




Abstract
Non-melanoma skin cancer, i.e. basal cell carcinoma (BCC) and squamous cell carcinoma (SCC) are the most frequent tumors and their number is still increasing worldwide (1). Furthermore, immunosuppression in organ transplant patients strongly contributes to the increase in skin cancer incidence—being 65–250 times more frequent than in the general population. Often these patients suffer from a second and third lesion and the severity of these tumors is linked to their number. SCCs in transplant recipients also appear to be more aggressive. They tend to grow rapidly, show a higher rate of local recurrences and metastasize in 5–8% of the patients (all reviewed in Ref. 2). This largely differs from BCCs which are more frequent in the general population—at a ratio of 4:1 as compared with SCCs—but the number is only increased by a factor of 10 in transplant recipients. This may suggest that ‘dormant’ SCC precursor cells/lesions are present at a high frequency in the population but they are well controlled by the immune system. BCC, on the other hand, may be less dependent on immune surveillance thereby underlining its different etiology. While for BCC development the genetic hallmark is abrogation of the ptch-sonic hedgehog pathway, little is known about the causal alterations of SCCs. However, the complexity of the genetic alterations (numerical and structural aberration profiles) in SCCs argues for several levels of genomic instability involved in the generation and progression of skin cancer.
The multi-stage model of skin carcinogenesis (SCC development)
While BCCs are believed to develop de novo , skin SCC development is viewed as a multistep process. In agreement with that, cumulative life time exposure to ultraviolet (UV) radiation (especially UV-B) is thought to be the primary cause of skin carcinogenesis. Early and potentially primary event mutations of p53 have been found in otherwise unsuspicious epidermis ( 3 – 5 ) and can be detected as p53 patches by immunohistochemistry. Indeed, 70% of these p53 patches were shown to have an underlying mutation in the p53 gene ( 4 , 6 ). It thus was reasoned that those are early precancerous lesions and may even serve as a risk marker for skin SCC development. However, a retrospective study of 250 cases did not establish statistically significant differences between skin from patients with solitary versus multiple skin carcinomas. Instead, this study highlighted an increased frequency of p53 patches with age ( 7 ).
Actinic keratoses (AK), on the other hand, are well established precancerous skin lesions and it is suggested that ∼10% of these sun-induced lesions will develop into skin SCCs ( 8 ). With reference to this it is not surprising that p53 mutations and in particular UV-type mutations are frequently found in AK. Bowen's disease (BD), also known as carcinoma in situ (CIS), represents a preinvasive stage of the invasive skin SCCs. AK and CIS can occur at multiple sites as well as in the vicinity of SCC, supporting the hypothesis of field cancerization already suggested from the immuno-positive p53 distribution ( 9 ). Further indications for their precursor state came from molecular and cytogenetic studies ( 10 – 12 ). In contrast, a Japanese study that investigated microsatellite instability (MSI) and loss of heterogeneity (LOH) in AK and SCC showed no MSI in neither AK nor SCC and LOH in only 7/37 AKs and 1/14 SCCs. Since LOH was also demonstrated in histologically normal skin they suggested that at least in Japan AK was not likely to precede SCC ( 13 ).
Another skin tumor that is still controversially discussed in the sequence of skin carcinogenesis is the keratoacanthoma (KA). KAs are benign cutaneous squamous neoplasias arising preferentially on sun-exposed skin. They are characterized by a rapid growth phase for the first 4–8 weeks and a possible spontaneous self-induced regression after 3–6 months. Due to the initially very similar growth rate and morphology of KAs and well differentiated SCCs ( 14 ), it is still a matter of debate whether KAs are a specific type of SCCs or are not within the same biological and diagnostical spectrum ( 15 ). Several architectural criteria were used to distinguish KAs from SCCs. However, even in combination, these were not satisfying ( 16 ). While a previous LOH study did not support the hypothesis that KAs are SCCs that regress as a result of external (host) influences ( 17 ) our present data argue in favour of KAs being genetically incomplete SCCs and therefore are likely still controlled environmentally (S. Popp, B. Jelinek and P. Boukamp, unpublished data). Thus, it remains to be seen whether KAs may still best be viewed as an aborted malignancy that only rarely progress into an invasive SCC as proposed by Schwartz ( 18 ) or are in most cases genetically incomplete versions of SCCs.
Abrogation of UV-damage response
A role for the p53 tumor suppressor gene
Which are the genetic lesions that characterize skin carcinoma development and progression? The most prominent and the best studied aberration in skin cancers is the mutation of the p53 tumor suppressor gene. While early studies suggested as many as >90% of SCCs and >50% of BCCs being mutated in the p53 gene ( 19 ), it is now well established that ∼50% of all skin cancers show mutations and this frequency rises to 90% in skin cancers with xeroderma pigmentosum, a recessive disorder associated with a defect in nucleotide excision repair that predispose to skin cancer (reviewed in Ref. 20 ). Most importantly, many p53 mutations are C to T transitions with a high frequency of CC to TT double base changes, thus being indicative of UV-radiation-induced mutations ( 21 , 22 ). For example, in colon cancer the second p53 allele is often lost during tumor progression ( 23 ), whereas it is intriguing to note that in skin carcinomas both the alleles can be mutated, with each allele carrying a different and mostly UV-type specific mutation ( 19 , 24 ). In addition, the mutations do not appear to be at random but show a pattern of hot spots different from that of internal malignancies ( 25 ). Giglia-Mari and Sarasin ( 20 ) recently proposed that from these mutational hot spots mutations in codon 177 are specific only for BCCs while mutations in codon 278, although mutated at a lower frequency also in certain internal cancers, seem to be specific for skin SCCs. Furthermore, since evidence is increasing that certain mutations may not only abrogate p53 function but may represent gain of function mutations (reviewed in Ref. 26 ), understanding the functional consequence of these specific p53 mutations still remains an important goal for a better understanding of the mechanisms involved in skin cancer development.
A role for the human papillomavirus (HPV)
Similar as by mutations, p53 function is also abrogated in human skin carcinomas by the HPV the action of which was extensively reviewed recently by Duensing and Münger ( 27 ). More than 100 subtypes have been identified but only a subgroup of so called high-risk mucosotropic HPV types, including HPV-16, 18, 31, 33, 35 and 58, are believed to be a causative agent in the development of cervical cancer. The E6 gene of these high risk HPV types is able to induce rapid proteasomal degradation of p53 thereby abolishing the induction of cell cycle arrest or apoptosis. As in cervical carcinomas, HPV DNA is also frequently detected in skin carcinomas. A high prevalence of HPV infection seems to be characteristic for immunocompetent (47%) and even more for immunosuppressed patients (75%). However, >40 different HPV types were identified ( 28 ) and the typical high risk HPV types (e.g. HPV 16 and 18) were not yet found in skin carcinomas (reviewed in Ref. 29 ). Therefore, it was suggested that mechanisms different from the activity of HPV oncoproteins in genital cancer may be involved in skin neoplastic transformation ( 30 ).
Recently it was shown that HPV38, which was detected in ∼50% of skin carcinomas but only in 10% of healthy skin, was able to actively support longevity/immortalization of cultured human skin keratinocytes ( 31 ). An Australian study further demonstrated HPV-38 DNA in 43% of solar keratoses, as well as 13 and 16% of SCCs and BCCs, respectively ( 32 ). In addition, HPV-8, thought to be involved in the rare inherited disorder of epidermodysplasia verruciformis that is characterized by the life-long occurrence of multiple flat warts and macular lesions, was expressed in the epidermis of mice (Keratin 14 promoter) and these HPV-8 transgenic mice spontaneously developed single or multiple benign skin tumors ( 33 ). Interestingly, HPV-16 transgenic mice (also under the K14 promoter) only developed hyperplasia associated with hyperkeratosis ( 34 ). All these still controversial findings together with the high frequency of HPV DNA in normal skin—particularly in hair follicles—and the fact that many different and often several HPV types are found concomitantly in normal and tumor tissues makes their role in skin carcinogenesis still elusive. Furthermore, a recent report that HPV DNA was frequently detected in swap samples collected from the superficial layers of skin tumors while only little HPV DNA was found in biopsies of the same tumors ( 35 ) strongly substantiates a rather critical attitude.
A role for cell cycle inhibitors
One cell cycle inhibitor that plays an important role in epithelial transformation is the cyclin-dependent kinase inhibitor p16 INK4 that specifically inhibits progression through G1 phase of the cell cycle by blocking the cyclin-dependent kinase 4 from phosphorylating the retinoblastoma protein ( 36 ). The INK4a locus encodes another structurally and functionally independent protein, p14 ARF , which is also believed to be a potent tumor suppressor (reviewed in Ref. 37 ). p14 ARF activates the p53 pathway in response to oncogenic signals, such as the c-myc or ras oncogene, by binding to the p53 negative regulator Mdm2 and preventing p53 degradation thereby inducing cell cycle arrest or apoptosis. Since p53 is often mutated in skin carcinomas and is likely to be an early event, elimination of p16 INK4 rather than p14 ARF seems to be involved in skin cancer development. Accordingly, p16 INK4 mutations were detected, although at a low frequency, in the general population and also in patients suffering from xeroderma pigmentosum ( 38 – 40 ), whereas p14 ARF mutations were not yet reported. It was discussed that p16 INK4 mutations may be late events in skin cancer development and therefore may have been missed in studies evaluating precancerous lesions or small tumors, i.e. microinvasive SCCs ( 40 ). On the other hand, loss of heterozygosity (LOH) as well as loss of parts or the entire short arm of chromosome 9—p16 INK4 maps to 9p21—are frequently observed in SCCs ( 24 , 41 ). This raises the question whether in skin carcinomas loss of the p16 INK4 function is rather a result of loss of one copy of chromosome 9p and silencing of the second copy by epigenetic mechanisms such as methylation. However, it also cannot be excluded entirely that inactivation of p16 INK4 may be less essential and another gene on chromosome 9p, which still needs to be identified, is crucial for skin cancer development.
Even though inhibition should lead to the absence of p16 INK4 protein, dysplastic and neoplastic epithelial cells from the cervix uteri were shown to frequently upregulate p16 INK4 as determined by immunostaining ( 42 ). Since cervical cancer is closely associated with HPV infection it is tempting to speculate that this upregulation may be causally related to HPV. Presently, two immunohistochemistry studies, evaluating p16 INK4 expression in AK, CIS (Bowen's disease), and SCCs of the skin, presented rather controversial results. Hodges et al . ( 43 ) found immunostaining in about all AKs and in situ SCCs but only in 30% of the invasive SCCs. Owing to an increase in staining intensity in AK to invasive SCC, they nevertheless proposed a positive correlation between expression of p16 INK4 and skin cancer progression. Salama and co-workers showed a high frequency of immunostaining for BD but very little for AK and none for SCC. From this, they concluded that p16 INK4 is a selective and specific marker to distinguish BD from KA/benign SCC ( 44 ). Our data further add to this confusion. We found p16 INK4 immunostaining in about all KAs but only few SCCs (B. Jelinek et al ., manuscript in preparation). Interestingly, all p16 INK4 positive SCCs were poorly differentiated. This may allow two interpretations. First, p16 INK4 expression is required for KAs to control their growth and thus allow tumor regression. Second, the differential expression of p16 INK4 in well versus poorly differentiated SCCs may highlight two different developmental pathways. Thus, contributions of the cell cycle inhibitor CDKNA2/p16 INK4a to skin cancer development is still far from being understood.
Alternatively to changes in 16 INK4 , abrogation of 14-3-3sigma, a member of a protein family that regulates cellular activity by binding and sequestering, e.g. cyclin B1 and cdc2, may substitute for an aberrant growth control in skin tumor cells. 14-3-3-sigma, which is controlled by p53, promotes premitotic cell cycle arrest following DNA damage and thereby prevents ‘mitotic catastrophy’ ( 45 ). Even though expressed at high levels in AKs and skin SCCs, 14-3-3sigma expression was partially or completely lost in BCCs mainly due to CpG-hypermethylation ( 46 ). It is generally believed that p16 INK4 remains wildtype in BCCs and accordingly (no methylation was found in the above study) it was suggested that 14-3-3sigma may substitute for p16 INK4 in BCCs, and the silencing of 14-3-3sigma may thus contribute to the evasion of senescence in BCC ( 46 ). Different from BCCs, abnormal 14-3-3sigma expression is not yet described for SCCs. Thus, further studies need to clarify whether or not a preference for 14-3-3sigma or p16 INK4 may be tumor (BCC versus SCC) type-specific.
Contribution of the ras oncogene
Another gene that is likely to be mutated by the UV radiation but is controversially discussed concerning its contribution to skin cancer development is the ras oncogene. From the three ras genes, Harvey- ( Ha ), Kirsten- ( Ki ) and N-ras , mutations in Ha-ras predominate in the general population with the mutations characteristically seen at codons 12, 13 and 61—all localized opposing UV-sensitive CC sites. Mutation frequencies were extensively analyzed in the early 1990s and at that time it was proposed that the ras oncogene significantly contributes to skin cancer development. Now, an overall mutation frequency of 10–20% is suggested for SCCs and BCCs ( 24 , 47 – 52 ). On the other hand, a high mutation frequency and prevalence of N-ras mutations was reported for xeroderma pigmentosum patients being consistent with an even more important role of UV-induced lesions and a different mutation profile in these patients ( 53 ).
This latter situation may be reflected by the best established and most frequently used mouse model for skin carcinogenesis, the initiation–promotion protocol. Using one treatment of the carcinogen di-methyl-benzanthracene (DMBA) followed by twice weekly application of the phorbole ester (TPA) causes a ras-dependent formation of papillomas that in part progress to SCCs (reviewed in Ref. 54 ). Although the tumors formed in this model show remarkable phenotypic similarities with human skin carcinogenesis, how far the molecular findings based on chemical carcinogenesis and on ‘ras-dependent’ skin carcinogenesis can be extrapolated is still an open question. The answers will help to unravel the as yet still poorly understood genetic pathway involved in human UV-induced skin carcinogenesis. For this, the SKH1 hairless mouse may represent a more relevant model (for a review see Ref. 55 ). With these mice, reproducible and quantitative data could be established on dose, time and wavelength of the UV radiation required for the SCC development ( 56 ). A high frequency of p53 mutations were found, whereas only 1 out of 32 tumors carried a ras mutation ( 57 ). Furthermore, the majority of the p53 mutations were located in a codon matching with a hotspot for p53 mutations in human skin carcinomas, thus largely mimicking the essentials of human UV-induced skin cancer. However, careful consideration is required when extrapolating results from these mice to humans. Differences exist, e.g. in the composition of melanin and with that in the protection against UV-radiation. Increased pheomelanin photosensitizes DNA and thus induces DNA damage. Depending on the skin type, various compositions of pheomelanin and an another form of melanin, the eumelanin, are observed. Eumelanin predominates in mouse melanocytes ( 58 ).
Contribution of the patched tumor suppressor gene
While the functional contribution of the above aberrations are still far from being understood, one aberration—loss of 9q—originally detected in the autosomal dominant disorder termed Gorlin or nevoid basal cell carcinoma (NBCC) syndrome ( 59 ) has significantly advanced our understanding of the origin of BCCs. NBCC syndrome, which is characterized by multiple BCCs at an early age ( 60 , 61 ), not only allowed indentification of the putative tumor suppressor gene, the PTCH gene ( 62 , 63 ), but also led to the identification of an important pathway, the hedgehog-patched-smoothened pathway, which is abrogated in >70% of BCCs.
In skin, sonic hedgehog (SHH) signaling has been implicated in hair follicle growth and morphogenesis. Patched1, the protein product of PTCH , is a cell surface receptor of the secreted signaling molecule SHH. In the absence of SHH, patched1 inhibits smoothened (SMO), a G-protein-coupled-like receptor. SMO is released upon binding of SHH to patched1 and can initiate a signal transduction cascade that causes activation of the transcription factor Gli1. Thus, dysregulation of this pathway by either the loss of PTCH or the forced expression of SMO results in elevated levels of the transcription factor Gli1 and, as a consequence, induces hair follicle tumors ( 64 – 67 ) by opposing cell cycle arrest and differentiation ( 68 ). Mutations of either PTCH or SMO have been found in >70% of sporadic human BCCs ( 69 , 70 ). Because of this high correlation it is generally accepted that abrogation of this pathway is the major cause of BCC development and that BCCs are hair follicle-derived tumors ( 71 ). Furthermore, Louro et al . recently performed a comparative gene expression profiling and suggested that Gli1-induced transcripts may convert normal keratinocytes into invasive tumor cells in a relatively direct fashion, without the need of other multiple genetic aberrations ( 72 ).
Accordingly, also mouse models demonstrated that abrogation of the ptch-sonic/hedgehog pathway (Ptch ± mice) is essential for basal cell tumorigenesis ( 65 ) and that over-expression of Gli1, the zink-finger transcription factor activated by hedgehog, is central and probably sufficient for tumor development ( 66 ). Interestingly, in mice, the over expression of Gli1 under the epithelial keratin 5 (K5) promoter developed predominantly follicle-derived tumors and only a few BCCs ( 66 ). However, K5-Gli2 transgenic mice exclusively developed BCCs ( 67 ). Thus, it will be of interest to determine whether a similar distribution will be seen in human tumors.
Also in SCCs, the chromosomal region 9q22 to q31 is frequently involved in aberrations. Some tumor cells show gain of the entire q-arm due to the formation of an iso-chromosome i(9q) [( 24 ) and unpublished data], whereas LOH in 9q22.3 was reported to be characteristic for carcinomas in situ and SCCs, indicating that these alterations are late events in the SCC development ( 73 ). Although it was suggested that interfollicular epidermal cells can express hair follicle markers when exposed to powerful morphogens such as SHH and WNTs ( 74 ), there is no indication to date that patched plays a role in SCC development. Even more, while BCCs are generally associated with hyperactivation of the sonic hedgehog signaling pathway causing a continuous activation of this growth promoting cascade (reviewed in Ref. 75 ), evidence for a specific signal transduction pathway forcing SCC development is still missing.
Molecular and cytogenetic changes
What is the difference between SCCs and BCCs that no specific developmental pathway could yet be established for SCCs? Which other aberrations could account for SCC development? Already in 1994, Rees and coworkers reported on LOH of 9q in as many as 77% of BCCs but only in 12% of SCCs. In contrast, LOH of 9p markers was frequent in SCC ( 41 ). In addition, a number of other chromosomes were shown to carry LOH in SCCs such as 3p, 13p, 17p and 17q ( 10 ). Comparison between AKs and KAs revealed that AKs shared many of the same loci as SCCs, whereas the frequency of LOH in KAs was low with only isolated losses at 9p, 9q and 10q ( 10 , 17 ).
By cytogenetic analysis and comparative genomic hybridization, which allows a genome wide screen of gains and losses ( 76 ), SCCs demonstrated a great karyotypic complexity and cytogenetic heterogeneity, whereas the aberration profile is much simpler in AKs and KAs ( 12 , 24 , 77 , 78 ). Jin and colleagues found that in SCCs many structural aberrations affecting centromeric regions, particularly those of chromosomes 3q, 8q, 9q and 5p, leads to whole-arm translocations and the duplication of one chromosome arm thereby causing the formation of iso-chromosomes ( 12 ). Additionally, they observed genetically unrelated clones within the same tumor, suggesting that a multi-focal development is rather frequent in skin cancer. AKs, on the other hand, only revealed few numerical changes, e.g. gain of chromosomes 7, 9 and 18, that were common, while recurrent iso-chromosomes were not seen ( 12 ). Our CGH studies suggest that gains and losses of the same chromosomes (in particular loss of 3p and 9p, and gain of 11q) are involved in KAs and SCCs. The important difference, however, is that the individual KA only carries one or two of these aberrations while they are commonly present as a combination in SCCs (S. Popp, B. Jelinek and P. Boukamp, unpublished). This further strengthens the hypothesis that, as discussed above, KAs may be genetically incomplete SCCs. Finally, multiplex fluorescence in situ hybridization (M-FISH), which can identify all chromosomes of a metaphase simultaneously ( 79 ), highlighted complex translocations in the SCC derived cell lines ( 78 ) substantiating the important role of genomic instability in the skin SCC development.
So far, the reason for the high frequency of chromosomal aberrations such as deletions, amplifications and translocations, causing the substantially deranged karyotypes of SCCs, is still elusive. Since BCCs ( 80 ) as well as Merkel cell carcinomas (MCC) ( 24 , 81 ), which are highly aggressive endocrine tumors of the skin that develop on the same sun exposed sites, exhibit significantly less chromosomal changes per tumor, the complex karyotype of SCCs cannot simply be attributed to the environment and to the recurrent damage by the UV radiation. It also does not correlate with tumor aggressiveness, because MCCs show a much higher frequency of recurrences and metastases than SCCs ( 82 ). However, the large spectrum of aberrations in SCCs and precursor lesions may provide a broad basis for the potentially tumorigenic cells that can more rapidly adapt to environmental changes, and thus may explain the high frequency of SCCs occurring in immune-suppressed patients after a relatively short latency period.
Human skin cancer progression models
Different from the initiation promotion skin cancer mouse model, two human model systems have been described that show the typical SCC aberration pattern and therefore are well suited to elucidate functional consequences of the above described genetic aberrations. The MET model consists of cell lines derived from a primary tumor, two recurrences, and a metastasis from the same patient ( 83 ). Genetic analysis of tumors and their derived cell lines provided evidence that the metastasis directly evolved from the primary tumor while the two recurrent tumors had developed independently from a precursor lesion. Furthermore, these studies demonstrated that the tumors and derived cell lines were genetically highly comparable and carried a number of aberrations typically seen in SCCs ( 78 ). Interestingly, these cells are devoid of p53 mutations and HPV-sequences ( 83 ) thus making this sequence of human SCC cells a unique in vivo progression model to study p53-independent changes in skin carcinogenesis.
Another model, frequently used for functional studies in human skin carcinogenesis, is based on the spontaneously immortalized HaCaT cells ( 84 ). In contrast to the MET cells, the HaCaT cells exhibit UV-type specific mutations in both alleles of the p53 gene ( 85 ). In addition, they carry chromosomal aberrations characteristically seen in SCCs ( 86 ). Introduction of the Harvey-ras oncogene (codon 12 mutation) allowed to establish benign and malignant tumorigenic variants ( 87 , 88 ), and in vivo selection finally led to metastatic conversion ( 89 ). With this sequence of different stages of skin carcinogenesis, the HaCaT cells already served as a suitable model to study the functional consequences of the over-expression and suppression of a number of genes associated with skin cancer progression ( 88 , 90 – 94 ).
Genomic instability as a driving force in skin cancer development
Studies using the HaCaT cells have shown that mutational inactivation of p53 and some other genetic aberrations typically found in skin carcinomas (loss of e3p, gain of 3q and loss of 9p—one copy each) were not sufficient to render the cells tumorigenic. Accordingly, tumorigenic conversion did not occur spontaneously during long-term propagation in vitro and this correlated with a rather stable genotype ( 86 ). However, exposing the cells to an increased temperature and thereby inducing an oxidative damage response caused formation of new chromosomal aberrations and tumorigenic conversion ( 90 ). Similarly, introduction of the ras oncogene per se did not induce tumor formation but required the cooperation of genetic changes that manifested in a step-wise fashion during the long-term propagation ( 87 , 88 ). In good agreement with that, the most consistent difference between precancerous skin lesions and SCCs is the increase in karyotypic complexity. This suggests that destabilizing the genome is an important driving force in skin cancer progression. Furthermore, there is increasing evidence that more and more factors are contributing to genomic instability and this provides the possibility for ever-new phenotypes including metastatic and drug-resistant ones ( 95 ).
How can p53 contribute to the accumulation of further genetic changes?
As discussed above, mutations in the p53 gene are believed to be a very early if not initial event in skin carcinogenesis ( 21 ). As a guardian of the genome ( 96 ), p53 is stabilized upon stress by phosphorylation and alters the expression of different sets of downstream target genes including those that cause a cell cycle arrest ( 97 ). Thus, the resulting damage, e.g. double strand (ds) breaks, can be repaired thereby preventing gross chromosomal changes including amplifications and deletions. In the p53 mutant cells, cell cycle arrest is abolished. This may allow, as a first step of genome destabilization, non-lethal chromosomal damage to be passed on to the daughter cells. Such a scenario is suggested in the case of human tumor cells that show a good correlation between mutation in the p53 gene and a genome wide instability as evidenced for colorectal cancers by karyotypic abnormalities, chromosomal amplifications and genome-wide allelic imbalances ( 98 – 100 ). Furthermore, Overholzer and colleagues recently showed that mutations in the p53 gene but not amplified HDM2, which inhibits the tumor suppressive properties of p53 by controlling p53 degradation, correlated with high levels of genomic instability in osteosarcomas ( 101 ). Therefore, they suggested a qualitative difference for direct mutational versus indirect HDM2-dependent inactivation in destabilizing the genome. On the other hand, Carroll et al . ( 102 ) provided evidence that p53 mutations as well as MDM2 overexpression, induced aneuploidy through centrosome amplification. Centrosome duplication, which has to occur with each cell cycle, is thought to be controlled by the phosphorylation status of the retinoblastoma protein, release of the E2F transcription factor and subsequent transcriptional activation of the cyclin-dependent kinase 2 late in G1 (reviewed in Ref. 103 ). From this it was proposed that the frequent abrogation of the Rb pathway may not only facilitate progression towards DNA replication but may also deregulate the centrosome duplication cycle ( 104 ) generating abnormal chromosome segregation and concomitantly aneuploidy.
Centrosome-mediated defects were also suggested for HPV-induced genomic instability (reviewed in Ref. 27 ). It is long known that HPV-16 E6 can cause structural chromosomal changes ( 105 ), whereas E7 is correlated with numerical chromosomal abnormalities resulting in aneuploidy ( 105 , 106 ). Duensing and colleagues further showed that E6 and E7 cooperate to induce genomic instability by uncoupling centrosome duplication from cell division ( 107 ) and that E7 is responsible for abnormal centrosome synthesis ( 108 ). In skin carcinomas, mutations in the p53 gene as well as HPV DNA sequences are frequently detected. It thus remains to be seen whether one or both may be responsible for the complex aberration profile by the initiation of genomic instability through deregulation of the centrosome duplication cycle.
Telomere dysfunction
Recently, a strong association of chromosomal rearrangements and telomere dysfunctions was proposed—thus referring to the original findings by Hermann Muller and Barbara McClintock who demonstrated in the 1930s that the ends of the chromosomes, the telomeres, play a crucial role in maintaining chromosomal stability. McClintock showed that following the breakage and fusion of maize chromosomes, loss of the ends of the chromosomes rendered the chromosomes highly recombinogenic ( 109 ).
As known today, these observations long preceded the genetic definition of a telomere that consists of thousands of repeats of the hexanucleotid TTAGGG and makes up to 15 kb in human cells. The double stranded telomeric DNA ends in a 3′ single stranded overhang that is believed to be required for a higher order structure (reviewed in Ref. 110 ). One important model is that the telomeres form loop structures, t-loops, and by invasion of the 3′ overhang into the duplex region of the double stranded telomeric DNA, protect the DNA against degradation and end-to-end fusion, thereby providing a protective cap for the chromosomes ( 111 ). These caps are, however, ‘finite’ owing to the failure of the DNA polymerase α to replicate linear DNA to the outermost end ( 112 ). This so-called end-replication problem causes a replication-dependent telomere loss, which in many normal somatic cells is manifested as a pattern of continuous telomere erosion and is now assumed to be the counting mechanism, the endogenous clock of cellular aging ( 113 ). In germ line and tumor cells a specialized ribonucleoprotein complex, telomerase, is expressed that can prevent telomere shortening by de novo synthesis of telomeric sequences and thus can maintain the capping function of the telomeres ( 110 ).
Studies with telomerase-deficient mice have shown that in late generations end-to-end chromosomal fusions were the most prominent aberrations ( 114 ). Since they frequently involved the chromosomes with the shortest telomeres, critically short and therefore unprotected telomeres were thought to be a primary cause of these end-to-end fusions ( 115 ). Similarly, uncapping of the telomeres by overexpressing a dominant-negative version of the telomere repeat binding factor TRF2 further substantiated that in human cells also uncapping of the telomeres resulted in chromosomal end-to-end fusions thereby generating dicentric chromosomes ( 116 ). If both the centromeres are active, dicentric chromosomes are highly unstable during mitosis. If each centromere is attached to a different pole these chromosomes form anaphase bridges, a situation commonly viewed as a marker for genomic instability. As a result of this bipolar tension, breakage can occur anywhere along the chromosome. This led to the proposal of a fusion-bridge-breakage cycle as a primary mechanism of genomic instability in cells that suffer from critically short telomeres ( 117 ). The highly recombinant telomere-free DNA ends may invade other chromosomes, perhaps based on areas of micro-homology, thereby yielding non-reciprocal translocations. This hypothesis, also known as Bridge-fusion-breakage model (BFB), is now extensively used to explain the various translocation chromosomes found in tumor cells (for a review see Ref. 118 ).
Alternatively, Greider and co-workers ( 119 ) suggest that end-to-end chromosome fusions may not initiate rearrangements but may rather be a secondary effect of end resection and thus represent stable byproducts. From the study of chromosomal rearrangements in diploid yeast strains, they found that chromosomal rearrangements predominantly occur in the terminal region of the chromosome. Since exonuclease Exo1p played a major role in the generation of these rearrangements, they suggested that end resection initiates genomic instability at dysfunctional telomeres.
Trying to unravel factors responsible for impaired telomere integrity, the telomerase-deficient mice were crossed with mice deficient in a number of genes implicated in the maintenance of the telomeres such as the repair proteins Ku86, the subunit of DNA-PK, DNA-PKcs and the poly(ADP)ribosyl polymerase PARP-1 ( 120 , 121 ). While it was shown that PARP-1 had no effect in telomere metabolism and therefore did not cause obvious changes, Ku86 and DNA-PKcs-deficiency caused accelerated loss of viability. This correlated with proliferative defects and age-related pathologies, but did not lead to an increased cancer incidence ( 122 ).
This may be unexpected because late generation telomerase knockout mice were shown to suffer from an increased rate of spontaneous tumors ( 123 ). Although it was proposed that the majority of tumor types originated from highly proliferative cell types, i.e. cells that were likely to sustain the highest degree of telomere shortening with increased age, the number of spontaneous skin carcinomas did not increase significantly. Gonzales-Suarez et al . ( 124 ) even reported on a dramatic inhibitory impact on chemical skin carcinogenesis (DMBA/TPA treatment) in the telomerase knockout mice providing evidence that short telomeres in the absence of telomerase activity could suppress tumor initiation. Along that line, absence of telomerase in transgenic mice expressing HPV16 had little effect. Genetic analyses of tumors from these mice did not provide evidence for telomere dysfunction or increased genomic instability ( 125 ).
Constitutive overexpression of telomerase (mTERT transgenic mice), on the other hand, promoted mammary carcinomas ( 126 ). While a skin cancer phenotype was not reported for these animals, transgenic mice over-expressing mTERT in an epithelia-specific manner—under the K5 promoter—revealed an increased number in papillomas when subjected to the DMBA/TPA protocol suggesting an increased tumor susceptibility upon telomerase activation. Interestingly, the conversion rate to carcinomas was not augmented ( 127 ). These mice also developed spontaneous tumors in the lung, mammary glands and the uterus with lymphomas being the most frequent tumor type ( 128 ). Stratified epithelia only showed hyperplasia and hyperkeratosis but no tumors arguing for telomerase up-regulation to be an early event in skin carcinogenesis and being related to proliferation rather than tumorigenic conversion. mTERT transgenic mice crossed into a p53 null background showed a similar tumor spectrum, though with a generally higher tumor yield ( 128 ). Since p53 mutations are frequent in skin cancers and as discussed above represent quite a specific mutation spectrum, it remains to be elucidated how far these mutations provide gain of function ( 26 ) and accordingly, whether abrogation of p53 can replace p53 mutations in order to elucidate the mechanism of human skin cancer development and progression.
Taken together, these findings rather argue in favour of telomerase as a driving force in tumor initiation than critically shortened telomeres being the basis of unstable and therefore susceptible chromosomes. If so, ‘telomere length-independent’ mechanisms of genomic instability are required.
Putative recombination models in telomerase-positive tumor cells
While uncapping of critically short telomeres predominantly should account for normal somatic and telomerase-negative cells and therefore would represent an initial stage in the transformation process, tumor cells generally express the telomerase and thus protect their telomere lengths. Nevertheless, there is ample evidence that these cells also suffer from genomic instability and that this is not a continuum of the telomere length-dependent BFB but can occur de novo upon induction of certain oncogenes e.g. the c-myc oncogene [for a review see Mai and Mushinski ( 129 )].
Genomic instability induced by the c-myc oncogene
One recurrent chromosomal aberration in SCCs the is gain of chromosome 8q through the formation if an iso-chromosome. Since 8q harbors the c-myc gene, located at 8q24, c-myc expression should be increased. As a result of this aberration, c-myc amplification was described for 50% of SCCs from renal transplant patients ( 130 ). In addition to its role in proliferation, there is increasing evidence that one important consequence of deregulated c-myc expression is the induction of genomic instability. c-Myc seems to favor gene amplification and gene rearrangements as well as karyotypic instability leading to numerical and structural chromosomal aberrations (for a review see Ref. 129 ). The most intriguing features of c-Myc-induced alterations are their reversibility. Using inducible systems, genomic instability proved to be transient upon a single induction of c-Myc in vitro and in vivo ( 131 , 132 ). Continued c-Myc activation, on the other hand, was described to be accompanied by numerical and structural chromosomal changes such as extra-chromosomal elements and chromosomal breakage but also centromere-telomere fusions. ( 133 ). Furthermore, in the absence of p53 the c-Myc effect seemed to be augmented ( 134 ) and aberrant centrosome duplication was discussed as a potential mechanism.
c-Myc may additionally contribute to genomic instability through a telomere organization-dependent mechanism. While in normal cells the telomeres are present as non-overlapping territories, tumor cells ( in vitro and in vivo ) with deregulated c-Myc as well as HaCaT cells constitutively expressing the c-myc oncogene, showed aggregation of telomeres in a high percentage of cells ( 135 , 136 ). Since these telomeric aggregates (TAs) could be detected in interphase nuclei as well as in mitotic figures and the percentage of apoptotic cells was not significantly increased as compared with the parental cells ( 136 ), their presence throughout the cell cycle is likely to contribute to unequal segregation of the chromosomes during mitosis, i.e. numerical aberrations. In addition, TAs also likely force the chromosomes to alter their location, thereby providing an explanation for how the different chromosomes come into such close vicinity that they are able to exchange chromosomal material and give rise to the in part very complex multi-chromosomal translocations characteristically seen in skin carcinoma cells ( 137 ).
Telomere-independent mechanisms of chromosomal instability
Fragile sites
Cuillo et al . provided convincing evidence that not only critically short telomeres initiate BFB. They showed that the PIP (prolactin-induced protein) gene which is overexpressed in 60–80% of primary and metastatic breast cancers ( 138 , 139 ) was duplicated in a breast carcinoma cell line due to one cycle of BFB. In their model, the first break was initiated at the fragile site FRA7I telomeric to the PIP gene ( 140 ). They proposed that fusion occurs after replication of the two sister chromatids leading to anaphase bridges. Breakage now results in one deleted and one amplified daughter cell. Although the mechanism responsible for activation of fragile sites in cancer cells is not fully understood there is evidence that stress resulting from oxygen starvation (hypoxia) and subsequent variations in tumor microenvironment can activate some chromosomal fragile sites. It is also possible that fragile site expression may result from spontaneous changes in chromatin organization and/or impairment of the rate of timing of DNA replication ( 141 ).
FRA3B, the most active common fragile site in the genome is mapped to chromosome 3p14.2, a chromosomal location frequently deleted in skin carcinomas. Furthermore, FRA3B is encompassed by the FHIT gene (fragile histidine triad), a putative tumor suppressor that is lost in various tumors (reviewed in Ref. 142 ). However, expression studies from different skin tumors as well as skin cancer cell lines exhibiting loss of one copy of chromosome 3p, demonstrated normal transcripts of the FITH gene ( 24 , 143 ). This suggests that the FITH gene is not a common target for deletions and that FRA3B is not a common break point in skin carcinomas.
DNA double strand breaks
While fragile sites favor the specific sites for DNA breaks and rearrangements, DNA ds breaks induced either in a replication-dependent manner or through exogenous insults e.g. reactive oxygen species produced by ionizing radiation or UV-A radiation are likely to occur throughout the genome and thus should cause a more random breakage. Interestingly, Rief ( 144 ) recently found that repair of ds breaks, which is dependent on non-homologous end-joining, is also dependent on the chromatin state. They demonstrated that restriction fragment reconstitution was considerably more efficient in centromeres than in the non-centromeric locations. Since there is some experimental evidence that chromosomal exchange aberrations are less frequent in genomic regions with highly condensed heterochromatin ( 145 – 147 ), this finding may support the hypothesis that the chromatin state is an important determinant of whether breaks are forced to rejoin correctly or can undergo non-homologous end-joining with DNA from other parts of the chromosome or even other chromosomes. The authors further discuss that not only the primary structure—the high number of repetitive elements—may increase the probability for homologous repair mechanisms but also the degree of chromatin condensation may limit the mobility of the broken ends. This could additionally lead to a much higher probability of properly rejoined breaks than at an average genomic location. Thus it is tempting to speculate that the intra-genomic heterogeneity of chromatin condensation may be one potential explanation for the tissue-specific prevalence of certain chromosomal alterations.
Centromeric break
The most common aberrations in skin SCCs are the centromeric breaks either leading to non-reciprocal translocation of different chromosome arms or to loss of one chromosome arm and duplication of the other, resulting in the formation of an iso-chromosome. The mechanism by which these aberrations evolve is still elusive. Recently Barbouti et al . ( 148 ) mapped the breakpoint region of i(17q), the most common iso-chromosome in human hematopoietic malignancies, and found it to be dicentric. They identified large, palindromic and low-copy repeats, and proposed that breakage at these palindome structures allows reunion between palindromes on sister chromatids and the formation of both dicentric and acentric structures. While the acentric part is lost, the dicentric part is retained through mitoses without disruption. This is possible because the centromeres are in such close proximity that one of them is inactivated. The authors further concluded that such somatic rearrangements are not random events but rather reflect susceptibility owing to the genomic structure ( 148 ).
Centromeres are the primary site for kinetochore formation, the protein complex that binds to the spindle microtubules in order to coordinate chromosome segregation to the opposite poles during mitosis. A second important activity of the centromeres is the establishment and maintenance of cohesion (for a review see Ref. 149 ). Therefore as an alternative hypothesis, forced cohesion of the two sister chromatids at the centromeres could favor breakage of the less adhesive arms and maintenance of the two tightly fixed sister copies which then could reestablish as iso-chromosomes. The chromosomes involved in iso-chromosome formation seem to be tissue-specific and in skin carcinomas often involve chromosomes 3q, 8q and 9q. It is now essential to determine whether the genomic structure of the centromeric region of these particular chromosomes differ in keratinocytes versus other cell types and therefore are part of the non-random chromosomal changes in skin SCCs.
Conclusions and further perspective
Comparison of the two non-melanoma skin cancers, BCC and SCC, clearly demonstrate that despite the increased knowledge about the role of a number of oncogenes, tumor suppressor genes and signal transduction pathways, the genetic mechanisms causing tumors such as skin SCCs are still poorly understood. On the one hand, BCCs exhibit a relatively simple genotype, with only few aberrations and a high degree of independence of the immune system. With the identification of an aberrant sonic hedgehog pathway, a major cause of BCCs development was identified. Correspondingly, the plant-derived teratogen cyclopamine has been shown to reverse the effects of oncogenic mutations of SMO and PTCH ( 150 ). Its efficacy in the BCC treatment in humans remains to be seen and thus its application as a successful therapeutic strategy to eliminate this highly destructive and most common tumor type.
On the other hand, skin SCCs develop, at least in part, from precursor lesions and accumulate a highly complex genotype ( Figure 1 ). While most of the genes causing tumor development and progression are still unknown as is a signal transduction pathway that can be causally related to SCC development, the most intriguing feature is the great number of chromosomal aberrations and the recurrent involvement of specific chromosomes in gains, losses and translocations, e.g. loss of 3p, loss of 9p and gain of 3q, 9q and 11q. With the increasing evidence that certain chromosomal aberrations and in particular certain combinations of aberrations are required for a fully malignant SCC, it is now of importance to unravel the mechanisms underlying the induction of these genetic alterations. Recent studies have shown that not only UV-B but also UV-A is involved in the UV-induced skin carcinogenesis (reviewed in Ref. 151 ). It now needs to be explored whether or not the oxidative stress generated by UV-A exposure may be a factor causing such chromosomal changes, i.e. inducing genomic instability, if so, how far ( 152 ). Thus, understanding and fighting genomic instability may be a promising future approach particularly for those tumors such as skin SCCs that suffer from numerous and highly complex aberrations.
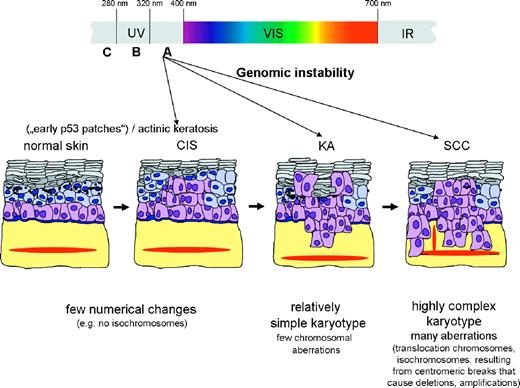
Schematic representation of the potential developmental stages during UV-induced skin carcinogenesis. Both UV-B and UV-A can contribute to skin cancer at different developmental stages by directly inducing DNA damage (e.g. UV-B-dependent p53 mutations) or indirectly by UV-A-induced oxidative stress generating genomic instability. Genomic instability is likely to participate in early development to generate the first chromosomal changes (relatively simple karyotype) as well as during later stages (transition to a malignant SCC) to contribute to the highly complex karyotype.
The author wishes to thank Drs Norbert Fusenig and Margareta Müller for helpful comments and Angelika Lampe for her help in editing the manuscript. This work was in part supported by the, European Union (LSHC-CT-2004-502943), as well as the Deutsche Krebshilfe eV.
Conflict of Interest Statement : None declared.
References
Euvrard,S., Kanitakis,J. and Claudy,A. (
Freedberg,I.M., Eisen,A., Wolff,K., Austen,K.F., Goldspith,L., Katz,S. and Fitzpatrick,T. (
Nakazawa,H., English,D., Randell,P.L., Nakazawa,K., Martel,N., Armstrong,B.K. and Yamasaki,H. (
Jonason,A.S., Kunala,S., Price,G.J., Restifo,R.J., Spinelli,H.M., Persing,J.A., Leffell,D.J., Tarone,R.E. and Brash,D.E. (
Ling,G., Persson,A., Berne,B., Uhlen,M., Lundeberg,J. and Ponten,F. (
Ren,Z.P., Ponten,F., Nister,M. and Ponten,J. (
Le Pelletier,F., Soufir,N., de la,S.P., Janin,A. and Basset-Seguin,N. (
Johnson,T.M., Rowe,D.E., Nelson,B.R. and Swanson,N.A. (
Kanjilal,S., Strom,S.S., Clayman,G.L., Weber,R.S., el Naggar,A.K., Kapur,V., Cummings,K.K., Hill,L.A., Spitz,M.R. and Kripke,M.L. (
Rehman,I., Takata,M., Wu,Y.Y. and Rees,J.L. (
Quinn,A.G., Sikkink,S. and Rees,J.L. (
Jin,Y., Jin,C., Salemark,L., Wennerberg,J., Persson,B. and Jonsson,N. (
Kushida,Y., Miki,H. and Ohmori,M. (
Billingsley,E.M., Davis,N. and Helm,K.F. (
Hurt,M.A. (
Cribier,B., Asch,P. and Grosshans,E. (
Waring,A.J., Takata,M., Rehman,I. and Rees,J.L. (
Brash,D.E., Ziegler,A., Jonason,A.S., Simon,J.A., Kunala,S. and Leffell,D.J. (
Giglia-Mari,G. and Sarasin,A. (
Ziegler,A., Jonason,A.S., Leffell,D.J., Simon,J.A., Sharma,H.W., Kimmelman,J., Remington,L., Jacks,T. and Brash,D.E. (
Tornaletti,S. and Pfeifer,G.P. (
Fearon,E.R. and Vogelstein,B. (
Popp,S., Waltering,S., Herbst,C., Moll,I. and Boukamp,P. (
Ziegler,A., Leffell,D.J., Kunala,S., Sharma,H.W., Gailani,M., Simon,J.A., Halperin,A.J., Baden,H.P., Shapiro,P.E. and Bale,A.E. (
Deppert,W., Gohler,T., Koga,H. and Kim,E. (
Duensing,S. and Münger,K. (
Meyer,T., Arndt,R., Nindl,I., Ulrich,C., Christophers,E. and Stockfleth,E. (
Pfister,H. (
Meyer,T., Arndt,R., Christophers,E., Nindl,I. and Stockfleth,E. (
Caldeira,S., Zehbe,I., Accardi,R., Malanchi,I., Dong,W., Giarre,M., de Villiers,E.M., Filotico,R., Boukamp,P. and Tommasino,M. (
Forslund,O., Ly,H., Reid,C. and Higgins,G. (
Schaper,I.D., Marcuzzi,G.P., Weissenborn,S.J., Kasper,H.U., Dries,V., Smyth,N., Fuchs,P. and Pfister,H. (
Kim,S.H., Kim,K.S., Lee,E.J. et al . (
Forslund,O., Lindelof,B., Hradil,E., Nordin,P., Stenquist,B., Kirnbauer,R., Slupetzky,K. and Dillner,J. (
Serrano,M., Hannon,G.J. and Beach,D. (
Kubo,Y., Urano,Y., Matsumoto,K., Ahsan,K. and Arase,S. (
Soufir,N., Daya-Grosjean,L., de La Salmoniere,P., Moles,J.P., Dubertret,L., Sarasin,A. and Basset-Seguin,N. (
Soufir,N., Ribojad,M., Magnaldo,T., Thibaudeau,O., Delestaing,G., Daya-Grosjean,L., Rivet,J., Sarasin,A. and Basset-Seguin,N. (
Quinn,A.G., Sikkink,S. and Rees,J.L. (
Klaes,R., Friedrich,T., Spitkovsky,D., Ridder,R., Rudy,W., Petry,U., Dallenbach-Hellweg,G., Schmidt,D. and von Knebel,D.M. (
Hodges,A. and Smoller,B.R. (
Salama,M.E., Mahmood,M.N., Qureshi,H.S., Ma,C., Zarbo,R.J. and Ormsby,A.H. (
Chan,T.A., Hermeking,H., Lengauer,C., Kinzler,K.W. and Vogelstein,B. (
Lodygin,D., Yazdi,A.S., Sander,C.A., Herzinger,T. and Hermeking,H. (
van der Schroeff,J.G., Evers,L.M., Boot,A.J. and Bos,J.L. (
Lieu,F.M., Yamanishi,K., Konishi,K., Kishimoto,S. and Yasuno,H. (
Pierceall,W.E., Goldberg,L.H., Tainsky,M.A., Mukhopadhyay,T. and Ananthaswamy,H.N. (
Campbell,C., Quinn,A.G. and Rees,J.L. (
Wilke,W.W., Robinson,R.A. and Kennard,C.D. (
Spencer,J.M., Kahn,S.M., Jiang,W., DeLeo,V.A. and Weinstein,I.B. (
Daya-Grosjean,L., Robert,C., Drougard,C., Suarez,H. and Sarasin,A. (
Dlugosz,A., Merlino,G. and Yuspa,S.H. (
van Kranen,H.J. and de Gruijl,F.R. (
de Gruijl,F.R. and Forbes,P.D. (
van Kranen,H.J., de Gruijl,F.R., de Vries,A., Sontag,Y., Wester,P.W., Senden,H.C., Rozemuller,E. and van Kreijl,C.F. (
Nishigori,C., Hattori,Y. and Toyokuni,S. (
Shanley,S., Ratcliffe,J., Hockey,A., Haan,E., Oley,C., Ravine,D., Martin,N., Wicking,C. and Chenevix-Trench,G. (
Kimonis,V.E., Goldstein,A.M., Pastakia,B., Yang,M.L., Kase,R., DiGiovanna,J.J., Bale,A.E. and Bale,S.J. (
Hahn,H., Wicking,C., Zaphiropoulous,P.G. et al . (
Johnson,R.L., Rothman,A.L., Xie,J. et al . (
Oro,A.E., Higgins,K.M., Hu,Z., Bonifas,J.M., Epstein,E.H.Jr and Scott,M.P. (
Aszterbaum,M., Epstein,J., Oro,A., Douglas,V., LeBoit,P.E., Scott,M.P. and Epstein,E.H.Jr (
Nilsson,M., Unden,A.B., Krause,D., Malmqwist,U., Raza,K., Zaphiropoulos,P.G. and Toftgard,R. (
Grachtchouk,M., Mo,R., Yu,S., Zhang,X., Sasaki,H., Hui,C.C. and Dlugosz,A.A. (
Fan,H. and Khavari,P.A. (
Gailani,M.R., Stahle-Backdahl,M., Leffell,D.J. et al . (
Xie,J., Murone,M., Luoh,S.M. et al . (
Jih,D.M., Lyle,S., Elenitsas,R., Elder,D.E. and Cotsarelis,G. (
Louro,I.D., Bailey,E.C., Li,X. et al . (
Ahmadian,A., Ren,Z.P., Williams,C., Ponten,F., Odeberg,J., Ponten,J., Uhlen,M. and Lundeberg,J. (
Oro,A.E. and Higgins,K. (
Kallioniemi,A., Kallioniemi,O.P., Sudar,D., Rutovitz,D., Gray,J.W., Waldman,F. and Pinkel,D. (
Jin,Y., Martins,C., Jin,C., Salemark,L., Jonsson,N., Persson,B., Roque,L., Fonseca,I. and Wennerberg,J. (
Popp,S., Waltering,S., Holtgreve-Grez,H., Jauch,A., Proby,C., Leigh,I.M. and Boukamp,P. (
Speicher,M.R., Gwyn,B.S. and Ward,D.C. (
Jin,Y., Martins,C., Salemark,L., Persson,B., Jin,C., Miranda,J., Fonseca,I. and Jonsson,N. (
Larramendy,M.L., Koljonen,V., Bohling,T., Tukiainen,E. and Knuutila,S. (
Hitchcock,C.L., Bland,K.I., Laney,R.G.III, Franzini,D., Harris,B. and Copeland,E.M.III (
Proby,C.M., Purdie,K.J., Sexton,C.J., Purkis,P., Navsaria,H.A., Stables,J.N. and Leigh,I.M. (
Boukamp,P., Petrussevska,R.T., Breitkreutz,D., Hornung,J., Markham,A. and Fusenig,N.E. (
Lehman,T.A., Modali,R., Boukamp,P., Stanek,J., Bennett,W.P., Welsh,J.A., Metcalf,R.A., Stampfer,M.R., Fusenig,N. and Rogan,E.M. (
Boukamp,P., Popp,S., Altmeyer,S., Hulsen,A., Fasching,C., Cremer,T. and Fusenig,N.E. (
Boukamp,P., Stanbridge,E.J., Foo,D.Y., Cerutti,P.A. and Fusenig,N.E. (
Boukamp,P., Peter,W., Pascheberg,U., Altmeier,S., Fasching,C., Stanbridge,E.J. and Fusenig,N.E. (
Mueller,M.M., Peter,W., Mappes,M., Huelsen,A., Steinbauer,H., Boukamp,P., Vaccariello,M., Garlick,J. and Fusenig,N.E. (
Boukamp,P., Popp,S., Bleuel,K., Tomakidi,E., Burkle,A. and Fusenig,N.E. (
Skobe,M. and Fusenig,N.E. (
Mueller,M.M. and Fusenig,N.E. (
Mudgil,A.V., Segal,N., Andriani,F., Wang,Y., Fusenig,N.E. and Garlick,J.A. (
Billings,S.D., Southall,M.D., Li,T., Cook,P.W., Baldridge,L., Moores,W.B., Spandau,D.F., Foley,J.G. and Travers,J.B. (
Duesberg,P.H. (
Vogelstein,B., Lane,D. and Levine,A.J. (
Georgiades,I.B., Curtis,L.J., Morris,R.M., Bird,C.C. and Wyllie,A.H. (
Eyfjord,J.E., Thorlacius,S., Steinarsdottir,M., Valgardsdottir,R., Ogmundsdottir,H.M. and Anamthawat-Jonsson,K. (
Primdahl,H., Wikman,F.P., von der,M.H., Zhou,X.G., Wolf,H. and Orntoft,T.F. (
Overholtzer,M., Rao,P.H., Favis,R., Lu,X.Y., Elowitz,M.B., Barany,F., Ladanyi,M., Gorlick,R. and Levine,A.J. (
Carroll,P.E., Okuda,M., Horn,H.F., Biddinger,P., Stambrook,P.J., Gleich,L.L., Li,Y.Q., Tarapore,P. and Fukasawa,K. (
Hinchcliffe,E.H. and Sluder,G. (
Kramer,A., Neben,K. and Ho,A.D. (
White,A.E., Livanos,E.M. and Tlsty,T.D. (
Hashida,T. and Yasumoto,S. (
Duensing,S., Lee,L.Y., Duensing,A., Basile,J., Piboonniyom,S., Gonzalez,S., Crum,C.P. and Munger,K. (
Duensing,S., Duensing,A., Crum,C.P. and Munger,K. (
McClintock,B. (
Griffith,J.D., Comeau,L., Rosenfield,S., Stansel,R.M., Bianchi,A., Moss,H. and de Lange,T. (
Olovnikov,A.M. (
Wright,W.E. and Shay,J.W. (
Blasco,M.A., Lee,H.W., Rizen,M., Hanahan,D., DePinho,R. and Greider,C.W. (
Hemann,M.T., Strong,M.A., Hao,L.Y. and Greider,C.W. (
van Steensel,B., Smogorzewska,A. and de Lange,T. (
Chang,S., Khoo,C. and DePinho,R.A. (
Murnane,J.P. and Sabatier,L. (
Hackett,J.A. and Greider,C.W. (
Goytisolo,F.A. and Blasco,M.A. (
Bailey,S.M. and Goodwin,E.H. (
Espejel,S., Klatt,P., Menissier-de Murcia,J., Martin-Caballero,J., Flores,J.M., Taccioli,G., de Murcia,G. and Blasco,M.A. (
Rudolph,K.L., Chang,S., Lee,H.W., Blasco,M., Gottlieb,G.J., Greider,C. and DePinho,R.A. (
Gonzalez-Suarez,E., Samper,E., Flores,J.M. and Blasco,M.A. (
Argilla,D., Chin,K., Singh,M., Hodgson,J.G., Bosenberg,M., de Solorzano,C.O., Lockett,S., DePinho,R.A., Gray,J. and Hanahan,D. (
Artandi,S.E., Alson,S., Tietze,M.K. et al . (
Gonzalez-Suarez,E., Samper,E., Ramirez,A., Flores,J.M., Martin-Caballero,J., Jorcano,J.L. and Blasco,M.A. (
Gonzalez-Suarez,E., Flores,J.M. and Blasco,M.A. (
Mai,S. and Mushinski,J.F. (
Pelisson,I., Soler,C., Chardonnet,Y., Euvrard,S. and Schmitt,D. (
Mai,S., Fluri,M., Siwarski,D. and Huppi,K. (
Felsher,D.W. and Bishop,J.M. (
Kuschak,T.I., Taylor,C., McMillan-Ward,E., Israels,S., Henderson,D.W., Mushinski,J.F., Wright,J.A. and Mai,S. (
McCormack,S.J., Weaver,Z., Deming,S., Natarajan,G., Torri,J., Johnson,M.D., Liyanage,M., Ried,T. and Dickson,R.B. (
Chuang,T.C., Moshir,S., Garini,Y. et al . (
Ermler,S., Krunic,D., Knoch,T.A., Moshir,S., Mai,S., Greulich-Bode,K.M. and Boukamp,P. (
Popp,S., Waltering,S., Holtgreve-Grez,H., Jauch,A., Proby,C., Leigh,I.M. and Boukamp,P. (
Murphy,G., Young,A.R., Wulf,H.C., Kulms,D. and Schwarz,T. (
Clark,W.H.Jr, Elder,D.E., Guerry,D., Braitman,L.E., Trock,B.J., Schultz,D., Synnestvedt,M. and Halpern,A.C. (
Ciullo,M., Debily,M.A., Rozier,L. et al . (
Hellman,A., Zlotorynski,E., Scherer,S.W., Cheung,J., Vincent,J.B., Smith,D.I., Trakhtenbrot,L. and Kerem,B. (
Huebner,K. and Croce,C.M. (
Sikkink,S.K., Rehman,I. and Rees,J.L. (
Rief,N. and Lobrich,M. (
Slijepcevic,P. and Natarajan,A.T. (
Muhlmann-Diaz,M.C. and Bedford,J.S. (
Surralles,J., Darroudi,F. and Natarajan,A.T. (
Barbouti,A., Stankiewicz,P., Nusbaum,C. et al . (
Dej,K.J. and Orr-Weaver,T.L. (
Taipale,J., Chen,J.K., Cooper,M.K., Wang,B., Mann,R.K., Milenkovic,L., Scott,M.P. and Beachy,P.A. (
Nishigori,C., Hattori,Y. and Toyokuni,S. (

{kind=link}