
Abstract
Objective
To report ‘posterior sympathetic ophthalmia’ in North Indian population as an early manifestation of sympathetic ophthalmia.
Methods
Forty consecutive patients with a diagnosis of sympathetic ophthalmia seen between 1989 and 2004 at our centre were studied for their clinical presentation and disease course. All received systemic corticosteroids and 12 patients, in addition, also received immunosuppressive agents.
Results
There were 28 male and 12 female patients with a median age of 29.4 years. In 22 of the 40 sympathizing eyes, the only presenting sign was the fundus lesions without any associated anterior segment inflammation. Only four eyes showed classically described granulomatous anterior uveitis at presentation. The fundus lesions predominately included exudative retinal detachment (29 eyes), yellowish-white mid-peripheral lesions (10 eyes), optic disc oedema (15 eyes), vasculitis (three eyes), and peripapillary choroidal neovascular membrane (two eyes). Over a median follow-up of 5.2 years, recurrences were seen in 12 of 40 (30%) eyes and were mainly in the anterior segment. Over a median follow-up of 5.2 years, a final visual acuity of 20/40 or better could be achieved in 29/36 (80.5%) eyes.
Conclusion
In the early stage, sympathetic ophthalmia may present only in the posterior segment without any associated anterior segment inflammation and carries a good visual prognosis. Anterior segment inflammation, however, maybe seen during recurrences.
Similar content being viewed by others

Introduction
Sympathetic ophthalmia is defined as bilateral granulomatous panuveitis that follows penetrating trauma or surgery in one eye.1, 2 The classical description of signs include granulomatous mutton fat keratic precipitates, anterior chamber, and vitreous inflammation with or without yellow–white lesions in the retinal periphery. Other fundus lesions like retinal detachment; papillitis, optic atrophy, and vasculitis are reported uncommonly and are generally seen in conjunction with anterior segment inflammation.2, 3 We observed that the classical presentation of the disease was rather uncommon in our population, majority of whom presented early with only fundus lesions, associated with minimal or no anterior segment inflammation. The atypical form of sympathetic ophthalmia with only posterior segment findings has been identified and termed as ‘posterior sympathetic ophthalmia’ in a report of two cases by Mcpherson et al.4 The present series aims to report the long-term follow-up and the disease course of 40 patients with posterior sympathetic ophthalmia.
Materials and methods
We studied 40 consecutive patients of sympathetic ophthalmia seen in the Uveitis Clinic of our tertiary care referral institute between June 1989 and August 2004. All except four patients were seen during the acute phase of the disease. All patients had a detailed history with complete ocular examination. In addition, the sympathizing eyes underwent investigations such as fundus fluorescein angiography (FFA) in 34 eyes, B-scan ultrasonography (USG) in 19 eyes, indocyanine green angiography (ICG) in eight eyes, and optical coherence tomography (OCT) in 10 eyes. Enucleation and histopathology of the exciting eye was carried out in 10 eyes.
The diagnosis of sympathetic ophthalmia was made if the patient had two or more of the following in the sympathizing eye, with history of trauma or surgery preceding the onset of uveitis:
-
1
Anterior segment inflammation
-
2
Posterior segment showing nummular chorioretinal lesions, exudative retinal detachment, optic disc oedema, vasculitis, and sunset glow fundus
-
3
Diffuse choroidal thickening in the posterior pole on USG
-
4
Multifocal pinpoint areas of hypo- or hyper fluorescence with late dye pooling, optic nerve head staining on fundus fluorescein angiography (FFA), and/or suggestive indocyanine green angiography (ICG), and/or multifocal serous retinal detachment on optical coherence tomography (OCT)
-
5
Characteristic histopathology of the exciting eye.
All patients seen in the acute phase of disease received systemic corticosteroids with a starting dose of 1.5–2 mg/kg body weight that was tapered slowly to a maintenance dose between 5 and 10 mg/day over the next 4–6 months, which was then continued for a period of 2–5 years. Of these, 20 patients received intravenous methylprednisolone in a dosage of 1 gm/day for the first 3–5 days and then continued on oral corticosteroids. In case of recurrences, the dose of oral corticosteroids was increased to 1.5–2 mg/kg body weight if the patient had new fundus lesions or severe anterior segment inflammation. The maintenance dose, this time, was not reduced below the level at which the recurrence had occurred. Topical corticosteroids and cycloplegics were given as per requirement. Twelve patients received immunosuppressive agents. All four patients seen in the chronic recurrent form received oral azathioprine in a dosage of 100–150 mg/day to begin with, which was gradually reduced over the next 6–12 months. Additionally, eight patients who initially responded to but were unable to continue corticosteroids due to either side effects (five patients) or recurrence (three patients) also received immunosuppressive therapy. Intravenous cyclophosphamide (750 mg/ g/week every 3–4 weeks with a maximum of seven doses) was given for managing acute recurrences while oral azathioprine (100–150 mg/day) or methotrexate (7.5–10.0 mg/day) was used as steroid sparer. The follow-up ranged from 2 to 14 years (median 5.2 years).
Results
There were 28 male and 12 female patients with an age ranging between 6 and 80 years (median 29.4 years). Trauma was the most common inciting event seen in 30 exciting eyes, of which 26 eyes had open globe injury and four had closed globe injury with no clinical evidence of full thickness or occult perforation on clinical examination alone. Ten exciting eyes had undergone surgery; the commonest being cataract surgery in five eyes (intracapsular cataract extraction in two and extracapsular cataract surgery in three); scleral buckle with subretinal fluid drainage in two eyes; pars-plana-vitrectomy in two eyes, and glaucoma filtration surgery in one eye. The interval between injury to presentation varied between 10 days to 6 years (mean 9.05±1.02 months).
The presenting visual acuity in the sympathizing eyes ranged from 20/200 to 20/40. Of the 40 sympathizing eyes, only 18 showed associated anterior segment inflammation, of which only four had granulomatous anterior uveitis. The remaining 14 eyes had nongranulomatous anterior uveitis that was mild in 10 and moderate in four eyes. Only three of the 18 eyes showed development of posterior synichae. Significantly, 22 of the 40 eyes had no associated anterior segment inflammation.
Fundus examination of the 36 eyes presenting in the acute phase showed vitritis in 30 eyes; exudative retinal detachment (29 eyes); yellow–white mid-peripheral lesions (14 eyes); optic disc oedema (15 eyes); vasculitis (three eyes) and peripapillary choroidal neovascular membrane (two eyes). Four eyes that presented in chronic phase showed sunset glow fundus and peripheral scars.
In the acute phase, the most common finding seen on FFA was initial hypo- to late hyperfluorescent dot lesions with dye pooling in 34 eyes. Only two eyes showed initial pinpoint hyperfluorescent dot lesions with progressive hyperfluorescence in the late phases. Associated optic disc hyperfluorescence was seen in 15 eyes, retinal vessel wall staining in three eyes, and peripapillary choroidal neovascular membrane in two eyes. All four eyes first seen during the chronic phase showed mottled fluorescence due to pigmentary disturbances. Diffuse choroidal thickening in the posterior pole was seen on ultrasonography in 12 of 19 eyes where it was done. ICG was done in eight eyes and showed hypofluorescent lesions in the intermediate and late phase in three eyes, early hypofluorescent lesions with late iso/hyperfluorescence in three eyes, and diffuse choroidal hyperfluorescence in two eyes. OCT showed multiple serous retinal detachments in all the 10 eyes where it was done.
Histopathology of the 10 exciting eyes was suggestive of sympathetic ophthalmia and showed granulomatous non-necrotizing inflammation in all with eosinophilic infiltration in four eyes; retinal perivasculitis in one eye, and retinal infiltrate in one eye.
Over a median follow-up of 5.2 years, a final visual acuity of 20/40 or better could be achieved in 29 eyes. Recurrences were seen in 12 eyes. Of these 12 eyes that showed recurrences, seven eyes had shown no anterior segment inflammation at presentation. All these patients were on 10–30 mg of oral prednisolone at the time of recurrence. Of these, 10 eyes showed severe anterior granulomatous inflammation, with associated vitritis in seven eyes and optic disc oedema in one eye. Two eyes showed posterior segment recurrence only with reappearance of exudative retinal detachment. Seven of the 12 eyes developed significant posterior synichae despite intensive topical and systemic corticosteroids. Twenty-four eyes showed development of sunset-glow fundus at follow-up, while three showed development of poliosis.
Subset analysis of 22 eyes that were seen with only posterior pole disease showed that the visual acuity in all these eyes was 20/100 or better at the time of initial presentation. Fundus examination of the 22 eyes presenting in the acute phase showed vitritis in all the 22 eyes; exudative retinal detachment (17 eyes); yellow–white mid-peripheral lesions (six eyes); optic disc oedema (10 eyes), and vasculitis (one eye). Three eyes had mild vitritis and optic disc oedema as the only presenting signs. All these patients were either hospitalized/or examined everyday from the day of diagnosis and were treated with systemic corticosteroids as mentioned. None of the eyes showed progression of lesions or the development of anterior segment inflammation during this episode. However, 19 of these 22 patients were receiving topical corticosteroids (3–6 times/day). Only three patients with mild vitritis and optic disc oedema were not receiving any topical corticosteroids. Seven of these 22 eyes had recurrences while on the tapering dose of 10–30 mg of oral prednisolone. At the time of recurrence, five eyes showed severe anterior granulomatous inflammation associated with vitritis in four eyes and optic disc oedema in one eye. Three of these five patients received intravenous cyclophosphamide while the remaining two received increased dosage of systemic corticosteroids. Two eyes showed posterior segment recurrence only with reappearance of exudative retinal detachment with no associated anterior segment inflammation and were managed with increased dosage of systemic corticosteroids. There was no difference in the outcome (final visual acuity and number of recurrences) between patients treated with immunosuppressive or corticosteroids over the follow-up. None of the three eyes that had only mild vitritis and optic disc oedema as initial presentation had recurrences over follow-up. Eighteen of 29 eyes that could achieve a final visual acuity of 20/40 had no anterior segment inflammation at presentation.
Case report
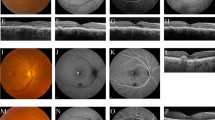
A-27-year-old-man underwent pars-plana vitrectomy in his left eye for combined retinal detachment. His postoperative best-corrected visual acuity in the left eye was 20/400. After 2 months, he complained of blurred vision in the right eye of 4 days duration. His best-corrected visual acuity in the right eye was 20/40. There was no anterior segment inflammation and vitreous showed 1+ cells. Right eye fundus showed serous retinal detachment with hypofluorescent lesions in the early phase and dye pooling in late phase of fluorescein angiography (Figure 1). Ultrasound B scan showed diffuse choroidal thickening in the posterior pole and OCT showed multifocal serous detachment. The left eye fundus showed an attached retina with no evidence of exudative detachment or disc oedema (Figure 2). He was diagnosed as sympathetic ophthalmia and received 1 gm/day of intravenous methyl prednisolone for 3 days followed by oral prednisolone of 1.5 mg/kg/day. After 48 h, his best-corrected visual acuity improved to 20/30 in the right eye and OCT showed decrease in the subretinal fluid. Over 3 years follow-up, during which he continued to receive 10 mg of prednisolone, the patient maintained a visual acuity of 20/20 in the right and 20/200 in the left eye with development of sunset glow fundus (Figure 3).

Fundus photograph R/E (a) showing clear media with serous retinal detachment in the posterior pole. Fundus fluorescein angiogram R/E (b–d) shows hypofluorescent choroidal lesions in the early phase and dye pooling in late phase.

Fundus photograph L/E (exciting eye) shows post pars plana vitrectomy attached retina.

Fundus photograph R/E (a) and L/E (b) 3 years later, showing sunset glow fundus.
Discussion
Ever since its first description by Mackenzie in 1840,5 who described the disease as ‘inflammation involving the whole of the internal structures of eyeball’, few atypical forms of the disease confined only to the posterior segment have been described.4, 6 The predominant posterior segment involvement as was seen in our series could represent either an atypical disease or conversely an early stage of the same disease. It is more likely to be the latter one as the anterior segment inflammation maybe absent in the early cases. In earlier days, probably the diagnosis was delayed and that is how majority of the patients have been reported to have anterior segment inflammation at presentation. However, now we are better at recognizing it and thus, many cases will be diagnosed at an early stage when they have absent/minimal anterior segment inflammation.
Duke-Elder and Perkins7 defined sympathetic ophthalmia as a specific bilateral inflammation of the entire uveal tract that follows a perforating wound and has an insidious onset and progressive course with exacerbations. Further, he mentioned that the disease varies in its clinical presentations and may start in the anterior or posterior segment, usually the former. He classified the disease into three phases, namely (a) prodrome characterized by accommodation failure and transient myopia; (b) early stage with mild aqueous or vitreous inflammation (that might be so slight that it could pass unnoticed), peripheral choroiditis, papillitis, and retinal oedema, and (c) late stage is seen after a month or two of the onset and is characterized by plastic iridocyclitis with large flat nodules seen on the iris surface. None of the patients in our series presented in the prodrome stage, while 36 of the 40 patients were seen in the acute stage with mostly posterior segment findings. This presentation differed from the classical form where granulomatous anterior uveitis has been described to be the main presenting clinical feature.1, 2, 3 Woods8 reported posterior form of the disease in 5% of all cases of sympathetic ophthalmia and described that posterior form is invariably accompanied by late development of an anterior uveitis and emphasized that this was neither clinically nor pathologically different from the classical form. In a subsequent report, however, Mcpherson and Dalton4 reported two cases of histopathologically confirmed sympathetic ophthalmia where they did not find any advancement of posterior segment inflammation to the anterior segment. This could have been due to the use of oral corticosteroids in their patients. In the present series, however, we found that while 22 eyes had no anterior segment inflammation at presentation, seven developed severe granulomatous inflammation during recurrences despite receiving adequate systemic corticosteroids. The recurrences were seen when these patients were on low-dose maintenance oral corticosteroids, thus indicating that anterior granulomatous uveitis probably represents the recurrent stage of the same disease of which posterior segment presentation represents the early form.
The similarities between sympathetic ophthalmia and Vogt–Koyanagi–Harada (VKH) have been previously reported.9, 10, 11 A report from our department has shown gradual evolution of sympathetic ophthalmia to VKH-like syndrome with the development of extraocular manifestations including poliosis and vitiligo.9 The development of sunset glow fundus in the present series indicates the strong resemblance between sympathetic ophthalmia and VKH syndrome. We feel that sympathetic ophthalmia, like VKH disease, has distinct stages with an early stage showing predominantly posterior segment findings associated with minimal or no anterior segment inflammation. Like in VKH disease, granulomatous anterior segment inflammation is seen either during late stage when these cases are not treated in the early phase or at the time of recurrences. Of the 12 patients who showed recurrences, 10 had only anterior segment inflammation at the time of recurrence with no signs of fundus involvement. Only two patients showed recurrence confined to the posterior segment.
Alternately, the disease behaviour could be variable owing to geographic, cultural, or ethnic influences including clinical phenotypes.12, 13, 14 The severity of the disease has been reported to be associated with HLA-DRB1*04.12 Jaini et al15 reported unusually high heterogeneity in DR4-DQB1 haplotype in North Indian population. They reported HLA DRB1*0405 to comprise only 11% of all DR4-positive patients in our population. Unlike that reported in most populations, we did not find significant association of HLA-DRB1*04 in our patients of VKH disease, majority of whom have incomplete or probable disease with mild course and good prognosis (unpublished data). Although we have not carried out HLA typing for our sympathetic ophthalmia patients, this could probably too be contributing towards atypical presentation in our population.
In conclusion, the present series describes a series of patients who, we feel, represent an early form of the disease with only fundus changes like exudative retinal detachment, yellowish–white nodules, papillitis, vasculitis, or peripapillary choroidal neovascular membrane. There may or may not be associated anterior segment or vitreous inflammation. Significant anterior segment inflammation maybe seen during recurrences only. It is important to recognize the disease at this stage and treat it as sympathetic ophthalmia with good visual prognosis.
References
Towler HMA, Lightman S . Sympathetic ophthalmia. Int Ophthalmol Clin 1995; 35: 31–42.
Rao NA, Forster DJ, Spalton DJ . Sympathetic ophthalmia.In: Podos SM, Yanoff M (eds). The Uvea Uveitis and Intraocular Neoplasms, Vol. 2, Chapter 8. Mosby-Wolfe: USA, 1995, pp 8.10–8.13.
Nussenblatt RB . Sympathetic Ophthalmia. In: Nussenblatt RB, Whitcup SM (eds). Uveitis Fundamentals and Clinical Practice, 3rd ed., Chapter 22. Elsevier: USA, 2004, pp 311–323.
Mcpherson S, Dalton T . Posterior form sympathetic ophthalmia. Trans Am Ophthalmol Soc 1975; LXXIII: 251–263.
Mackenzie W . Treatise on the Diseases of the Eye, 3rd ed., Longmans: London, 1840, pp 523–534.
Muller-Hermelink HK, Mackiw EK, Daus W . Early stage of human sympathetic ophthalmia. Arch Ophthalmol 1984; 102: 1353–1357.
Duke-Elder S, Perkins ES . Sympathetic ophthalmitis. Diseases of the Uveal Tract, Vol. IX, Chapter III Henry Kimpton Publishers: London, 1977, pp 558–593.
Woods AC . Sympathetic ophthalmia. Arch Ophthal 1936; 19: 9–15.
Jain IS, Chander B . Sympathetic ophthalmia simulating Vogt–Koyanagi syndrome. Orient Arch Ophthal 1964; 2: 36–39.
Fine BS, Gilligan JH . The Vogt–Koyanagi syndrome. A variant of sympathetic ophthalmia: report of two cases. Am J Ophthalmol 1957; 43: 433–440.
Rao NA, Marak GE . Sympathetic ophthalmia simulating Vogt–Koyanagi–Harada's disease: a clinicopathologic study of four cases. Jpn J Ophthalmol 1983; 27: 506–511.
Kilmartin DJ, Wilson D, Liversidge J, Dick AD, Bruce J, Acheson RW et al. Immunogenetics and clinical phenotype of sympathetic ophthalmia in British and Irish patients. Br J Ophthalmol 2001; 85: 281–286.
Mann I . Bowmann Lecture: climate, culture and disease. Trans Ophthalmol Soc UK 1961; 81: 261–282.
Albert DM, Rohena R . A historical review of sympathetic ophthalmia and its epidemiology. Surv Ophthalmol 1989; 34: 1–14.
Jaini R, Kaur G, Mehra NK . Heterogeneity of HLA DRB1*04 and its associated haplotypes in North Indian population. Hum Immunol 2002; 63: 24–29.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gupta, V., Gupta, A. & Dogra, M. Posterior sympathetic ophthalmia: a single centre long-term study of 40 patients from North India. Eye 22, 1459–1464 (2008). https://doi.org/10.1038/sj.eye.6702927
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.eye.6702927
Keywords
This article is cited by
-
Clinical classification, visual outcomes, and optical coherence tomographic features of 48 patients with posterior sympathetic ophthalmia
Orphanet Journal of Rare Diseases (2022)
-
The clinical importance of uveomeningeal syndromes
Spektrum der Augenheilkunde (2021)
-
Intraocular surgery under adalimumab therapy in patients with refractory uveitis: a single center study of 23 eyes
Japanese Journal of Ophthalmology (2021)
-
Postsurgical sympathetic ophthalmia: retrospective analysis of a rare entity
International Ophthalmology (2018)
