
Abstract
Purpose
The EPIRET3 retinal prosthesis was implanted in six volunteers legally blind from retinitis pigmentosa (RP) and removed after 4 weeks. Two years later, these subjects were re-examined to investigate ocular side effects and potential changes to quality of life.
Methods
Vision-related quality of life was recorded using the NEI-VFQ-25 questionnaire. Clinical data including interval history, visual acuity, and intraocular pressure were obtained. Anterior and posterior segments of the study eyes were examined and photographed; this included fluorescein angiography and optical coherence tomography (OCT).
Results
Data from five patients could be analysed. Life-quality score was consistent with results obtained at baseline. No unexpected structural alteration could be found in the study eyes. A moderate epiretinal gliosis was present in areas where the epiretinal stimulator had been fixated using retinal tacks. Angiography revealed no leakage or neovascularisation; OCT showed no generalised increase of central retinal thickness.
Conclusions
Vision-related quality of life is low in patients suffering from end-stage RP. No further deterioration of life quality could however be detected within our monitoring period. Surgery was well tolerated by both patients and their eyes, without adverse events occurring during the follow-up period. Epiretinal gliosis is known to occur with retinal tacks, but seems of no major concern to the integrity of the study eyes. However, it may potentially interfere with functional aspects of active implants. Hence, alternative, possibly biochemical, fixation methods merit further research.
Similar content being viewed by others
Introduction
Despite ongoing research, no treatment has thus far been established that could alter the natural course of retinitis pigmentosa (RP), which may lead to severe visual impairment and eventually complete loss of vision.1, 2, 3 However, it has been shown that by electrical stimulation of retinal ganglion cells, phosphenes can be elicited in blind RP patients.4 This evidence encouraged the construction of an entirely intraocular retinal prosthesis, EPIRET3, which is equipped with electrodes to be placed on the retinal surface for epiretinal stimulation. We previously reported data from a pilot-study testing the EPIRET3 prosthesis in humans.5 The device had been implanted in six subjects legally blind from RP. Following functional tests over a period of 4 weeks, the implants were ultimately removed from the study eyes. For the evaluation of long-term safety, follow-up visits are a crucial part of clinical studies on active visual implants.6 Therefore, in this extension study of the EPIRET3 trial, we re-investigated the structural and functional integrity of the study eyes. In addition, data on vision-related quality of life that was obtained throughout the study was completed with measurements from the follow-up visit and analysed as a whole.
Methods
The EPIRET3 retinal prosthesis comprises extraocular and intraocular components. Extraocular components include a portable computer system to determine the stimulation patterns, a transmitter module, and a transmitter coil mounted on a spectacle frame. The intraocular implant contains a receiver coil for wireless acquisition of power and data from the transmitter via induction. The coil is contained within an intraocular lens together with two microchips, which are responsible for extracting the stimulation signal and generating the stimulation pulses. A flexible micro cable links this assembly to 25 stimulation electrodes. These electrodes are 100 μm in diameter and 25 μm in height; they are arranged in a hexagonal pattern on a polyimide foil. The carrier foil is equipped with small eyelets for fixation on the retinal surface using retinal tacks. Although the implant body is coated with parylene C to ensure biocompatibility, the electrodes are sputter coated with iridium oxide to maximise the charge-delivery capacity.
Implantation surgery was performed under general anaesthesia. Phacoemulsification of the lens was followed by a complete 20-gauge vitrectomy via the pars plana. Using the vitrectome, an excentric posterior capsulotomy was created. The receiver unit was inserted into the capsular bag through an 11-mm corneal incision. This incision was partially closed using single 10-0 nylon sutures. The micro cable and stimulator foil were then pushed through the remaining aperture and through the posterior lens capsule defect into the vitreous cavity. The electrodes were lowered onto the macular region by the progressive removal of a perfluorcarbon cushion. Subsequently, they were attached to the posterior pole by the use of titanium retinal tacks (‘Heimann’ Retina Tack, Geuder AG, Heidelberg, Germany). At the end of the surgery, the eye was filled with balanced salt solution.
Being considered experimental devices, by request of the ethics committee, the implants were not permitted to remain in the study eyes for periods exceeding 1 month. Hence, at the end of the acute testing period, the surgical steps described above were reversed to remove the devices. Briefly, the electrode array was loosened from its fixation; the retinal tacks however remained in situ, unless they were found to be loose. Subsequently, the implant was removed in one piece through the re-opened corneal incision.
Six patients had been included in the initial implantation trial. Of these, five patients attended the follow-up visit, which took place 27–29 months after implantation. One patient was deceased before this visit, because of breast cancer. At the time of implantation surgery, the five patients reviewed here had a mean age of 52.8 years (range 35–69 years). Visual acuity ranged from no light perception to hand movements. On average, these patients had been legally blind for 5 years (range 3–8 years) when the device was implanted. Common features of the study population as defined by the inclusion/exclusion criteria were absence of concomitant ocular disease, no history of intraocular surgery (except cataract surgery), and no other active implant.
Upon presentation, assessment of vision-related quality of life was performed first, using the NEI-VFQ-25.7 This questionnaire had been administered before implantation, as well as during the acute testing period and shortly after removal of the device. The global score was calculated as described by Papageorgiou et al.8 To assess changes to the mean global score during the course of the study, statistical analysis was performed using repeated measures ANOVA. Subsequently, an interval history was taken, visual acuity was measured, and intraocular pressure (IOP) measurements were performed using Goldmann applanation tonometry. Clinical evaluation of the anterior and posterior segments was done by slit-lamp biomicroscopy and indirect ophthalmoscopy. Photographs of the anterior segment and fundus were obtained and fluorescein angiography was performed following injection of 500 mg (100 mg/ml) sodium fluorescein (Fluorescein 10%, Alcon Pharma GmbH, Freiburg, Germany), using a monocular retina camera (FF 450plus, Carl Zeiss Meditec, Jena, Germany/EOS 5D, Canon Deutschland GmbH, Krefeld, Germany). Optical coherence tomography (OCT) scans were performed using the Stratus OCT (Carl Zeiss Meditec).
We certify that all applicable institutional and governmental regulations concerning the ethical use of human volunteers were followed during this research.
Results
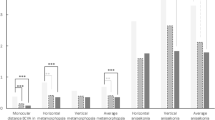
In Figure 1, an overview is given of the changes in overall scores obtained in the life-quality questionnaire throughout the entire study period. The NEI-VFQ-25 had been administered before, during, and shortly after the prosthesis was in place, as well as during the follow-up visit. Repeated measures ANOVA yielded a P-value of 0.63, suggesting that no statistically significant changes to the global life-quality score have occurred throughout the study period.

Evolution of composite scores obtained in the NEI-VFQ25 throughout the study. The maximum composite score that can be achieved using this instrument is 100. Measurement 1: 1–2 days before implantation. Measurement 2: 3 weeks after implantation. Measurement 3: 6 months after implantation. Measurement 4: 27–29 months after implantation. Error bars: ±1 SD.
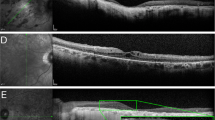
Table 1 provides a synopsis of preoperative findings. All patients showed typical fundus signs of RP before the interventions. Table 2 gives the corresponding morphological findings for each patient on the follow-up visit. The patients had been left aphakic following the explantation procedure. Their cornea was clear and the retina was attached in all cases. This was the case, regardless of whether or not retinal tacks had been left in place. Invariably, mild-to-moderate epiretinal gliosis was seen around the tack fixation sites. Figure 2 demonstrates the photographical aspect of the posterior segments at the time of follow-up. Compared with previous post-operative pictures, they suggest a stable situation without clinically relevant progression of gliosis. In Figure 3, results of the fluorescein angiography can be found. Here, alterations typically produced by RP could be discerned; however, no vascular abnormalities or leakage were seen. Mean retinal thickness at the posterior pole as determined by OCT was 162.5 μm (range 85–236 μm). In agreement with the funduscopic appearance and fluorescein angiography, OCT shows epiretinal gliosis particularly around tack fixation sites; however, intraretinal fluid was not detected (data not shown). No angiography or OCT data could be obtained for patient ES-01, as hypersensitivity prevented mydriasis. Data extracted from the patients’ history and individual findings are presented briefly in the following sections.

Fundus photographs taken at the follow-up visit. No significant change could be observed in comparison with images taken 6 months after explantation (see Roessler et al5).

Fluorescein angiography was performed during the follow-up visit. The top row shows images obtained at approximately 20 s; the bottom row shows images taken at 5–6 min. RP-related changes can be seen; however, vascular abnormalities or leakage are absent.
Patient AC-01
This patient had undergone treatment for a non-specific conjunctivitis in the study eye more than 1 year after the surgery, which had responded to topical medication without complications. At the time of follow-up, there were no complaints; ocular or general medications (or history) had not changed since the study interventions. The clinical examination did not reveal any signs of an adverse reaction. There was however a moderate degree of non-progressive epiretinal gliosis at the tack fixation site.
Patient AC-03
The patient reported a recent inflammatory reaction in the study eye due to the corneal sutures, which had still been in place. The sutures had been removed by the patient's ophthalmologist; dexamethasone/neomycine/polymyxine-B eye-drops (Isopto-Max, Alcon Pharma GmbH) were administered following the removal. At the time of follow-up, there was no ocular medication and the patient expressed no complaints. In the general medical history and medications, no changes or events were reported. The clinical examination was largely without any changes compared with prior visits. Upon fundoscopy, we observed a mild epiretinal gliosis at one of the tack sites. This gliosis had been previously recorded at 6 months after explantation; no further progression was seen.
Patient AC-04
This patient reported a slight decline in his residual visual perception on both sides since the interventions, but no further complaints. Otherwise, there were no changes in medication and no events concerning ocular or general medical history. Clinical examination revealed some minor choroidal atrophy within the area where the retinal tacks had been placed. On fluorescein angiography, marked central atrophy of the retinal pigment epithelium becomes evident, which was however present before implantation. In combination with some additional atrophy resulting from laser photocoagulation at the posterior pole, a heterogeneous image is formed which may resemble mild leakage. A similar picture was however seen in the preoperative angiographies and was interpreted as an exposure artefact, a notion that is supported also by the OCT data obtained.
Patient AC-05
In this patient, removal of the implant had caused a retinal break, which was treated with laser coagulation and silicone oil tamponade. Three months later, the silicone oil had been removed. Since then, the patient had noted a slight decline in visual perception in the study eye. No additional ocular or general complaints were reported. Upon clinical examination, the epiretinal gliosis, which had been reported around the tack fixation site already before silicone oil removal, was found to be stable and non-progressive.
Patient ES-01
The patient reported no additional ocular complaints since the beginning of the trial. Remaining visual perception had remained stable. There had been no need for any intervention in the study eye throughout the follow-up period. Slit-lamp examination revealed no signs of inflammation, the anterior segment showed no unexpected changes. Funduscopically, a mild and non-progressive gliosis could be seen surrounding the retinal tacks. The tacks had remained firmly attached, as has the retina itself.
Discussion
Adding to findings from the acute clinical trial, the follow-up data reported herein provide knowledge on medium to long-term effects of the implantation and explantation surgery, as well as the intraocular retention of retinal tacks.
When interpreting the NEI-VFQ-25 scores, it needs to be taken into account that the German language version of this questionnaire has not been validated for use in RP patients.9 We also hasten to point out that although in our study the patients were equipped with a functional epiretinal device, this was only activated to conduct a specific testing programme. Patients were not continuously fitted with the extraocular system components; therefore, a gain in everyday life quality was not to be expected. The low overall score in all the patients indicates that blindness due to RP results in substantial overall loss of vision-related quality of life. Further loss of life quality during or after the study could not be detected. This may however be due to reduced sensitivity to minor changes at the lower end of the testing scale. Other tools such as the Veterans Affairs Low Vision Visual Functioning Questionnaire may provide a suitable add-on to increase sensitivity in future studies.10
Short-term findings from the EPIRET3 clinical trial were reported by Roessler et al.5 This report includes the description of two adverse events: acute sterile hypopyon after implantation in one patient, and a retinal defect in another. Otherwise, implants and surgery had been tolerated well. On the long-term follow-up visits, we found a stable situation in all the study eyes. Visual perception present at baseline had been preserved, with the possible exception of patient AC-05, in whom a retina defect had been treated with laser and silicon oil. Some patients presented minor anterior synechiae; however, the IOP remained within normal range. Fluorescein angiography revealed typical RP patterns, but areas of excessive dye leakage or choroidal neovascularisation were absent. No changes were observed in angiographic findings compared with those obtained during the acute trial.11 Alterations found at the posterior pole from causes other than RP were epiretinal gliosis around retinal tacks. These tacks had been left in place upon explantation of the device, as their removal may cause tissue damage. Only where tacks were found to be loose, were they taken out. Retinal tacks have been shown to be suitable for bringing the microelectrode array in close contact to the retinal surface.12, 13 Stable positioning is a prerequisite to ensure successful activation. From clinical applications in the repair of giant retinal tears, it was reported that a glial reaction adjacent to the tack may occur.14, 15 It remains to be determined whether glial proliferation, albeit non-progressive, may compromise the function of the prosthesis. In addition, the retinal defect which occurred in patient AC-05 was prompted by the removal of a loose tack.5 Together, these findings suggest that alternative fixation methods such as bioadhesives warrant further research. Plasma polymerised N-isopropyl acrylamide (pNIPAM) is one substance for which results from both in vitro and in vivo testing are available.16, 17 Although tack fixation is currently the standard used in all epiretinal devices that have thus far attained clinical testing, bioadhesives such as pNIPAM may in future become a more atraumatic, while still reversible, means of fixation.
The use of OCT as a means of assessing retinal implant biocompatibility has recently been suggested in the literature and was therefore included in this follow-up study.18 Because of a lack of data from the initial clinical trial, we matched measurements obtained from our patients with normative values from RP patients obtained by Huang et al.19 During acute trials with epiretinal electrode arrays in dogs, an increase in retinal thickness under the arrays in the first month after implantation has been recorded.18 In our human volunteers, there is remarkable variation in central retinal thickness. However, no significant oedema can be observed 24 months post implantation.
Our data suggest that despite the fact that the entirely intraocular approach of the epiretinal implant, EPIRET3, entails extensive intraocular surgery, acute adverse reactions can be managed and do not negatively affect long-term stability. Therefore, surgery for implantation and explantation of the device may be considered sufficiently safe and with an acceptably low complication profile to justify further in-vivo testing. Subsequent long-term trials would not only generate more knowledge about safety and biocompatibility issues, but also provide new opportunities to conduct functional testing and improve encoding of the stimulation pattern. However, improvements to the fixation of the device are warranted.

References
Smith AJ, Bainbridge JW, Ali RR . Prospects for retinal gene replacement therapy. Trends Genet 2009; 25 (4): 156–165.
Bhatia B, Singhal S, Jayaram H, Khaw PT, Limb GA . Adult retinal stem cells revisited. Open Ophthalmol J 2010; 4: 30–38.
Abe T, Yoshida M, Yoshioka Y, Wakusawa R, Tokita-Ishikawa Y, Seto H et al. Iris pigment epithelial cell transplantation for degenerative retinal diseases. Prog Retin Eye Res 2007; 26 (3): 302–321.
Humayun MS, de Juan E, Dagnelie G, Greenberg RJ, Propst RH, Phillips DH . Visual perception elicited by electrical stimulation of retina in blind humans. Arch Ophthalmol 1996; 114 (1): 40–46.
Roessler G, Laube T, Brockmann C, Kirschkamp T, Mazinani B, Goertz M et al. Implantation and explantation of a wireless epiretinal retina implant device: observations during the EPIRET3 prospective clinical trial. Invest Ophthalmol Vis Sci 2009; 50 (6): 3003–3008.
Cohen ED . Safety and effectiveness considerations for clinical studies of visual prosthetic devices. J Neural Eng 2007; 4 (1): S124–S129.
Mangione CM, Lee PP, Gutierrez PR, Spritzer K, Berry S, Hays RD et al. Development of the 25-item National Eye Institute Visual Function Questionnaire. Arch Ophthalmol 2001; 119 (7): 1050–1058.
Papageorgiou E, Hardiess G, Schaeffel F, Wiethoelter H, Karnath HO, Mallot H et al. Assessment of vision-related quality of life in patients with homonymous visual field defects. Graefes Arch Clin Exp Ophthalmol 2007; 245 (12): 1749–1758.
Franke G, Esser J, Voigtlaender A, Maehner N . [The National Eye Institute Visual Function questionnaire (NEI-VFQ), an inventory to assess quality of life in the visually impaired]. Zeitschr Med Psychol 1998; 7: 178–184.
Franke GH, Gall C . [Quality of life—methodology and clinical practice aspects with a focus on ocular medicine]. Ophthalmologe 2008; 105 (8): 727–734.
Roessler G, Laube T, Brockmann C, Kirschkamp T, Mazinani B, Menzel-Severing J et al. Angiographic findings following tack fixation of a wireless epiretinal retina implant device in blind RP patients. Graefes Arch Clin Exp Ophthalmol 2011; 249 (9): 1281–1286.
Walter P, Szurman P, Vobig M, Berk H, Lüdtke-Handjery HC, Richter H et al. Successful long-term implantation of electrically inactive epiretinal microelectrode arrays in rabbits. Retina (Philadelphia, PA) 1999; 19 (6): 546–552.
Majji AB, Humayun MS, Weiland JD, Suzuki S, D’Anna SA, de Juan Jr E . Long-term histological and electrophysiological results of an inactive epiretinal electrode array implantation in dogs. Invest Ophthalmol Vis Sci 1999; 40 (9): 2073–2081.
Ando F, Kondo J . A plastic tack for the treatment of retinal detachment with giant tear. Am J Ophthalmol 1983; 95 (2): 260–261.
de Juan E, Hickingbotham D, Machemer R . Retinal tacks. Am J Ophthalmol 1985; 99 (3): 272–274.
Tunc M, Humayun M, Cheng X, Ratner BD . A reversible thermosensitive adhesive for retinal implants: in vivo experience with plasma-deposited poly(N-isopropyl acrylamide). Retina (Philadelphia, PA) 2008; 28 (9): 1338–1343.
Tunc M, Cheng X, Ratner BD, Meng E, Humayun M . Reversible thermosensitive glue for retinal implants. Retina (Philadelphia, PA) 2007; 27 (7): 938–942.
Güven D, Weiland JD, Maghribi M, Davidson JC, Mahadevappa M, Roizenblatt R et al. Implantation of an inactive epiretinal poly(dimethyl siloxane) electrode array in dogs. Exp Eye Res 2006; 82 (1): 81–90.
Huang Q, Chowdhury V, Coroneo MT . Evaluation of patient suitability for a retinal prosthesis using structural and functional tests of inner retinal integrity. J Neural Eng 2009; 6 (3): 035010.
Acknowledgements
This work has been supported by the following grants from the German Ministry of Education and Research (BMBF): 01IN501B, 01IN501F9, 01IN501K7, 01KP0004, 01KP0006, 01KP0402, 01KP0404, 01KP0405, 01PK0001.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Menzel-Severing, J., Laube, T., Brockmann, C. et al. Implantation and explantation of an active epiretinal visual prosthesis: 2-year follow-up data from the EPIRET3 prospective clinical trial. Eye 26, 501–509 (2012). https://doi.org/10.1038/eye.2012.35
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/eye.2012.35
Keywords
This article is cited by
-
An update on visual prosthesis
International Journal of Retina and Vitreous (2023)
-
Attitudes of potential recipients toward emerging visual prosthesis technologies
Scientific Reports (2023)
-
Full gaze contingency provides better reading performance than head steering alone in a simulation of prosthetic vision
Scientific Reports (2021)
-
Validation of a vision-related activity scale for patients with retinitis pigmentosa
Health and Quality of Life Outcomes (2020)
-
Contemporary approaches to visual prostheses
Military Medical Research (2019)
