




Abstract
We report seven patients, six from a single institution, who developed subacute limbic encephalitis initially considered of uncertain aetiology. Four patients presented with symptoms of hippocampal dysfunction (i.e. severe short-term memory loss) and three with extensive limbic dysfunction (i.e. confusion, seizures and suspected psychosis). Brain MRI and [18F]fluorodeoxyglucose (FDG)-PET complemented each other but did not overlap in 50% of the patients. Combining both tests, all patients had temporal lobe abnormalities, five with additional areas involved. In one patient, FDG hyperactivity in the brainstem that was normal on MRI correlated with central hypoventilation; in another case, hyperactivity in the cerebellum anticipated ataxia. All patients had abnormal CSF: six pleocytosis, six had increased protein concentration, and three of five examined had oligoclonal bands. A tumour was identified and removed in four patients (mediastinal teratoma, thymoma, thymic carcinoma and thyroid cancer) and not treated in one (ovarian teratoma). An immunohistochemical technique that facilitates the detection of antibodies to cell surface or synaptic proteins demonstrated that six patients had antibodies to the neuropil of hippocampus or cerebellum, and one to intraneuronal antigens. Only one of the neuropil antibodies corresponded to voltage-gated potassium channel (VGKC) antibodies; the other five (two with identical specificity) reacted with antigens concentrated in areas of high dendritic density or synaptic-enriched regions of the hippocampus or cerebellum. Preliminary characterization of these antigens indicates that they are diverse and expressed on the neuronal cell membrane and dendrites; they do not co-localize with VGKCs, but partially co-localize with spinophilin. A target autoantigen in one of the patients co-localizes with a cell surface protein involved in hippocampal dendritic development. All patients except the one with antibodies to intracellular antigens had dramatic clinical and neuroimaging responses to immunotherapy or tumour resection; two patients had neurological relapse and improved with immunotherapy. Overall, the phenotype associated with the novel neuropil antibodies includes dominant behavioural and psychiatric symptoms and seizures that often interfere with the evaluation of cognition and memory, and brain MRI or FDG-PET abnormalities less frequently restricted to the medial temporal lobes than in patients with classical paraneoplastic or VGKC antibodies. When compared with patients with VGKC antibodies, patients with these novel antibodies are more likely to have CSF inflammatory abnormalities and systemic tumours (teratoma and thymoma), and they do not develop SIADH-like hyponatraemia. Although most autoantigens await characterization, all share intense expression by the neuropil of hippocampus, with patterns of immunolabelling characteristic enough to suggest the diagnosis of these disorders and predict response to treatment.
Introduction
There is evidence that subacute limbic encephalitis can be immune mediated. In these patients, the rapid clinical presentation and lack of symptom specificity lead to a wide differential diagnosis including, among others, viral infections, inflammatory or autoimmune disorders (lupus, Sjögren's, Hashimoto thyroiditis and CNS vasculitis), toxic and metabolic encephalopathies, and paraneoplastic syndromes. While detection of onconeuronal antibodies to intraneuronal antigens (i.e. Hu, CV2/CRMP5 and Ma2) indicates a paraneoplastic origin and limited response to treatment (Honnorat et al., 1996; Alamowitch et al., 1997; Dalmau et al., 2004), the detection of antibodies to cell surface antigens such as voltage-gated potassium channels (VGKCs) suggests a lesser risk for an underlying cancer and better response to immunosuppression (Pozo-Rosich et al., 2003; Vincent et al., 2004). However, in at least 20% of patients with subacute limbic encephalitis, initial diagnostic tests are negative (Bien et al., 2000; Gultekin et al., 2000; Watanabe et al., 2003). Because the CSF of these patients often shows abnormalities similar to those found in immune-mediated disorders, and because some patients improve with immunotherapy, we postulated the presence of immune mechanisms that are missed by conventional studies. To test this hypothesis, we used an immunohistochemical method that enhances detection of serum or CSF antibodies to the neuropil, including cell surface antigens and other proteins expressed in synaptic-enriched regions or areas with a dense dendritic network. We report the clinical features, MRI and metabolic neuroimaging, and immunological correlates of seven such patients. A remarkable finding is that all the patients with antibodies specifically reacting with the neuropil had dramatic neurological response to treatment regardless of the identity of the antigens.
Patients and methods
Six patients were seen by the authors at the Hospital of the University of Pennsylvania over a 24 month period, and one patient (seen by A.V.) was diagnosed elsewhere; all patients have had clinical follow-up since symptom presentation. Patients had in common the subacute onset of confusion, memory problems, abnormal behaviour or seizures that led to the presumed diagnosis of limbic encephalitis. None of the patients had a history of cancer, AIDS, immunosuppression or bone marrow transplant. In all instances, HSV encephalitis and other infectious aetiologies were excluded with appropriate serum and CSF tests. All patients had brain MRI or [18F]fluorodeoxyglucose (FDG)-PET, and CT of the chest, abdomen and pelvis or body FDG-PET scan. Other tests included blood cell count and general chemistry, B12, folic acid, thyroid function test, thyroglobulin and thyroperoxidase antibodies, RPR, Lyme serology, antinuclear antibodies, antibodies to double-stranded DNA, Sjogren's serology (SSA or SSB), angiotensin-converting enzyme (ACE), and CSF analysis and cytology. Sera from patients 2, 3, 4 and 6 were negative for VGKC antibodies using an α-dendrotoxin radioimmunoassay; the other patients' sera were not tested. Serological studies for antineuronal antibodies were performed at symptom presentation and during follow-up. All subjects gave informed written consent and the studies were approved by the Institutional Review Board at the University of Pennsylvania.
Patients
See Supplementary material available at Brain on-line.
Sera and CSF
Patients' sera and CSF were kept frozen until use. Control samples included 13 sera from patients with suspected or confirmed limbic encephalitis seen by the authors during the same time period (described later), and archived frozen sera from 50 patients with confirmed paraneoplastic limbic encephalitis, 25 patients with encephalitis of unclear aetiology and 11 patients with limbic encephalitis and radioimmunoassay-positive VGKC antibodies (10 seen at other institutions). Sera from patients with antibodies to glutamic acid decarboxylase (GAD) and amphiphysin were used for analysis of distribution of neuropil reactivity.
Brain tissue processing
Paraformaldehyde (PFA)-fixed tissue
Rats were anaesthesized and euthanized by decapitation without tissue perfusion; brains were removed and kept for 10 days in 4% PFA at 4°C. Subsequently, brains were cryoprotected with 30% sucrose for 48 h, embedded in freezing medium, and snap-frozen in isopentane chilled with liquid nitrogen.
Other tissue processing
Brains from rats perfused with 4% PFA were removed and kept in 4% PFA for 1 h, and subsequently cryoprotected and embedded in freezing medium as above. Non-perfused rat brains were removed and directly embedded in freezing medium without fixative.
Immunoblot and immunohistochemistry
Sera (diluted 1 : 500) and CSF (1 : 10) were examined for antibodies using an immunoblot avidin–biotin peroxidase assay, as reported (Bataller et al., 2003). Immunoblots included protein extracts (100 µg/ml) from purified human cortical neurons, Purkinje cells and the recombinant proteins, HuD, Cdr2, Nova, Ma1, Ma2, CRMP5 and amphiphysin.
Immunohistochemistry was performed with cryostat-cut 7 µm thick sections mounted directly on slides. Non-pre-fixed tissue was incubated for 10 min with acetone or methanol–acetone at 4°C. Subsequently, all tissue sections were serially incubated with 0.25% H2O2 for 20 min, 10% goat serum for 30 min, the patient's serum or CSF at the indicated dilutions in 10% goat serum overnight at 4°C, biotinylated goat anti-human IgG (1 : 2000) for 2 h and avidin–biotin peroxidase for 1 h, and the reactivity developed with diaminobenzidine. Other primary antibodies used in consecutive tissue sections included: polyclonal rabbit antibodies to VGKCs Kv1.1, Kv1.2 and Kv1.6 (dilution 1 : 50; all from Sigma, St Louis, MO); a monoclonal antibody to Kv1.2 (1 : 50; Upstate Laboratories, Lake Placid, NY); a polyclonal antibody to synaptophysin (1 : 1000, Sigma); a monoclonal antibody to spinophilin (1 : 50; Upstate Laboratories); and human control serum with amphiphysin and Hu antibodies (1 : 500).
Intrathecal synthesis of antibodies was determined using serum and CSF samples normalized with the same concentration of IgG and serially diluted in parallel. Patients were considered to have intrathecal synthesis of antibodies if the end dilution point of CSF showed reactivity that was no longer present in the paired serum containing the same amount of IgG.
Immunocompetition assay
To determine whether patients' antibodies targeted similar epitopes, immunocompetition assays were used, as reported (Dalmau et al., 1992). In brief, tissue sections pre-incubated with patient's serum (or control normal serum) were subsequently incubated with IgG isolated from the serum of another patient and labelled with biotin. Abrogation of reactivity indicated that the patient's serum contained antibodies similar to those present in the biotinylated IgG.
Double immunolabelling of hippocampal neurons
Hippocampal rat neuronal cell cultures were prepared as reported (Buchhalter and Dichter, 1991). Neurons were grown on coverslides, fixed with PFA, and serially incubated with serum from one patient (diluted 1 : 250) for 1 h, and goat anti-human IgG labelled with fluorescein for 30 min. After washing, slides were incubated with biotinylated IgG from another patient with neuropil antibodies (or VGKC antibodies, or IgG from a normal individual), or the indicated monoclonal or polyclonal antibodies to VGKCs, spinophilin or synaptophysin for 1 h. The reactivity of biotinylated human IgG was demonstrated with avidin–rhodamine for 30 min, and the reactivity of antibodies made in mouse or rabbit with the appropriate rhodamine-labelled secondary antibodies. Coverslides containing the neurons were mounted using aqueous medium.
Results
Clinical features
Patients included three men and four women, median age 47 years (Table 1). Four patients (nos 1, 2, 5 and 7) presented with a classical picture of limbic encephalitis with dominant hippocampal or medial temporal lobe dysfunction characterized by severe short-term memory loss, and agitation and behavioural changes (no. 2). Two of these four patients (nos 1 and 2) remained with dominant short-term memory deficits, and case 2 with persistent but intermittent mood swings and personality changes. Case 5 had rapid progression of short-term memory loss to a more severe and extensive encephalitis, and case 7 had stable short-term memory loss for 4 months and subsequently developed progressive cerebellar ataxia and mild brainstem dysfunction.
Clinical features, CSF and neuroimaging findings
| Case | Sex/age | Limbic encephalitis (LE): presenting symptoms | CSF | Initial brain MRI | Brain FDG-PET |
|---|---|---|---|---|---|
| 1 | M/60 | Hippocampal syndrome: short-term memory loss, confusion, partial complex seizures, diaphoresis, and palinopsia | 0 WBC, protein 63, glucose 64, OB not examined, no ISAb IgG index not examined | FLAIR, T2 abnormalities in medial temporal lobes. No contrast enhancement | Hypermetabolism in medial temporal lobes |
| Other: akathisia, fasciculations, hyponatraemia | |||||
| 2 | F/65 | Hippocampal syndrome: short-term memory loss, confusion and irritability Other: downbeat nystagmus, abnormal behaviour, increased fluid intake | 30 WBC, protein 97, glucose 59, positive OB, positive ISAb IgG index 1.24 | Mild FLAIR abnormalities in medial temporal lobes. No contrast enhancement | Diffuse decreased metabolism |
| 3 | F/44 | Extensive LE: psychiatry admission for acute agitation, personality change, memory loss, generalized tonic–clonic seizures | 15 WBC, protein 18, glucose 90, no OB, ISAb not examined IgG index <0.66 | Non-specific, scattered T2 foci of hyperintensity in frontal lobes. No contrast enhancement | Hypermetabolism in left frontal–temporal region |
| Other: word finding difficulty, mild right hemiparesis | |||||
| 4 | F/26 | Extensive LE: psychiatry admission for acute change in behaviour, confusion, generalized tonic–clonic seizures | 49 WBC, protein 67, glucose 66, OB not examined, positive ISAb IgG index not examined | FLAIR and T2 abnormalities in cerebral cortex and cerebellum with cerebellar contrast enhancement | Increased metabolism in right temporal lobe and brainstem; decreased metabolism in occipital lobes |
| Other: central hypoventilation, ankle clonus and left Babinski sign | |||||
| 5 | F/44 | Hippocampal syndrome: pure short-term memory loss Evolved to extensive LE, focal motor seizures | 44 WBC, protein 91, glucose 70, no OB, positive ISAb IgG index <0.66 | Initial MRI normal. Follow-up 5 days later: mild temporal lobe FLAIR abnormalities (right > left). No contrast enhancement | Increased metabolism in right temporal lobe and left cerebellum |
| 6 | M/38 | Extensive LE: confusion, agitation, short-term memory loss, generalized tonic–clonic seizures (prior episode of LE 5 years earlier) Evolved to hippocampal and multifocal encephalitis, and stiff-person syndrome | 7 WBC, protein 50, glucose 92, positive OB, positive ISAb IgG index not examined | FLAIR abnormalities in right medial and lateral temporal lobe, right frontal, left insular and left occipital regions. No contrast enhancement | Not done |
| 7 | M/55 | Hippocampal syndrome: short-term memory loss Evolved to cerebellar and brainstem dysfunction; partial complex seizures | 81 WBC, protein 150, glucose 59, positive OB, positive ISAb IgG index 40.1 | Persistent FLAIR, T2 abnormalities in medial temporal lobes. Positive contrast enhancement. Evolving hippocampal atrophy | Increased metabolism in temporal lobes, right insula and cerebellum |
| Case | Sex/age | Limbic encephalitis (LE): presenting symptoms | CSF | Initial brain MRI | Brain FDG-PET |
|---|---|---|---|---|---|
| 1 | M/60 | Hippocampal syndrome: short-term memory loss, confusion, partial complex seizures, diaphoresis, and palinopsia | 0 WBC, protein 63, glucose 64, OB not examined, no ISAb IgG index not examined | FLAIR, T2 abnormalities in medial temporal lobes. No contrast enhancement | Hypermetabolism in medial temporal lobes |
| Other: akathisia, fasciculations, hyponatraemia | |||||
| 2 | F/65 | Hippocampal syndrome: short-term memory loss, confusion and irritability Other: downbeat nystagmus, abnormal behaviour, increased fluid intake | 30 WBC, protein 97, glucose 59, positive OB, positive ISAb IgG index 1.24 | Mild FLAIR abnormalities in medial temporal lobes. No contrast enhancement | Diffuse decreased metabolism |
| 3 | F/44 | Extensive LE: psychiatry admission for acute agitation, personality change, memory loss, generalized tonic–clonic seizures | 15 WBC, protein 18, glucose 90, no OB, ISAb not examined IgG index <0.66 | Non-specific, scattered T2 foci of hyperintensity in frontal lobes. No contrast enhancement | Hypermetabolism in left frontal–temporal region |
| Other: word finding difficulty, mild right hemiparesis | |||||
| 4 | F/26 | Extensive LE: psychiatry admission for acute change in behaviour, confusion, generalized tonic–clonic seizures | 49 WBC, protein 67, glucose 66, OB not examined, positive ISAb IgG index not examined | FLAIR and T2 abnormalities in cerebral cortex and cerebellum with cerebellar contrast enhancement | Increased metabolism in right temporal lobe and brainstem; decreased metabolism in occipital lobes |
| Other: central hypoventilation, ankle clonus and left Babinski sign | |||||
| 5 | F/44 | Hippocampal syndrome: pure short-term memory loss Evolved to extensive LE, focal motor seizures | 44 WBC, protein 91, glucose 70, no OB, positive ISAb IgG index <0.66 | Initial MRI normal. Follow-up 5 days later: mild temporal lobe FLAIR abnormalities (right > left). No contrast enhancement | Increased metabolism in right temporal lobe and left cerebellum |
| 6 | M/38 | Extensive LE: confusion, agitation, short-term memory loss, generalized tonic–clonic seizures (prior episode of LE 5 years earlier) Evolved to hippocampal and multifocal encephalitis, and stiff-person syndrome | 7 WBC, protein 50, glucose 92, positive OB, positive ISAb IgG index not examined | FLAIR abnormalities in right medial and lateral temporal lobe, right frontal, left insular and left occipital regions. No contrast enhancement | Not done |
| 7 | M/55 | Hippocampal syndrome: short-term memory loss Evolved to cerebellar and brainstem dysfunction; partial complex seizures | 81 WBC, protein 150, glucose 59, positive OB, positive ISAb IgG index 40.1 | Persistent FLAIR, T2 abnormalities in medial temporal lobes. Positive contrast enhancement. Evolving hippocampal atrophy | Increased metabolism in temporal lobes, right insula and cerebellum |
OB = oligoclonal bands; ISAb = intrathecal synthesis of antibodies; IgG index (<0.66). CSF normal values: glucose 55–80 mg/dl; protein 16–46 mg/dl; white blood cells (WBC) <4/µl.
Clinical features, CSF and neuroimaging findings
| Case | Sex/age | Limbic encephalitis (LE): presenting symptoms | CSF | Initial brain MRI | Brain FDG-PET |
|---|---|---|---|---|---|
| 1 | M/60 | Hippocampal syndrome: short-term memory loss, confusion, partial complex seizures, diaphoresis, and palinopsia | 0 WBC, protein 63, glucose 64, OB not examined, no ISAb IgG index not examined | FLAIR, T2 abnormalities in medial temporal lobes. No contrast enhancement | Hypermetabolism in medial temporal lobes |
| Other: akathisia, fasciculations, hyponatraemia | |||||
| 2 | F/65 | Hippocampal syndrome: short-term memory loss, confusion and irritability Other: downbeat nystagmus, abnormal behaviour, increased fluid intake | 30 WBC, protein 97, glucose 59, positive OB, positive ISAb IgG index 1.24 | Mild FLAIR abnormalities in medial temporal lobes. No contrast enhancement | Diffuse decreased metabolism |
| 3 | F/44 | Extensive LE: psychiatry admission for acute agitation, personality change, memory loss, generalized tonic–clonic seizures | 15 WBC, protein 18, glucose 90, no OB, ISAb not examined IgG index <0.66 | Non-specific, scattered T2 foci of hyperintensity in frontal lobes. No contrast enhancement | Hypermetabolism in left frontal–temporal region |
| Other: word finding difficulty, mild right hemiparesis | |||||
| 4 | F/26 | Extensive LE: psychiatry admission for acute change in behaviour, confusion, generalized tonic–clonic seizures | 49 WBC, protein 67, glucose 66, OB not examined, positive ISAb IgG index not examined | FLAIR and T2 abnormalities in cerebral cortex and cerebellum with cerebellar contrast enhancement | Increased metabolism in right temporal lobe and brainstem; decreased metabolism in occipital lobes |
| Other: central hypoventilation, ankle clonus and left Babinski sign | |||||
| 5 | F/44 | Hippocampal syndrome: pure short-term memory loss Evolved to extensive LE, focal motor seizures | 44 WBC, protein 91, glucose 70, no OB, positive ISAb IgG index <0.66 | Initial MRI normal. Follow-up 5 days later: mild temporal lobe FLAIR abnormalities (right > left). No contrast enhancement | Increased metabolism in right temporal lobe and left cerebellum |
| 6 | M/38 | Extensive LE: confusion, agitation, short-term memory loss, generalized tonic–clonic seizures (prior episode of LE 5 years earlier) Evolved to hippocampal and multifocal encephalitis, and stiff-person syndrome | 7 WBC, protein 50, glucose 92, positive OB, positive ISAb IgG index not examined | FLAIR abnormalities in right medial and lateral temporal lobe, right frontal, left insular and left occipital regions. No contrast enhancement | Not done |
| 7 | M/55 | Hippocampal syndrome: short-term memory loss Evolved to cerebellar and brainstem dysfunction; partial complex seizures | 81 WBC, protein 150, glucose 59, positive OB, positive ISAb IgG index 40.1 | Persistent FLAIR, T2 abnormalities in medial temporal lobes. Positive contrast enhancement. Evolving hippocampal atrophy | Increased metabolism in temporal lobes, right insula and cerebellum |
| Case | Sex/age | Limbic encephalitis (LE): presenting symptoms | CSF | Initial brain MRI | Brain FDG-PET |
|---|---|---|---|---|---|
| 1 | M/60 | Hippocampal syndrome: short-term memory loss, confusion, partial complex seizures, diaphoresis, and palinopsia | 0 WBC, protein 63, glucose 64, OB not examined, no ISAb IgG index not examined | FLAIR, T2 abnormalities in medial temporal lobes. No contrast enhancement | Hypermetabolism in medial temporal lobes |
| Other: akathisia, fasciculations, hyponatraemia | |||||
| 2 | F/65 | Hippocampal syndrome: short-term memory loss, confusion and irritability Other: downbeat nystagmus, abnormal behaviour, increased fluid intake | 30 WBC, protein 97, glucose 59, positive OB, positive ISAb IgG index 1.24 | Mild FLAIR abnormalities in medial temporal lobes. No contrast enhancement | Diffuse decreased metabolism |
| 3 | F/44 | Extensive LE: psychiatry admission for acute agitation, personality change, memory loss, generalized tonic–clonic seizures | 15 WBC, protein 18, glucose 90, no OB, ISAb not examined IgG index <0.66 | Non-specific, scattered T2 foci of hyperintensity in frontal lobes. No contrast enhancement | Hypermetabolism in left frontal–temporal region |
| Other: word finding difficulty, mild right hemiparesis | |||||
| 4 | F/26 | Extensive LE: psychiatry admission for acute change in behaviour, confusion, generalized tonic–clonic seizures | 49 WBC, protein 67, glucose 66, OB not examined, positive ISAb IgG index not examined | FLAIR and T2 abnormalities in cerebral cortex and cerebellum with cerebellar contrast enhancement | Increased metabolism in right temporal lobe and brainstem; decreased metabolism in occipital lobes |
| Other: central hypoventilation, ankle clonus and left Babinski sign | |||||
| 5 | F/44 | Hippocampal syndrome: pure short-term memory loss Evolved to extensive LE, focal motor seizures | 44 WBC, protein 91, glucose 70, no OB, positive ISAb IgG index <0.66 | Initial MRI normal. Follow-up 5 days later: mild temporal lobe FLAIR abnormalities (right > left). No contrast enhancement | Increased metabolism in right temporal lobe and left cerebellum |
| 6 | M/38 | Extensive LE: confusion, agitation, short-term memory loss, generalized tonic–clonic seizures (prior episode of LE 5 years earlier) Evolved to hippocampal and multifocal encephalitis, and stiff-person syndrome | 7 WBC, protein 50, glucose 92, positive OB, positive ISAb IgG index not examined | FLAIR abnormalities in right medial and lateral temporal lobe, right frontal, left insular and left occipital regions. No contrast enhancement | Not done |
| 7 | M/55 | Hippocampal syndrome: short-term memory loss Evolved to cerebellar and brainstem dysfunction; partial complex seizures | 81 WBC, protein 150, glucose 59, positive OB, positive ISAb IgG index 40.1 | Persistent FLAIR, T2 abnormalities in medial temporal lobes. Positive contrast enhancement. Evolving hippocampal atrophy | Increased metabolism in temporal lobes, right insula and cerebellum |
OB = oligoclonal bands; ISAb = intrathecal synthesis of antibodies; IgG index (<0.66). CSF normal values: glucose 55–80 mg/dl; protein 16–46 mg/dl; white blood cells (WBC) <4/µl.
Three patients (nos 3, 4 and 6) presented with a syndrome suggesting more extensive involvement of the limbic system with or without dysfunction of other brain regions, characterized by severe confusion, inappropriate behaviour and seizures that initially limited the memory evaluation. Patients 3 and 4 were admitted to the Psychiatry Unit for suspicion of psychosis, and had rapid progression of symptoms to a more severe encephalopathy with extreme agitation, inability to follow commands, stupor, mild hemiparesis (no. 3) and central hypoventilation requiring intubation (no. 4). Patient 6 had short-term memory loss that became detectable after his confusion and seizures improved; his syndrome resulted from limbic and multifocal cortical encephalitis that overlapped with symptoms of stiff-person syndrome.
In five patients, the symptoms had a rapid monophasic development, reaching the maximal deficit in 2–4 weeks. Patient 2 had three episodes of symptom recurrence while being tapered from prednisone. Patient 6 had a similar disorder (short-term memory loss, confusion, agitation and medial temporal lobe MRI abnormalities) that resolved spontaneously 5 years earlier.
EEG and CSF studies
Six patients had clinical seizures: three generalized tonic–clonic (nos 3, 4 and 6), two partial complex seizures (nos 1 and 7) and one (no. 5) focal motor. Multiple EEGs obtained in all patients at different time points showed diffuse slowing without epileptic activity; in two instances, epileptic activity was recorded in patients 3 and 5, both in the left temporal lobe.
The CSF was abnormal in all seven patients (Table 1): five had increased protein concentration and lymphocytic pleocytosis, one had increased protein concentration, and one had lymphocytic pleocytosis. The abnormal total protein concentration ranged from 50 to 150 mg/dl (median 79 mg/dl), and the pleocytosis from seven to 81 white blood cells/µl (median 37). Three of five patients tested had CSF oligoclonal bands; and two of four had an elevated IgG index.
Brain MRI and FDG-PET correlates
The number of brain MRIs obtained for each patient ranged from two to seven (median four). All four patients with predominant hippocampal dysfunction had MRI abnormalities restricted to one or both medial temporal lobes (Table 1, Figs 1 and 2). In two of these patients (nos 2 and 5), the abnormalities were mild and only visible with FLAIR (fluid attenuated inversion recovery) sequences (Fig. 1). The other two patients (nos 1 and 7) had bilateral medial temporal lobe hyperintensities in FLAIR and T2 sequences, with normal or mild hypointensity in T1 sequences and positive contrast enhancement (no. 7) (Figs 1 and 2).
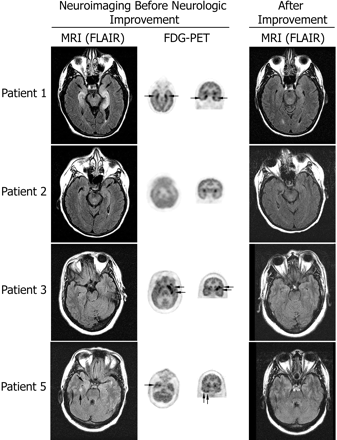
MRI and FDG-PET at symptom presentation and MRI after improvement. Axial MRI FLAIR sequences have been matched with axial and coronal FDG-PET to show the areas of maximal involvement. The same MRI region is shown after symptom improvement. Patient 1 had bilateral medial temporal lobe FLAIR abnormalities that correlated with the areas of FDG hyperactivity; the MRI findings are remarkably improved 4 months later. Patient 2 had mild FLAIR abnormalities in the medial temporal lobes with generalized FDG hypoactivity (attributed to antidepressants); the MRI was normal 3 months later. Patient 3 had bilateral non-specific foci of T2 and FLAIR abnormalities in frontal lobes (not shown) and normal temporal lobe findings; the FDG-PET showed hyperactivity in the left frontotemporal region; follow-up MRI 22 months later was unchanged (non-specific abnormalities in frontal lobes; normal temporal lobes) without evolving atrophy. Patient 5 had mild FLAIR abnormalities in the temporal lobes that predominated in the right side, and correlated with the indicated areas of FDG hyperactivity in the right temporal lobe (FDG hyperactivity was also present in the left cerebellum, not shown); the follow-up MRI 27 months later was normal.
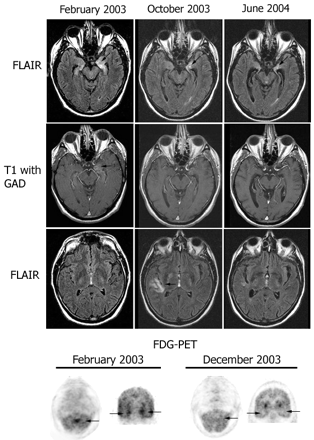
Follow-up MRI and FDG-PET in patient 7. MRI FLAIR sequences and T1 with gadolinium (T1 with GAD) obtained at three time points during the clinical course of patient 7. Note the presence of bilateral medial temporal lobe FLAIR hyperintensities with progressive evolving atrophy (arrows point to the progressive volume loss in the left hippocampus), and transient FLAIR abnormality in the right insula (October 2003). After gadolinium administration (T1 with GAD), there was mild enhancement in the left medial temporal lobe (February 2003) that resolved in the follow-up studies. The FDG-PET obtained in February 2003 showed hyperactivity in the vermis of the cerebellum (arrow) that preceded by 4 months the development of gait ataxia, and hyperactivity in the medial temporal lobes (bilateral arrows in coronal sections) that correlated with the limbic dysfunction and MRI FLAIR abnormalities in the temporal lobes. The FDG-PET in December 2003 shows resolution of the hyperactivity that correlated with transient stabilization of symptoms and decreased CSF pleocytosis after treatment with cyclophosphamide and corticosteroids.
The three patients with a syndrome suggesting extensive limbic dysfunction had abnormal MRI findings (Table 1), but only one (no. 6) had typical medial temporal lobe FLAIR and T2 abnormalities in addition to discrete multifocal cortical brain FLAIR hyperintensities (Figs 1 and 3; patient 4 not shown).
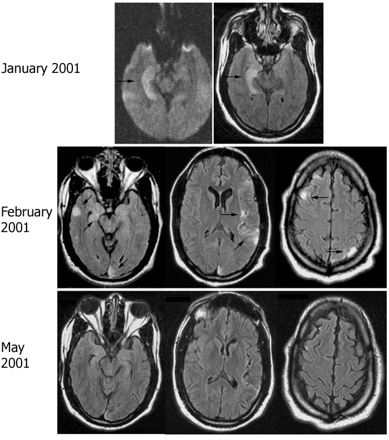
Follow-up MRI in patient 6. MRI FLAIR sequences obtained at different time points of symptom recurrence in patient 6. Note the discrete cortical areas of hyperintensity involving the medial and lateral right temporal region, right frontal lobe, left insular and left parietal and occipital lobes (arrows). Significant improvement was noted in the MRI obtained in May 2001, correlating with clinical improvement of the multifocal encephalitis.
All patients except no. 6 had FDG-PET at symptom presentation (Figs 1 and 2): all but no. 2 showed hypermetabolism in one or both temporal lobes. Patient 4 had additional hyperactivity in the brainstem and hypoactivity in the occipital lobes (data not shown), and patients 5 and 7 had additional activity in the cerebellum (patient 7 shown in Fig. 2).
Overall, combining the MRI and FDG-PET findings, all patients had predominant temporal lobe abnormalities, three had additional involvement of the cerebellum and three of the cerebral cortex. The MRI and FDG-PET partially correlated in patients 1, 5 and 7, and did not correlate in patients 2, 3 and 4.
Tumours
All patients underwent CT of the chest, abdomen and pelvis, and, if negative, body FDG-PET. A tumour was found in five patients, four demonstrated with CT and one with FDG-PET (papillary carcinoma of the thyroid). In four patients, the tumour was completely removed: one mature teratoma of the mediastinum, one thymic carcinoma, one invasive thymoma, and one papillary carcinoma of the thyroid. One patient had an ovarian mass composed of fat and calcium, consistent with a dermoid tumour, that was not biopsied.
Antibodies
Immunohistochemical studies were optimized to identify antibodies to cell surface antigens or synaptic-enriched regions. This was accomplished using non-perfused rat brain fixed for 10 days in PFA and cryoprotected with sucrose. Sections of this tissue allowed unequivocal detection of antibodies to GAD, amphiphysin and VGKCs in human control sera previously tested positive with radioimmunoassay (GAD or VGKCs) or immunoblot (amphiphysin). All 11 VGKC radioimmunoassay-positive sera, but no other sera, produced an intense and characteristic pattern of reactivity with rat hippocampus, and to a lesser degree cerebellum and brain, that allowed their identification among blinded samples.
Six patients had serum and CSF antibodies that were optimally detected with the indicated non-perfused PFA-fixed tissue. The antibodies of patient 1 had the characteristic immunolabelling of VGKC antibodies (compare patient 1 in Fig. 4 with the positive control for VGKC antibodies in Fig. 5); in immunocompetition assays, this serum completely blocked the reactivity of all control patients with VGKC antibodies, confirming the presence of these antibodies (data not shown). The other five patients (2–6) had diverse patterns of reactivities suggesting different antibodies, but all sharing the property of strong immunolabelling of areas of dense dendritic network and synaptic-enriched regions in the neuropil of hippocampus or cerebellum, sparing most neuronal cell bodies (Table 2; Figs 4 and 5). Two of these five sera (patients 2 and 6) completely blocked the neuropil reactivity of each other and partially blocked the reactivity of sera 3 and 5, but none of the five sera competed for VGKC antibodies (data not shown). Furthermore, in double immunolabelling of hippocampal neuronal cultures, the antibody reactivity of these five patients did not co-localize with the reactivity of VGKCs (Kv1.1, Kv 1.2 and Kv 1.6) or with the control patients with VGKCs determined with radioimmunoassay (Fig. 6).
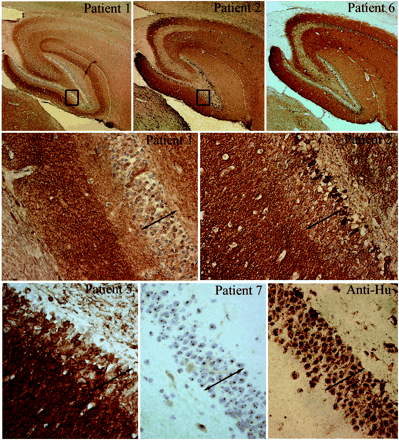
Immunohistochemical analysis of neuropil antibodies. Upper row: sagittal sections of rat brain immunoreacted with sera of patients 1, 2 and 6. Note that the three sera show intense reactivity with the neuropil of hippocampus. Patients 2 and 6 harboured the same novel neuropil antibody and abrogated the reactivity of each other in immunocompetition assays; patient 2 had an additional antibody reacting with a subset of neurons in the inner part of the dentate gyrus (shown at high magnification in the middle row). Middle and lower rows show at high magnification the reactivity of sera from patients 1, 2, 5 and 7 and a control anti-Hu. Compare the intracellular reactivity of sera 7 and anti-Hu with the predominant neuropil reactivity of sera 1, 2 and 5 that spare the nuclei and cytoplasm of neurons (except for serum 2 that reacts with a subset of cells). Note that serum 1 has identical reactivity to the serum of a control patient with radioimmunoassay-positive VGKC antibodies shown at low and high magnifications in Fig. 5 (slides mildly counterstained with haematoxylin; upper row ×5; middle and lower rows ×200).
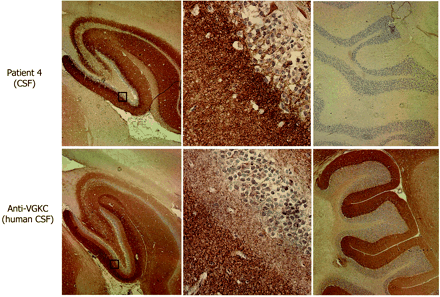
Immunohistochemical comparison of the neuropil antibodies of patient 4 with human VGKC antibodies. Sagittal sections of rat brain immunoreacted with CSF of patient 4 and CSF from a control patient with VGKC antibodies determined with the α-dendrotoxin radioimmunoassay. Note the different patterns of hippocampal reactivity (left panels). The neuropil antibody of patient 4 preferentially reacts with the inner aspect of the molecular layer adjacent to the granular cells of the dentate gyrus, while the VGKC antibodies predominantly react with the outer aspect of the molecular layer distant from the granular cells. These differences are better demonstrated in the squares magnified in the middle panels. The neuropil antibody of patient 4 does not react with cerebellum, while VGKC antibodies react with the molecular layer of cerebellum (right panels). Slides mildly counterstained with haematoxylin. Magnification: left panels ×5; middle panels ×400, right panels ×10.
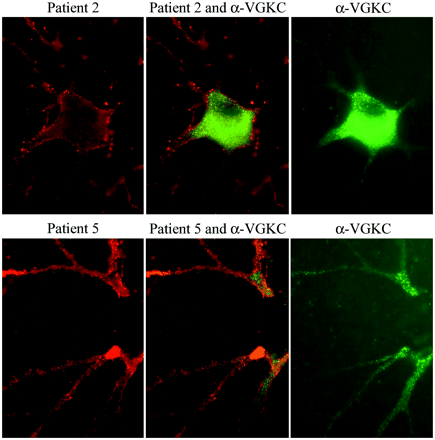
Double immunolabelling of hippocampal neuronal cultures with patients' sera and VGKC antibodies. Using rat hippocampal neurons, the antibodies from patients 2 and 5 (red, left panels) produce widespread labelling of the surface of neurons and dendrites without co-localization (middle panels) with VGKC antibodies (green, right panels). Note that in cultured rat hippocampal neurons, the VGKC antibodies predominantly label the proximal aspect of the neuronal processes and cytoplasm. Sera of 10 other patients with radioimmunoassay-positive VGKC, and monoclonal and polyclonal antibodies to Kv1.2 produced identical reactivity to the VGKC serum used here (data not shown). All panels ×800 (oil).
Tumour association, antibodies, treatment and outcome
| Case | Tumour | Antibody reactivity | Treatment | Outcome* | Outcome+ | Clinical outcome |
|---|---|---|---|---|---|---|
| 1 | – | VGKC; neuropil of hippocampus = cerebellum > cortex | IVIg, corticosteroids, phenytoin | MRI improved; no evolving atrophy (4) | Initial, 1 : 8000; follow-up 1 : 2000 (4) | Returned to baseline |
| 2 | – | (1) Neuropil hippocampus > cerebellum > cortex. (2) Small subset of neurons in the dentate gyrus, cortex and cerebellum | At presentation, plasma exchange and corticosteroids; at relapse, IVIg and corticosteroids | MRI normal; no evolving atrophy (3) | Initial, after 2nd recurrence, 1 : 32 000; follow-up, after 3rd recurrence, 1 : 4000 (4) | Residual behavioural problem and short-term memory deficit |
| 3 | Mature teratoma in the mediastinum | Neuropil of molecular layer of hippocampus | Tumour removal | Stable non-specific, scattered T2 foci of hyperintensity in frontal lobes (22) | Initial, 1 : 500; follow-up, not detectable (22) | Returned to baseline |
| 4 | CT: ovarian mass with fat and calcification (dermoid cyst) | Neuropil of inner molecular layer of hippocampus | Corticosteroids | MRI normal (5) FDG-PET: minimally decreased activity in the left temporoparietal region (5) | Initial, 1 : 1000; follow-up, not detectable (5) | Returned to baseline |
| 5 | Thymic carcinoma | Neuropil of hippocampus > cerebellum > cortex | Tumour removal. Partial treatment with IVIg and corticosteroids | MRI normal (27) | Initial, 1 : 32 000; follow-up, not detectable (27) | Returned to baseline |
| 6 | Thymoma | (1) Neuropil of hippocampus > cerebellum > cortex (2) GAD antibodies | Tumour removal Corticosteroids for encephalitis. Plasma exchange, IVIg, corticosteroids for stiff-person syndrome | MRI improved (5 months after relapse) | Initial, 1 : 16 000; follow-up, 1 : 4000 (5) GAD antibody titres unchanged during the 5 month follow-up | Mild residual memory deficits and steroid-dependent muscle spasms and rigidity |
| 7 | Papillary carcinoma of the thyroid | Nuclear antigens expressed by all CNS neurons and the thyroid cancer. No neuropil antibodies | Corticosteroids, plasma exchange, IVIg, rituximab, oral cyclophosphamide | MRI: progressive atrophy of hippocampi and cerebellum. Transient FLAIR abnormality in the right insula (19) FDG-PET: no hyperactivity (13) | Initial, 1 : 8000; follow-up, 1 : 8000 (13) | Progressive deterioration and death |
| Case | Tumour | Antibody reactivity | Treatment | Outcome* | Outcome+ | Clinical outcome |
|---|---|---|---|---|---|---|
| 1 | – | VGKC; neuropil of hippocampus = cerebellum > cortex | IVIg, corticosteroids, phenytoin | MRI improved; no evolving atrophy (4) | Initial, 1 : 8000; follow-up 1 : 2000 (4) | Returned to baseline |
| 2 | – | (1) Neuropil hippocampus > cerebellum > cortex. (2) Small subset of neurons in the dentate gyrus, cortex and cerebellum | At presentation, plasma exchange and corticosteroids; at relapse, IVIg and corticosteroids | MRI normal; no evolving atrophy (3) | Initial, after 2nd recurrence, 1 : 32 000; follow-up, after 3rd recurrence, 1 : 4000 (4) | Residual behavioural problem and short-term memory deficit |
| 3 | Mature teratoma in the mediastinum | Neuropil of molecular layer of hippocampus | Tumour removal | Stable non-specific, scattered T2 foci of hyperintensity in frontal lobes (22) | Initial, 1 : 500; follow-up, not detectable (22) | Returned to baseline |
| 4 | CT: ovarian mass with fat and calcification (dermoid cyst) | Neuropil of inner molecular layer of hippocampus | Corticosteroids | MRI normal (5) FDG-PET: minimally decreased activity in the left temporoparietal region (5) | Initial, 1 : 1000; follow-up, not detectable (5) | Returned to baseline |
| 5 | Thymic carcinoma | Neuropil of hippocampus > cerebellum > cortex | Tumour removal. Partial treatment with IVIg and corticosteroids | MRI normal (27) | Initial, 1 : 32 000; follow-up, not detectable (27) | Returned to baseline |
| 6 | Thymoma | (1) Neuropil of hippocampus > cerebellum > cortex (2) GAD antibodies | Tumour removal Corticosteroids for encephalitis. Plasma exchange, IVIg, corticosteroids for stiff-person syndrome | MRI improved (5 months after relapse) | Initial, 1 : 16 000; follow-up, 1 : 4000 (5) GAD antibody titres unchanged during the 5 month follow-up | Mild residual memory deficits and steroid-dependent muscle spasms and rigidity |
| 7 | Papillary carcinoma of the thyroid | Nuclear antigens expressed by all CNS neurons and the thyroid cancer. No neuropil antibodies | Corticosteroids, plasma exchange, IVIg, rituximab, oral cyclophosphamide | MRI: progressive atrophy of hippocampi and cerebellum. Transient FLAIR abnormality in the right insula (19) FDG-PET: no hyperactivity (13) | Initial, 1 : 8000; follow-up, 1 : 8000 (13) | Progressive deterioration and death |
Brain MRI or FDG-PET (no. of months after symptom presentation)
antibody titres (no. of months after symptom presentation). IVIg = intravenous immunoglobulin G.
Tumour association, antibodies, treatment and outcome
| Case | Tumour | Antibody reactivity | Treatment | Outcome* | Outcome+ | Clinical outcome |
|---|---|---|---|---|---|---|
| 1 | – | VGKC; neuropil of hippocampus = cerebellum > cortex | IVIg, corticosteroids, phenytoin | MRI improved; no evolving atrophy (4) | Initial, 1 : 8000; follow-up 1 : 2000 (4) | Returned to baseline |
| 2 | – | (1) Neuropil hippocampus > cerebellum > cortex. (2) Small subset of neurons in the dentate gyrus, cortex and cerebellum | At presentation, plasma exchange and corticosteroids; at relapse, IVIg and corticosteroids | MRI normal; no evolving atrophy (3) | Initial, after 2nd recurrence, 1 : 32 000; follow-up, after 3rd recurrence, 1 : 4000 (4) | Residual behavioural problem and short-term memory deficit |
| 3 | Mature teratoma in the mediastinum | Neuropil of molecular layer of hippocampus | Tumour removal | Stable non-specific, scattered T2 foci of hyperintensity in frontal lobes (22) | Initial, 1 : 500; follow-up, not detectable (22) | Returned to baseline |
| 4 | CT: ovarian mass with fat and calcification (dermoid cyst) | Neuropil of inner molecular layer of hippocampus | Corticosteroids | MRI normal (5) FDG-PET: minimally decreased activity in the left temporoparietal region (5) | Initial, 1 : 1000; follow-up, not detectable (5) | Returned to baseline |
| 5 | Thymic carcinoma | Neuropil of hippocampus > cerebellum > cortex | Tumour removal. Partial treatment with IVIg and corticosteroids | MRI normal (27) | Initial, 1 : 32 000; follow-up, not detectable (27) | Returned to baseline |
| 6 | Thymoma | (1) Neuropil of hippocampus > cerebellum > cortex (2) GAD antibodies | Tumour removal Corticosteroids for encephalitis. Plasma exchange, IVIg, corticosteroids for stiff-person syndrome | MRI improved (5 months after relapse) | Initial, 1 : 16 000; follow-up, 1 : 4000 (5) GAD antibody titres unchanged during the 5 month follow-up | Mild residual memory deficits and steroid-dependent muscle spasms and rigidity |
| 7 | Papillary carcinoma of the thyroid | Nuclear antigens expressed by all CNS neurons and the thyroid cancer. No neuropil antibodies | Corticosteroids, plasma exchange, IVIg, rituximab, oral cyclophosphamide | MRI: progressive atrophy of hippocampi and cerebellum. Transient FLAIR abnormality in the right insula (19) FDG-PET: no hyperactivity (13) | Initial, 1 : 8000; follow-up, 1 : 8000 (13) | Progressive deterioration and death |
| Case | Tumour | Antibody reactivity | Treatment | Outcome* | Outcome+ | Clinical outcome |
|---|---|---|---|---|---|---|
| 1 | – | VGKC; neuropil of hippocampus = cerebellum > cortex | IVIg, corticosteroids, phenytoin | MRI improved; no evolving atrophy (4) | Initial, 1 : 8000; follow-up 1 : 2000 (4) | Returned to baseline |
| 2 | – | (1) Neuropil hippocampus > cerebellum > cortex. (2) Small subset of neurons in the dentate gyrus, cortex and cerebellum | At presentation, plasma exchange and corticosteroids; at relapse, IVIg and corticosteroids | MRI normal; no evolving atrophy (3) | Initial, after 2nd recurrence, 1 : 32 000; follow-up, after 3rd recurrence, 1 : 4000 (4) | Residual behavioural problem and short-term memory deficit |
| 3 | Mature teratoma in the mediastinum | Neuropil of molecular layer of hippocampus | Tumour removal | Stable non-specific, scattered T2 foci of hyperintensity in frontal lobes (22) | Initial, 1 : 500; follow-up, not detectable (22) | Returned to baseline |
| 4 | CT: ovarian mass with fat and calcification (dermoid cyst) | Neuropil of inner molecular layer of hippocampus | Corticosteroids | MRI normal (5) FDG-PET: minimally decreased activity in the left temporoparietal region (5) | Initial, 1 : 1000; follow-up, not detectable (5) | Returned to baseline |
| 5 | Thymic carcinoma | Neuropil of hippocampus > cerebellum > cortex | Tumour removal. Partial treatment with IVIg and corticosteroids | MRI normal (27) | Initial, 1 : 32 000; follow-up, not detectable (27) | Returned to baseline |
| 6 | Thymoma | (1) Neuropil of hippocampus > cerebellum > cortex (2) GAD antibodies | Tumour removal Corticosteroids for encephalitis. Plasma exchange, IVIg, corticosteroids for stiff-person syndrome | MRI improved (5 months after relapse) | Initial, 1 : 16 000; follow-up, 1 : 4000 (5) GAD antibody titres unchanged during the 5 month follow-up | Mild residual memory deficits and steroid-dependent muscle spasms and rigidity |
| 7 | Papillary carcinoma of the thyroid | Nuclear antigens expressed by all CNS neurons and the thyroid cancer. No neuropil antibodies | Corticosteroids, plasma exchange, IVIg, rituximab, oral cyclophosphamide | MRI: progressive atrophy of hippocampi and cerebellum. Transient FLAIR abnormality in the right insula (19) FDG-PET: no hyperactivity (13) | Initial, 1 : 8000; follow-up, 1 : 8000 (13) | Progressive deterioration and death |
Brain MRI or FDG-PET (no. of months after symptom presentation)
antibody titres (no. of months after symptom presentation). IVIg = intravenous immunoglobulin G.
Patient 4 with diffuse encephalitis harboured antibodies that produced the most restricted immunolabelling of hippocampus, with barely visible immunolabelling of other areas of the brain or cerebellum (Fig. 5). The reactivity of this patient's antibodies co-localized with EFA6 (data not shown).
Consecutive sections of rat brain immunolabelled with the patients' sera or CSF and several markers of synapses (synaptophysin and amphiphysin) and dendrites (spinophilin) demonstrated in all instances neuropil reactivity (Fig. 7), but the patients' antibodies had more hippocampal specificity (not shown). Double immunolabelling of hippocampal neuronal cultures with patients' antibodies and synaptophysin or spinophilin showed partial co-localization with both; some sera showed better segmental co-localization with spinophilin than others (Fig. 7). Overall, a common feature of the novel neuropil antibodies was that in rat neuronal cultures, all showed reactivity unevenly distributed on the cell membrane with clustered regions of immunolabelling; this reactivity clearly differed from the evenly distributed, fine dot-like reactivity of VGKC antibodies, which in cultured neurons is not restricted to the cell surface (Figs 6 and 7).
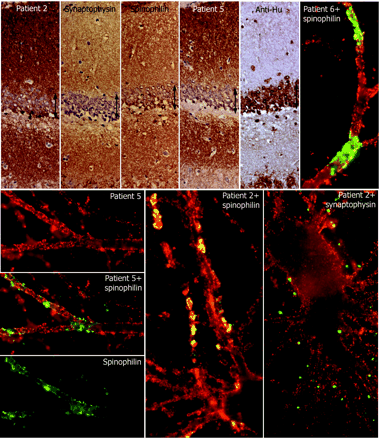
Comparative reactivity of patients' antibodies with synaptic and dendritic markers. Top row, first five panels: consecutive coronal sections of rat hippocampus reacted with the indicated antibodies. Note the intense and diffuse reactivity of all antibodies (except anti-Hu) with the neuropil of hippocampus, relatively sparing the cell bodies of the neurons of the dentate gyrus (arrows). Sections mildly counterstained with haematoxylin ×400. Other panels: single and double immunolabelling of neuronal cultures with sera of the indicated patients (red) and spinophilin (green) or synaptophysin (green). Note the predominant surface localization of the antigens targeted by all patients' sera; there is imperfect co-localization (yellow) with the segmental expression of spinophilin, and less frequent co-localization with synaptophysin. All panels ×800 (oil).
Two patients with neuropil antibodies had additional antibodies in serum and CSF: patient 2 had antibodies reacting with a discrete subset of cells lining the internal granular layer of the dentate gyrus of hippocampus and scattered neuronal and glial cells of brain and cerebellum (Fig. 4). Patient 6 had additional antibodies to GAD (data not shown).
Only one patient (case 7) had serum and CSF antibodies against intracellular antigens expressed by all neurons and the tumour (thyroid carcinoma) (Fig. 4). The reactivity spared the neuropil and was detectable using acetone- or methanol–acetone-fixed tissue.
None of the antibodies of the seven patients identified any antigen in immunoblot studies. Intrathecal synthesis of antibodies was examined in five patients with neuropil antibodies: four had intrathecal synthesis and one was negative (patient with VGKC antibodies) (data not shown). The novel neuropil antibodies were not identified in any of the indicated control samples from patients seen by the authors or in banked sera.
Clinical, radiological and neuro-immunological correlates
Table 2 shows the treatment, neuroimaging and clinical outcomes, and the antibody titre follow-up. Six patients had dramatic clinical improvement, five with associated MRI improvement and one with stable findings (Figs 1 and 3). Specifically, four of these patients (nos 1, 3, 4 and 5) were able to return to their jobs. Patient 2 has returned to some activities at home, but has residual behavioural abnormalities and short-term memory deficits. Patient 6 recovered from the two episodes of multifocal encephalitis that occurred 5 years apart; the striking recovery following the second episode allowed him to get married a few weeks later; he has mild residual memory deficits, and muscle rigidity and spasms that are controlled with 10 mg of daily prednisone.
Patient 7 had onconeuronal antibodies against intracellular antigens and developed progressive neurological deterioration over 22 months that eventually resulted in death. Despite multiple immunotherapies and tumour resection, his symptoms evolved from classic limbic encephalopathy to predominant cerebellar and brainstem dysfunction. Seven follow-up MRIs obtained during the course of the disease showed progressive hippocampal and cerebellar atrophy and a transient FLAIR abnormality in the right insula (Fig. 2). A correlation was also found with the brain FDG-PET findings that in the early stage showed uptake in the medial temporal lobes and vermis of the cerebellum (anticipating by 4 months the cerebellar syndrome), and in the more advanced stage showed hypoactivity paralleling transient symptom stabilization and decreased pleocytosis after treatment with cyclophosphamide and corticosteroids (data not shown).
Discussion
We report the clinical, neuroimaging (MRI and FDG-PET) and immunological studies undertaken in seven patients with subacute limbic encephalitis for which after an initial comprehensive evaluation no aetiology had been identified. The most remarkable finding was that six patients had antibodies that shared the property of intense reactivity with areas of dense dendritic network or synaptic-enriched regions in the neuropil of hippocampus or cerebellum, and one patient had antibodies to intraneuronal antigens. Regardless of the identity of the antigens (in one case VGKCs), these findings are important because all patients with neuropil antibodies had remarkable clinical and neuroimaging improvement with treatment. In contrast, the patient with antibodies to intraneuronal antigens had symptom progression and worsening neuroimaging refractory to most treatments, similar to what has been reported in most patients with paraneoplastic antibodies to intraneuronal antigens (Alamowitch et al., 1997; Gultekin et al., 2000).
The fact that five patients with novel neuropil antibodies were identified at our institution in a 24 month period suggests that these disorders are frequent. In the same time period, we have seen 13 additional patients with limbic encephalitis: eight paraneoplastic (five associated with small cell lung carcinoma, two thyroid cancer and one testicular cancer), one related to Sjögren's syndrome, three HSV encephalitis and one recently diagnosed with VGKC antibodies for whom no follow-up is yet available. There are three possible explanations for why most of the neuropil antibodies have not been identified previously. First, in conventional immunohistochemistry as used for classical paraneoplastic antibodies, the neuropil antibodies are negative or show faint reactivity that is difficult to interpret. Secondly, some investigators use rat cerebellum to determine the presence of antibodies; this approach would have missed the antibodies of patients 3 and 4. Thirdly, while the classical paraneoplastic antibodies remain detectable for many months or years (Rojas et al., 2000; Llado et al., 2004), the titres of the neuropil antibodies decrease or become undetectable as the patient's symptoms improve or resolve.
A critical step in identifying the neuropil antibodies is the use of non-perfused rat brain tissue fixed for an extended time (10 days) with PFA; the reactivity is less optimal with PFA-perfused rat tissue and absent or poorly detected with other fixatives. This method also allows detection of the recently reported VGKC antibodies in patients with limbic encephalitis or Morvan's syndrome (Buckley et al., 2001; Liguori et al., 2001). Sera of all 11 control patients with VGKC antibodies produced an identical pattern of reactivity that was identified in only one of our patients (case 1); these 12 sera abrogated the reactivity of each other in an immunocompetition assay, indicating that they targeted the same epitopes. In contrast, the sera of the other five patients with neuropil antibodies (cases 2–6) produced patterns of reactivity different from that seen with VGKC antibodies, and neither competed with them nor co-localized in neuronal double labelling assays, verifying that they targeted different antigens.
In addition to neuropil antibodies, two patients had other antibodies reacting with a subset of neurons and glial cells (no. 2) and with GAD (no. 6). The neuropil antibodies of these two patients competed for the same epitopes and therefore correlated with their common syndrome of limbic dysfunction. In patient 6, the presence of GAD antibodies correlated with the recent onset of diabetes mellitus and stiff-person syndrome that manifested while the multifocal encephalitis with limbic dysfunction was resolving. After corticosteroids, the serum titre of each antibody paralleled the corresponding syndrome; the neuropil antibodies decreased significantly with striking improvement of the encephalitis, whereas the GAD antibodies remained elevated, with partial but steroid-dependent improvement of the muscle spasms and rigidity.
When compared with the CSF findings reported in two studies of patients with encephalitis and VGKC antibodies (combined frequency of pleocytosis 31%; median white blood cells 6/µl; intrathecal synthesis of antibodies 0%) (Thieben et al., 2004; Vincent et al., 2004), the CSF of the five patients with novel neuropil antibodies had more frequent and intense abnormalities (pleocytosis 100%; median white blood cells 44/µl; intrathecal synthesis of antibodies 100%), suggesting an ongoing immunological process within the CNS. In agreement with those studies, the only patient of our series without CSF pleocytosis was case 1 who harboured VGKC antibodies and developed limbic encephalitis and SIADH-like hyponatraemia that resolved with immunotherapy.
The antibody that showed the most predominant reactivity with hippocampus was identified in patient 4 who developed acute onset personality change, extreme agitation, central hypoventilation, seizures, and brain MRI and FDG-PET findings compatible with encephalitis of the limbic system and other brain regions. CT of the abdomen and pelvis demonstrated an ovarian mass with fat and calcium, suggesting a dermoid tumour (benign teratoma) that was not removed. Subsequently, we have identified the same antibody in serum and CSF of two other patients with a similar syndrome and pathologically confirmed ovarian teratoma. The encephalitis of these three patients was subacute and severe; two recovered, but one died as a result of the disorder.
To our knowledge, this is the first study that compares brain MRI and FDG-PET at symptom presentation in a series of patients with antibody-associated encephalitis, although individual cases have been reported previously (Kassubek et al., 2001; Dadparvar et al., 2003; Dalmau et al., 2004; Lee et al., 2004; Scheid et al., 2004). Both techniques complemented each other, but their findings overlapped in only 50% of the patients. Combining brain MRI and FDG-PET, all seven patients had predominant abnormalities in the temporal lobes that correlated with the symptoms of limbic dysfunction. Additionally, the FDG-PET more often than the MRI revealed abnormalities in the brainstem, cerebral cortex or cerebellum, some of them clinically silent. These findings, coupled with the presence of pleocytosis in the patients' CSF, probably represent inflammatory changes in the abnormal brain regions. Prior clinicopathological studies support this concept by the demonstration of frequent inflammatory infiltrates distant from the limbic system, usually in the brainstem and cerebellum (see review of 41 autopsy studies by Gultekin et al., 2000). Overall, the neuroimaging data suggest that in the appropriate context (i.e. patients with encephalopathy suspected to be autoimmune, EEG without epileptic activity and normal MRI), FDG-PET helps to demonstrate focal areas of encephalitis and may forewarn of serious complications (i.e. the patient with FDG hyperactivity in the brainstem developed central hypoventilation). Conversely, brain MRI and FDG-PET can be normal or have minimal changes in patients with limbic encephalitis and CSF inflammatory abnormalities, as occurred in patient 2.
The neuroimaging follow-up also correlated with the clinical outcome. Five of six patients with clinical improvement had MRI or FDG-PET normalization or improvement, and one had stable small T2 hyperintensities in the frontal lobes of unclear significance. The most remarkable correlation was found in patient 7 whose chronic disorder allowed long-term follow-up. This patient was monitored with eight CSF studies, seven brain MRIs and three FDG-PETs obtained over 22 months until his death. The correlation of findings, with increased FDG activity in the hippocampi and cerebellar vermis (anticipating gait ataxia by 4 months), and decreased FDG activity paralleling a transient slowing of symptom progression and CSF improvement, suggest pursuing further studies to assess the utility of FDG-PET for prognosis and treatment planning of paraneoplastic disorders.
As far as treatment is concerned, six patients received immunotherapy [corticosteroids, plasma exchange or intravenous immunoglobulin G (IVIg)] and four of five cases had tumour resection. Excluding the patient with VGKC antibodies, the efficacy of immunotherapy was confirmed in four instances where only immunosuppression was used (one patient without tumour, one whose dermoid tumour was not treated and two at neurological relapse).
Based on the current findings, we propose a classification of antibody-associated encephalitis with implications for prognosis and treatment (Table 3). One group comprises patients with antibodies to intracellular antigens including most of the previously characterized paraneoplastic antibodies; patient 7 would pertain to this group (Honnorat et al., 1996; Alamowitch et al., 1997; Dalmau et al., 2004). A common feature of all these antibodies is that they react with most neurons of the CNS without regional specificity. Cytotoxic T-cell mechanisms appear to play a major pathogenic role (Benyahia et al., 1999; Bernal et al., 2002), and clinical experience indicates that the neurological and oncological outcomes are usually poor, except for a subgroup of young patients with anti-Ma2 encephalitis and testicular tumours (Graus et al., 2001; Dalmau et al., 2004). The major concern in managing these disorders is the prompt treatment of the tumour and immunotherapy mainly targeting cytotoxic T-cells (Bataller and Dalmau, 2003).
Clinical and immunological subgroups of limbic encephalitis
| Neuronal antibodies: Hu, Ma2, CV2/CRMP5, atypical | VGKC antibodies | ‘Novel neuropil antibodies’ | |
|---|---|---|---|
| Hippocampal specificity of antibodies | No; antibodies react with neurons of any part of the neuraxis. | Mild; all patients with similar pattern of antibody reactivity | Intense; different patterns (some with pure limbic reactivity) |
| CSF abnormalities (pleocytosis, elevated IgG index, oligoclonal bands) | Frequent | Infrequent (normal CSF or with mild abnormalities) | Frequent |
| Intrathecal synthesis of antibodies | Frequent | Infrequent/absent | Frequent |
| Hyponatraemia | No (except some patients with SCLC) | Frequent | No |
| Clinical phenotypes other than limbic encephalitis | Several according to type of antibody (Bataller and Dalmau, 2004) | Neuromyotonia; Morvan syndrome (Vincent et al., 2004) | Prominent behavioural abnormalities, psychiatric symptoms and seizures; central hypoventilation may occur* |
| Brain MRI | Frequent medial temporal lobe FLAIR/T2 hyperintensities (classical findings) | Frequent classical findings | Infrequent classical findings, but frequent temporal lobe involvement |
| Tumour association | SCLC, non-SCLC, testicular tumours, thymoma, other | Infrequent: SCLC, thymoma | Frequent: teratoma, thymoma* |
| Response to treatment (tumour and/or immunosuppression) | Rare; except for patients with testicular tumours and Ma2 encephalitis | Frequent (corticosteroids, IVIg, plasma exchange) | Frequent (tumour and/or corticosteroids, IVIg, plasma exchange) |
| Clinical course | Progressive until stabilization or death (Hu, CV2/CRMP5); relapses are rare | Relapses may occur and are treatable | Relapses may occur and are treatable |
| Outcome of antibody titres | Usually detectable for months or years | Decrease or disappear in months | Decrease or disappear in months |
| Neuronal antibodies: Hu, Ma2, CV2/CRMP5, atypical | VGKC antibodies | ‘Novel neuropil antibodies’ | |
|---|---|---|---|
| Hippocampal specificity of antibodies | No; antibodies react with neurons of any part of the neuraxis. | Mild; all patients with similar pattern of antibody reactivity | Intense; different patterns (some with pure limbic reactivity) |
| CSF abnormalities (pleocytosis, elevated IgG index, oligoclonal bands) | Frequent | Infrequent (normal CSF or with mild abnormalities) | Frequent |
| Intrathecal synthesis of antibodies | Frequent | Infrequent/absent | Frequent |
| Hyponatraemia | No (except some patients with SCLC) | Frequent | No |
| Clinical phenotypes other than limbic encephalitis | Several according to type of antibody (Bataller and Dalmau, 2004) | Neuromyotonia; Morvan syndrome (Vincent et al., 2004) | Prominent behavioural abnormalities, psychiatric symptoms and seizures; central hypoventilation may occur* |
| Brain MRI | Frequent medial temporal lobe FLAIR/T2 hyperintensities (classical findings) | Frequent classical findings | Infrequent classical findings, but frequent temporal lobe involvement |
| Tumour association | SCLC, non-SCLC, testicular tumours, thymoma, other | Infrequent: SCLC, thymoma | Frequent: teratoma, thymoma* |
| Response to treatment (tumour and/or immunosuppression) | Rare; except for patients with testicular tumours and Ma2 encephalitis | Frequent (corticosteroids, IVIg, plasma exchange) | Frequent (tumour and/or corticosteroids, IVIg, plasma exchange) |
| Clinical course | Progressive until stabilization or death (Hu, CV2/CRMP5); relapses are rare | Relapses may occur and are treatable | Relapses may occur and are treatable |
| Outcome of antibody titres | Usually detectable for months or years | Decrease or disappear in months | Decrease or disappear in months |
Based on two additional cases of patients with teratoma referred from other institutions and tested for neuropil antibodies. SCLC = small cell lung carcinoma; IVIg = intravenous immunoglobulin G.
Clinical and immunological subgroups of limbic encephalitis
| Neuronal antibodies: Hu, Ma2, CV2/CRMP5, atypical | VGKC antibodies | ‘Novel neuropil antibodies’ | |
|---|---|---|---|
| Hippocampal specificity of antibodies | No; antibodies react with neurons of any part of the neuraxis. | Mild; all patients with similar pattern of antibody reactivity | Intense; different patterns (some with pure limbic reactivity) |
| CSF abnormalities (pleocytosis, elevated IgG index, oligoclonal bands) | Frequent | Infrequent (normal CSF or with mild abnormalities) | Frequent |
| Intrathecal synthesis of antibodies | Frequent | Infrequent/absent | Frequent |
| Hyponatraemia | No (except some patients with SCLC) | Frequent | No |
| Clinical phenotypes other than limbic encephalitis | Several according to type of antibody (Bataller and Dalmau, 2004) | Neuromyotonia; Morvan syndrome (Vincent et al., 2004) | Prominent behavioural abnormalities, psychiatric symptoms and seizures; central hypoventilation may occur* |
| Brain MRI | Frequent medial temporal lobe FLAIR/T2 hyperintensities (classical findings) | Frequent classical findings | Infrequent classical findings, but frequent temporal lobe involvement |
| Tumour association | SCLC, non-SCLC, testicular tumours, thymoma, other | Infrequent: SCLC, thymoma | Frequent: teratoma, thymoma* |
| Response to treatment (tumour and/or immunosuppression) | Rare; except for patients with testicular tumours and Ma2 encephalitis | Frequent (corticosteroids, IVIg, plasma exchange) | Frequent (tumour and/or corticosteroids, IVIg, plasma exchange) |
| Clinical course | Progressive until stabilization or death (Hu, CV2/CRMP5); relapses are rare | Relapses may occur and are treatable | Relapses may occur and are treatable |
| Outcome of antibody titres | Usually detectable for months or years | Decrease or disappear in months | Decrease or disappear in months |
| Neuronal antibodies: Hu, Ma2, CV2/CRMP5, atypical | VGKC antibodies | ‘Novel neuropil antibodies’ | |
|---|---|---|---|
| Hippocampal specificity of antibodies | No; antibodies react with neurons of any part of the neuraxis. | Mild; all patients with similar pattern of antibody reactivity | Intense; different patterns (some with pure limbic reactivity) |
| CSF abnormalities (pleocytosis, elevated IgG index, oligoclonal bands) | Frequent | Infrequent (normal CSF or with mild abnormalities) | Frequent |
| Intrathecal synthesis of antibodies | Frequent | Infrequent/absent | Frequent |
| Hyponatraemia | No (except some patients with SCLC) | Frequent | No |
| Clinical phenotypes other than limbic encephalitis | Several according to type of antibody (Bataller and Dalmau, 2004) | Neuromyotonia; Morvan syndrome (Vincent et al., 2004) | Prominent behavioural abnormalities, psychiatric symptoms and seizures; central hypoventilation may occur* |
| Brain MRI | Frequent medial temporal lobe FLAIR/T2 hyperintensities (classical findings) | Frequent classical findings | Infrequent classical findings, but frequent temporal lobe involvement |
| Tumour association | SCLC, non-SCLC, testicular tumours, thymoma, other | Infrequent: SCLC, thymoma | Frequent: teratoma, thymoma* |
| Response to treatment (tumour and/or immunosuppression) | Rare; except for patients with testicular tumours and Ma2 encephalitis | Frequent (corticosteroids, IVIg, plasma exchange) | Frequent (tumour and/or corticosteroids, IVIg, plasma exchange) |
| Clinical course | Progressive until stabilization or death (Hu, CV2/CRMP5); relapses are rare | Relapses may occur and are treatable | Relapses may occur and are treatable |
| Outcome of antibody titres | Usually detectable for months or years | Decrease or disappear in months | Decrease or disappear in months |
Based on two additional cases of patients with teratoma referred from other institutions and tested for neuropil antibodies. SCLC = small cell lung carcinoma; IVIg = intravenous immunoglobulin G.
The other two groups comprise patients with antibodies that react with the brain neuropil (sparing the cytoplasm and nuclei of neurons), and include VGKC antibodies and the collectively termed ‘novel neuropil antibodies’ (Table 3). Preliminary studies suggest that the neuropil antigens localize on the cell surface of neurons and dendrites; therefore, partial co-localization was seen with synaptophysin and spinophilin. In some patients (nos 2 and 6), the autoantigens appeared intensely expressed in the segments that co-localized with spinophilin. It is tempting to speculate that the immune response caused hippocampal dendritic pathology, an abnormality that with reduced spinophilin has been reported recently in patients with schizophrenia and mood disorders (Law et al., 2004).
Overall, the autoantigens of the novel neuropil antibodies are more concentrated in the hippocampus than the VGKCs. We have noted distinctive intrahippocampal variability in the patterns of staining, but the intense labelling of the neuropil is common to all, and characteristic enough to allow their identification. Because the radioimmunoassay used in the clinical analysis of VGKC antibodies identifies a limited number of subunits (Kv1.1, Kv1.2 and Kv1.6), it is reasonable to speculate that antibodies to other subunits, K(+) channel families or ion channels may also associate with encephalitis (Vincent et al., 2004). In support of this theory, the antibodies of one patient (case 4) produced a pattern of reactivity that perfectly co-localized with that from another patient whose serum served to isolate EFA6, a protein involved in hippocampal dendritic development (Sakagami et al., 2004; manuscript in preparation). Although the antibodies of patient 4 did not react with recombinant EFA6, the findings suggest that they target a related protein. A recent study demonstrates that at the plasma membrane EFA6 complexes with ARF6 and TWIK1, a member of a new family of K(+) channels involved in neuronal excitability (Decressac et al., 2004).
The common phenotype associated with all the novel neuropil antibodies includes dominant behavioural and psychiatric symptoms (often obscuring the associated short-term memory deficits), seizures and brain MRI abnormalities that are less frequently restricted to the hippocampus than in patients with classical limbic encephalitis. Patients with the novel antibodies are more likely to have CSF inflammatory findings and underlying tumours (thymoma and teratoma) than patients with VGKC antibodies, and do not develop SIADH-like hyponatraemia. In contrast to patients with classical paraneoplastic antibodies, the encephalitis of these patients improves with immunotherapy and, if present, treatment of the associated tumour. Future studies will focus on the individual characterization of the autoantigens and possible subsyndromes that may associate with each autoimmunity or underlying tumour.
These authors contributed equally to this work
We wish to thank Ms Adriana Zupa-Fernandez and Ms Margaret Maronski for their excellent technical assistance, Dr Francesc Graus (Hospital Clinic, University of Barcelona, Spain) and Dr Luis Bataller (Hospital La Fe, University of Valencia, Spain) for critical review of the manuscript, and Dr Graus (Barcelona, Spain), Dr Karen C. Bloch (Vanderbilt University, TN, USA) and D. Amelia Evoli (Catholic University, Rome, Italy) for providing sera of 10 patients with VGKC antibodies. Supported in part by RO1CA89054 and RO1CA107192 (J.D).
References
Alamowitch S, Graus F, Uchuya M, Reñé R, Bescansa E, Delattre JY. Limbic encephalitis and small cell lung cancer—clinical and immunological features.
Bataller L, Dalmau J. Paraneoplastic disorders of the central nervous system: update on diagnostic criteria and treatment.
Bataller L, Rosenfeld MR, Graus F, Vilchez JJ, Cheung NK, Dalmau J. Autoantigen diversity in the opsoclonus–myoclonus syndrome.
Benyahia B, Liblau R, Merle-Beral H, Tourani JM, Dalmau J, Delattre JY. Cell-mediated autoimmunity in paraneoplastic neurological syndromes with anti-Hu antibodies.
Bernal F, Graus F, Pifarre A, Saiz A, Benyahia B, Ribalta T. Immunohistochemical analysis of anti-Hu-associated paraneoplastic encephalomyelitis.
Bien CG, Schulze-Bonhage A, Deckert M, Urbach H, Helmstaedter C, Grunwald T, et al. Limbic encephalitis not associated with neoplasm as a cause of temporal lobe epilepsy.
Buchhalter JR, Dichter MA. Electrophysiological comparison of pyramidal and stellate nonpyramidal neurons in dissociated cell culture of rat hippocampus.
Buckley C, Oger J, Clover L, Tuzun E, Carpenter K, Jackson M, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis.
Dadparvar S, Anderson GS, Bhargava P, Guan L, Reich P, Alavi A, et al. Paraneoplastic encephalitis associated with cystic teratoma is detected by fluorodeoxyglucose positron emission tomography with negative magnetic resonance image findings.
Dalmau J, Furneaux HM, Cordon-Cardo C, Posner JB. The expression of the Hu (paraneoplastic encephalomyelitis/sensory neuronopathy) antigen in human normal and tumor tissues.
Dalmau J, Graus F, Villarejo A, Posner JB, Blumenthal D, Thiessen B, et al. Clinical analysis of anti-Ma2-associated encephalitis.
Decressac S, Franco M, Bendahhou S, Warth R, Knauer S, Barhanin J, et al. ARF6-dependent interaction of the TWIK1 K+ channel with EFA6, a GDP/GPT exchange factor for ARF6.
Graus F, Keime-Guibert F, Reñé R, Benyahia B, Ribalta T, Ascaso C, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients.
Gultekin SH, Rosenfeld MR, Voltz R, Eichen J, Posner JB, Dalmau J. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients.
Honnorat J, Antoine JC, Derrington E, Aguera M, Belin MF. Antibodies to a subpopulation of glial cells and a 66 kDa developmental protein in patients with paraneoplastic neurological syndromes.
Kassubek J, Juengling FD, Nitzsche EU, Lucking CH. Limbic encephalitis investigated by 18FDG-PET and 3D MRI.
Law AJ, Weickert CS, Hyde TM, Kleinman JE, Harrison PJ. Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines.
Lee BY, Newberg AB, Liebeskind DS, Kung J, Alavi A. FDG-PET findings in patients with suspected encephalitis.
Liguori R, Vincent A, Clover L, Avoni P, Plazzi G, Cortelli P, et al. Morvan's syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels.
Llado A, Mannucci P, Carpentier AF, Paris S, Blanco Y, Saiz A, et al. Value of Hu antibody determinations in the follow-up of paraneoplastic neurologic syndromes.
Pozo-Rosich P, Clover L, Saiz A, Vincent A, Graus F. Voltage-gated potassium channel antibodies in limbic encephalitis.
Rojas I, Graus F, Keime-Guibert F, Reñé R, Delattre JY, Ramon JM, et al. Long-term clinical outcome of paraneoplastic cerebellar degeneration and anti-Yo antibodies.
Sakagami H, Matsuya S, Nishimura H, Suzuki R, Kondo H. Somatodendritic localization of the mRNA for EFA6A, a guanine nucleotide exchange protein for ARF6, in rat hippocampus and involvement in dendritic formation.
Scheid R, Lincke T, Voltz R, von Cramon DY, Sabri O. Serial 18F-fluoro-2-deoxy-d-glucose positron emission tomography and magnetic resonance imaging of paraneoplastic limbic encephalitis.
Thieben MJ, Lennon VA, Boeve BF, Aksamit AJ, Keegan M, Vernino S. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody.
Vincent A, Buckley C, Schott JM, Baker I, Dewar BK, Detert N, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis.
Author notes
1Department of Neurology and 2Division of Nuclear Medicine, Department of Radiology, Hospital of the University of Pennsylvania, Philadelphia, PA and 3Department of Neurosurgery, Medical College of Georgia, Augusta, GA, USA
4Present address: Department of Radiology, University of Iowa, Iowa City, IA, USA
5Present address: Department of Neurology, University of California, Los Angeles, CA, USA


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}