, 7) | 0.030 |
| Placebo | 132 (130, 133) | 139 (137, 141)a | 7 (6, 8) |
| Lipid parameter and treatment group | Baseline value | Six week value | Percentage change from baseline at 6 weeks | P-value for percentage change treatment comparison |
|---|---|---|---|---|
| Total cholesterol (mmol/L) | ||||
| Atorvastatin | 5.33 (5.29, 5.38) | 3.54 (3.48, 3.59) | −33 (−34, −32) | <0.001 |
| Placebo | 5.38 (5.33, 5.43) | 5.54 (5.49, 5.62) | 4 (4, 5) | |
| LDL-C (mmol/L) | ||||
| Atorvastatin | 3.20 (3.15, 3.25) | 1.69 (1.64, 1.72) | −46 (−48, −45) | <0.001 |
| Placebo | 3.25 (3.20, 3.28) | 3.46 (3.41, 3.51) | 10 (8, 11) | |
| HDL-C (mmol/L) | ||||
| Atorvastatin | 1.22 (1.20, 1.22) | 1.20 (1.17, 1.20) | −1 (−2, 0) | 0.431 |
| Placebo | 1.20 (1.17, 1.22) | 1.17 (1.17, 1.20) | 0 (−1, 1) | |
| Triglycerides (mmol/L) | ||||
| Atorvastatin | 1.99 (1.94, 2.05) | 1.43 (1.39, 1.47) | −23 (−25, −21) | <0.001 |
| Placebo | 2.05 (1.98, 2.09) | 2.02 (1.96, 2.09) | 5 (3, 7) | |
| ApoB (mg/dL) | ||||
| Atorvastatin | 132 (131, 134) | 87 (86, 89)a | −33 (−34, −32) | <0.001 |
| Placebo | 133 (131, 135) | 138 (136, 140)a | 6 (5, 7) | |
| ApoA-1 (mg/dL) | ||||
| Atorvastatin | 133 (131, 134) | 137 (136, 139)a | 6 (4, 7) | 0.030 |
| Placebo | 132 (130, 133) | 139 (137, 141)a | 7 (6, 8) |
Data are represented as mean (95% CI); percentage change means CI and treatment comparison P-values from ANCOVA models with effects owing to treatment and baseline value.
aApplies to 16 weeks.
Baseline and 6 weeks on-treatment lipid, lipoprotein, and apolipoprotein concentrations
| Lipid parameter and treatment group | Baseline value | Six week value | Percentage change from baseline at 6 weeks | P-value for percentage change treatment comparison |
|---|---|---|---|---|
| Total cholesterol (mmol/L) | ||||
| Atorvastatin | 5.33 (5.29, 5.38) | 3.54 (3.48, 3.59) | −33 (−34, −32) | <0.001 |
| Placebo | 5.38 (5.33, 5.43) | 5.54 (5.49, 5.62) | 4 (4, 5) | |
| LDL-C (mmol/L) | ||||
| Atorvastatin | 3.20 (3.15, 3.25) | 1.69 (1.64, 1.72) | −46 (−48, −45) | <0.001 |
| Placebo | 3.25 (3.20, 3.28) | 3.46 (3.41, 3.51) | 10 (8, 11) | |
| HDL-C (mmol/L) | ||||
| Atorvastatin | 1.22 (1.20, 1.22) | 1.20 (1.17, 1.20) | −1 (−2, 0) | 0.431 |
| Placebo | 1.20 (1.17, 1.22) | 1.17 (1.17, 1.20) | 0 (−1, 1) | |
| Triglycerides (mmol/L) | ||||
| Atorvastatin | 1.99 (1.94, 2.05) | 1.43 (1.39, 1.47) | −23 (−25, −21) | <0.001 |
| Placebo | 2.05 (1.98, 2.09) | 2.02 (1.96, 2.09) | 5 (3, 7) | |
| ApoB (mg/dL) | ||||
| Atorvastatin | 132 (131, 134) | 87 (86, 89)a | −33 (−34, −32) | <0.001 |
| Placebo | 133 (131, 135) | 138 (136, 140)a | 6 (5, 7) | |
| ApoA-1 (mg/dL) | ||||
| Atorvastatin | 133 (131, 134) | 137 (136, 139)a | 6 (4, 7) | 0.030 |
| Placebo | 132 (130, 133) | 139 (137, 141)a | 7 (6, 8) |
| Lipid parameter and treatment group | Baseline value | Six week value | Percentage change from baseline at 6 weeks | P-value for percentage change treatment comparison |
|---|---|---|---|---|
| Total cholesterol (mmol/L) | ||||
| Atorvastatin | 5.33 (5.29, 5.38) | 3.54 (3.48, 3.59) | −33 (−34, −32) | <0.001 |
| Placebo | 5.38 (5.33, 5.43) | 5.54 (5.49, 5.62) | 4 (4, 5) | |
| LDL-C (mmol/L) | ||||
| Atorvastatin | 3.20 (3.15, 3.25) | 1.69 (1.64, 1.72) | −46 (−48, −45) | <0.001 |
| Placebo | 3.25 (3.20, 3.28) | 3.46 (3.41, 3.51) | 10 (8, 11) | |
| HDL-C (mmol/L) | ||||
| Atorvastatin | 1.22 (1.20, 1.22) | 1.20 (1.17, 1.20) | −1 (−2, 0) | 0.431 |
| Placebo | 1.20 (1.17, 1.22) | 1.17 (1.17, 1.20) | 0 (−1, 1) | |
| Triglycerides (mmol/L) | ||||
| Atorvastatin | 1.99 (1.94, 2.05) | 1.43 (1.39, 1.47) | −23 (−25, −21) | <0.001 |
| Placebo | 2.05 (1.98, 2.09) | 2.02 (1.96, 2.09) | 5 (3, 7) | |
| ApoB (mg/dL) | ||||
| Atorvastatin | 132 (131, 134) | 87 (86, 89)a | −33 (−34, −32) | <0.001 |
| Placebo | 133 (131, 135) | 138 (136, 140)a | 6 (5, 7) | |
| ApoA-1 (mg/dL) | ||||
| Atorvastatin | 133 (131, 134) | 137 (136, 139)a | 6 (4, 7) | 0.030 |
| Placebo | 132 (130, 133) | 139 (137, 141)a | 7 (6, 8) |
Data are represented as mean (95% CI); percentage change means CI and treatment comparison P-values from ANCOVA models with effects owing to treatment and baseline value.
aApplies to 16 weeks.
Sharret AR, Ballantyne CM, Coady SA, Heiss G, Sorlie PD, Catellier D, Patsch W. Coronary heart disease prediction from lipoprotein cholesterol levels, triglycerides, lipoprotein(a), apolipoproteins A-I and B, and HDL density subfractions: The Atherosclerosis Risk in Communities (ARIC) Study.
Pedersen T, Olsson A, Faergeman O, Kjekshus J, Wedel H, Berg K, Wilhelmsen L, Haghfelt T, Thorgeirson G, Pyörälä K, Miettinen T, Christophersen B, Tobert JA, Musliner TA, Cook TJ. Lipoprotein changes and reduction in the incidence of major coronary heart disease events in the Scandinavian Simvastatin Survival Study (4S).
Cannon CP, Weintraub WS, Demopoulos LA, Vicari R, Frey MJ, Lakkis N, Neumann FJ, Robertson DH, DeLucca PT, DiBattiste PM, Gibson CM, Braunwald E. Comparison of early invasive and conservative strategies in patients with unstable coronary syndromes treated with the glycoprotein IIb/IIIa inhibitor tirofiban.
Schwartz G, Oliver M, Ezekowitz M, Ganz P, Waters D, Kane J, Texter M, Pressler ML, Black D, Chaitman BR, Olsson AG. Rationale and design of the myocardial ischemia reduction with aggressive cholesterol lowering (MIRACL) study that evaluates atorvastatin in unstable angina pectoris and in non-Q-wave acute myocardial infarction.
Friedewald W, Levy R, Fredrickson D. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge.
Faulkner M, Hilleman D, Destache C, Mooss A. Potential influence of timing of low-density lipoprotein cholesterol evaluation in patients with acute coronary syndrome.
Nissen SE, Tsunoda T, Tuzcu EM, Schoenhagen P, Cooper CJ, Yasin M, Eaton GM, Lauer MA, Sheldon WS, Grines CL, Halpern S, Crowe T, Blankenship JC, Kerensky R. Effect of recombinant apoA-I milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial.
Tsimikas S, Witztum JL, Miller ER, Sasiela WJ, Szarek M, Olsson AG, Schwartz GG. High-dose atorvastatin reduces total plasma levels of oxidized phospholipids and immune complexes present on apolipoprotein B-100 in patients with acute coronary syndromes in the MIRACL trial.


{kind=link}