
Abstract
Fractures across the stages of chronic kidney disease (CKD) could be due to osteoporosis, some form of renal osteodystrophy defined by specific quantitative histomorphometry or chronic kidney disease–mineral and bone disorder (CKD–MBD). CKD–MBD is a systemic disease that links disorders of mineral and bone metabolism due to CKD to either one or all of the following: abnormalities of calcium, phosphorus, parathyroid hormone or vitamin D metabolism; abnormalities in bone turnover, mineralization, volume, linear growth or strength; or vascular or other soft-tissue calcification. Osteoporosis, as defined by the National Institutes of Health, may coexist with renal osteodystrophy or CKD–MBD. Differentiation among these disorders is required to manage correctly the correct disorder to reduce the risk of fractures. While the World Health Organization (WHO) bone mineral density (BMD) criteria for osteoporosis can be used in patients with stages 1–3 CKD, the disorders of bone turnover become so aberrant by stages 4 and 5 CKD that neither the WHO criteria nor the occurrence of a fragility fracture can be used for the diagnosis of osteoporosis. The diagnosis of osteoporosis in stages 4 and 5 CKD is one of the exclusion—excluding either renal osteodystrophy or CKD–MBD as the cause of low BMD or fragility fractures. Differentiations among the disorders of renal osteodystrophy, CKD–MBD or osteoporosis are dependent on the measurement of specific biochemical markers, including serum parathyroid hormone (PTH) and/or quantitative bone histomorphometry. Management of fractures in stages 1–3 CKD does not differ in persons with or without CKD with osteoporosis assuming that there is no evidence for CKD–MBD, clinically suspected by elevated PTH, hyperphosphatemia or fibroblast growth factor 23 due to CKD. Treatment of fractures in persons with osteoporosis and stages 4 and 5 CKD is not evidence-based, with the exception of post-hoc analysis suggesting efficacy and safety of specific osteoporosis therapies (alendronate, risedronate and denosumab) in stage 4 CKD. This review also discusses how to diagnose and manage fragility fractures across the five stages of CKD.
Similar content being viewed by others

Introduction
The kidney and the skeleton have intimate biological relationships that can affect bone strength as well as renal physiological functions (Figure 1).1,2 The individual or collective roles of each of these interactions will be detailed in this manuscript, but from the figure, one can see how molecules regulated or produced by the kidney, the parathyroid glands, the bone and the vasculature all contribute to bone metabolism. Both intrinsic primary renal diseases such as diabetic nephropathy, as well as changes in renal function associated with the aging-kidney, affect systemic (including bone) metabolism which, in the case of skeletal interactions with the kidney, can lead to a heterogeneous group of bone diseases all of which are associated with skeletal fragility and increased risk for fractures.

The physiological interactions between bone, the kidney, parathyroid glands and vasculature. Miller PD, Sprague S, Shane E. (original) 2014.
For the clinician confronted with an older patient who has decreased glomerular filtration rate (GFR) and low trauma fractures, the diagnostic challenge is discriminating fractures due to osteoporosis vs the heterogeneous bone diseases traditionally associated with chronic kidney disease (CKD): renal osteodystrophy (a histomorphometric classification) or CKD–mineral and bone disorder (CKD–MBD).1,2 CKD–MBD includes the systemic interactions among all of the tissues affected by the pathophysiological processes that accompany CKD.
The National Kidney Foundation (NKF) has moved to incorporate estimated (calculated) glomerular filtration rate (eGFR) on all routine biochemical profiles done by all physicians during the annual health examinations. The eGFR inclusion is an attempt to identify (e.g., screen) the largest growing segment of the CKD population (stage 3) eGFR 60–30 mL·min−1.3–8 It is forecasted that by identifying earlier stages of CKD and treating those factors that may lead to progressive renal damage, that the more advanced stages of CKD can be mitigated.
The NKF classifies five stages of CKD (Table 1).4,8 Stages 1 and 2 CKD must have associated urinary microscopic changes (either hematuria and/or proteinuria) in addition to an eGFR <110 mL·min−1 to fulfill the diagnostic criteria for stage 1 or 2 CKD. However, based on reduced eGFR alone, without needing accompanying proteinuria or hematuria, CKD can be coded by the International Classification of Diseases (ICD codes) as stage 3–5 CKD (stage 3: eGFR <60–30 mL·min−1; stage 4: eGFR 30–15 mL·min−1 and stage 5: eGFR <15 mL·min−1 and/or end-stage renal disease (ESRD)). Stage 5D stands for ESRD in patients on dialysis. In part, the requirement of proteinuria or hematuria to fulfill the diagnosis of stage 1–2 CKD is predicated on the observations that age-related reductions in eGFR (as opposed to intrinsic renal disease associated reductions in eGFR) will not carry the same concerned prognosis as CKD with proteinuria, since the absence of proteinuria implies the lack of intrinsic renal damage, which may be less onerous than lower eGFR with proteinuria.
More recently, the NKF has subdivided stage 3 CKD (eGFR 60–30 mL·min−1) into 2 subtypes: stage 3A (eGFR 60–45 mL·min−1) and 3B (eGFR 45–30 mL·min−1).9 The purpose of this division is to acknowledge that stage 3 CKD encompasses a broad range of renal function and that there may be clear biological distinctions among the renal–bone–vascular calcification pathophysiology (e.g. CKD–MBD) between 3A and 3B CKD. Derangements in the biochemical regulatory process that accompany CKD become more profound the greater the severity of CKD. Early CKD is associated with a progressive rise in osteocyte-derived fibroblast growth factor 23 (FGF-23) and, later, a progressive rise in endogenous parathyroid hormone production (PTH) that are higher in stage 3A vs. 3B CKD (Figure 1).9–11 The progressions of these metabolic abnormalities are also seen in the quantitative histomorphometric analysis of bone biopsies qssociated with progressive CKD: e.g., osteitis fibrosa cystica (severe hyperparathyroid bone disease) is more severe the higher the PTH (especially PTH values >9× the ULN). Additionally, sustained hyperphosphatemia is more persistent and is associated with increased vascular calcification as reductions in GFR become greater. Adynamic renal bone disease, characterized by very low or absent bone turnover, is suggested in those with low PTH and low tissue-specific alkaline phosphatase activity. It is seen more often at the lower stages of the NKF classification (3B and lower).89,12–15
A major clinical challenge when faced with a CKD patient who also has fragility fractures lies in the discrimination between osteoporosis and the various forms of renal bone disease, the latter either defined by quantitative histormophometry on bone biopsy, or by the term CKD–MBD: chronic kidney disease–mineral and bone disorder. Management of fractures differs if a low trauma fracture is due to osteoporosis as opposed to fractures due to renal bone disease.1,2
Osteoporosis and CKD–MBD
Osteoporosis is defined as a condition of reduced bone strength leading to an increase risk of fracture.16 Implicit in this definition is a reduction in bone density and bone quality. Since bone quality cannot be measured in clinical practice, the clinical diagnosis of osteoporosis rests either in the bone mineral density (BMD) determination or in the low trauma (fragility) fracture in women or men 50 years of age and older once other causes of bone fragility have been excluded (e.g., osteomalacia, osteogenesis imperfecta).17 The diagnostic value of BMD was codified in 1994, by the World Health Organization (WHO).18 Osteoporosis could be defined by a T-score (the number of standard deviations a person's BMD is below the mean BMD for the young normal health population). The cut-point for the diagnosis, based upon epideminological considerations of fracture risk, was set at a T-score of −2.5 or lower. This cutoff level was chosen on the relationship between the life-time risk of hip fracture in Caucasian women (20%), and the average T-score at the hip between the ages of 50–85 years (20%—2.5 or lower).19 The initial purpose of the BMD WHO-derived classification was to determine the prevalence of osteoporosis in the world's population in order to aid in health-economic planning. Soon after 1994, the T-score made its way into clinical use and also became an ICD to be a second means of diagnosing osteoporosis in individual patients who have not yet had a fragility fracture. The clinical utility of the T-score lies in its use as a risk factor for osteoporotic fracture. Fracture risk approximately doubles for each standard deviation; the BMD is below −2.5 in untreated post-menopausal women as compared to the same population of the same age with a T-score of zero.
One limitation of the T-score is that it does not define the etiology of low BMD; nor should it be used as a stand-alone risk factor for making management decisions.20 Since low BMD captures approximately 50% of the bone strength, and, at the current time, bone quality (the contributing factor for the other 50% of bone strength) cannot be measured clinically,21,22 the T-score must be applied along with other validated risk factors for fracture risk that contribute to fracture risk independent of the BMD level. Thus, the WHO also supported the development of the largest and most robust validated risk model to predict absolute 10-year risk for major fracture (colles, humerus, vertebrae, hip and tibia) and/or hip fracture in untreated post-menopausal women. Nine validated risk factors, each independent but differently weighted, risk factor for fracture were identified and statistically validated in FRAXTM acronym: fracture risk assessment modeling.23 The website link to FRAX is: http://www.shef.ac.uk/FRAX/tool.jsp. One can also access the FRAX calculator via the International Society for Clinical Densitometry (ISCD.ORG), the National Osteoporosis Foundation (NOF.ORG) or the International Osteoporosis Foundation (IOF.ORG) websites.
However, the WHO working groups could not validate the level of GFR or the derived equation, eGFR in the FRAXTM model. The sample size from the population studies was not large enough to validate a GFR–BMD relationship to risk. It is important to stress, however, that since FRAXTM data were completed and implemented, additional independent risk fractures for fracture have been identified such that in clinical practice adding fracture risk beyond that risk quantitated by FRAXTM alone is an important adjunct in management decisions.24
As FRAXTM application has evolved, so has the implementation of additional risk factors not originally validated in FRAXTM but applied in a clinical manner utilizing risk factors identified in separate population studies (e.g., the Garvan Model) that had adjustments for fracture risk according to fall frequency. In addition, the level of bone turnover, identified in specific population studies, is also an independent risk factor for fracture (EPIOS).25 Type 2 diabetes mellitus has also recently been identified as an independent risk factor for fracture. These latter risk factors, like CKD as a risk factor, were not validated in the original FRAXTM model since the statistical power was inadequate in the original FRAXTM cohort to be included in the validated model.
Newer measurements of bone strength, by QCT-derived finite element analysis or DXA derived trabecular bone score, were also not included in the FRAXTM model, since these technologies postdated FRAXTM.26–29
CKD has been identified as a risk factor for increased fracture risk. A number of population studies and authoritative editorials provide evidence that CKD, even stage 3 CKD, may be associated with a greater fracture risk than observed in age-matched and BMD-matched patients without CKD.30–39 This greater risk may be related to the interactions among the multitude of pathophysiological biological changes that accompany CKD such as secondary hyperparathyroidism, abnormalities in 1,25 dihydroxyvitamin D synthesis, phosphorus retention, chronic metabolic acidosis, elevated sclerostin and/or FGF 23.40–43 These accompanying biochemical changes may independently or collectively alter bone remodeling (turnover), or mineralization. Before the Kidney Disease Improving Global Outcome working group coined the term CKD–MBD to embrace the systemic pathology that accompanies altered bone turnover in this population, the classification of the bone diseases accompanying CKD was defined by quantitative bone histomorphometry and collectively termed renal osteodystrophy.12 These histomorphometric classifications are still scientific and valid.44,45 Quantitative histomorphometry requires double tetracycline labeling in order to define dynamic bone turnover parameters, which have specific criteria for the specific type of renal osteodystrophy.46–51 In contrast, CKD–MBD is difficult to define in clinical practice and does not have any specific ICD-9 (or soon, ICD-10) diagnostic code or known relationship to fracture risk. While it is known that the biochemical abnormalities accompanying CKD–MBD alter bone turnover or mineralization which can influence bone strength, the operational clinical differentiation among the diseases accompanying CKD lies in distinguishing between adynamic, hyperparathyroid, mixed renal bone disease, osteomalacia and osteoporosis—all of which may have a low BMD and/or be associated with fragility (including hip) fractures.1,2,46,52–56 The challenge for physicians managing fragility fractures in patients with CKD is discriminating fractures due to osteoporosis from fractures due to the traditional bone diseases accompanying CKD.1,2
BMD and use of FRAXTM at different stages of CKD
Stage 1–3 CKD
The WHO criteria for diagnosing osteoporosis can be used across the spectrum of stage 1–3 CKD (GFR 110–30 mL·min−1). All of the clinical trials for the Food and Drug Administration (FDA) registration of treatments for post-menopausal osteoporosis randomized subjects using WHO criteria. In all of these registration trials, some form of renal function assessment was used to include or exclude subjects. The exclusion criteria ranged from a baseline serum creatinine concentration below 1.27 mg·dL−1 for the alendronate trials; 1.1× the upper limit of normal for the risedronate registration trials; <2.4 mg·dL−1 for the ibandronate trials; or eGFR >30 mL·min−1 (zoledronic acid and denosumab trials)1,2,40,52). The serum creatinine concentration is a poor indicator of GFR since the source of serum creatinine is from muscle and persons with low muscle mass or low BMI may have a serum creatinine in the normal laboratory reference range yet have a low GFR.6–8 Another justification of why the WHO criteria for the diagnosis of osteoporosis can be used in stage 1–3 CKD is that the measurable derangements in bone and mineral metabolism suggesting the presence of CKD–MBD (secondary hyperparathyroidism or hyperphosphatemia) are less pronounced at a GFR >30 mL·min−1, unless there are also non-renal related causes of secondary hyperparathyroidism8,9,11,41,54–57 (Table 2); Third, neither the serum PTH nor serum phosphorus were systematically measured at randomization in the whole populations constituting the osteoporosis registration trials. As compared to placebo, all of the therapies approved for the treatment of PMO have efficacy across the range of renal function defined by the randomization criteria for each trial. Thus there seems to be an understanding among the metabolic bone community that the WHO criteria for the diagnosis of osteoporosis can be applied in a similar manner in patients with stage 1–3 CKD as long as there are no renal related biochemical abnormalities suggesting CKD-MBD.1,2,12 Likewise, there is agreement that given the same list of risk factors, FRAXTM can be applied to stage 1–3 CKD in a similar manner as it is in the PMO populations without known CKD, recognizing that more severe stage 3 (e.g. 3B) CKD may have a greater risk for fracture than seen earlier stages of stage 3A CKD9). The biology of bone in patients with CKD is clearly in a different milieu than patients with osteoporosis without CKD. Since measurable changes in molecules that affect bone metabolism (PTH, FGF-23, serum phosphorus) may be seen in early, even stage 2 CKD.12,42,56,57 However, to what extent these early rises in serum phosphorus regulatory peptides have on the clinical profiling between osteoporosis and CKD-MBD remains elusive.
Stage 4–5 CKD
By the time patient progresses to stage IV–V CKD, the disorders in bone metabolism become so dominant that the WHO criteria for the diagnosis of osteoporosis or use of FRAXTM become invalid. The WHO working group (1994)18 and the analysis of risk factors for fracture (FRAXTM) (2001)23 were confined to the condition of post-menopausal osteoporosis; and, Kidney Disease Improving Global Outcomes CKD–MBD (2009)12 was conceived 17 years after the T-score was conceived (1992). In addition, it took another decade after the pivotal WHO PMO population studies for clinical societies, led by the International Society for Clinical Densitometry (ISCD) and the International Osteoporosis Foundation (IOF) (Bucharest, Romania, October 2011) to acquire opinion based agreement that the clinical capacity of WHO diagnostic criteria may be applied to specific populations other than PMO—elderly men and persons under the age of 50 years if secondary conditions influencing bone strength were co-existent.20,24 The most recent ISCD Position Development Conference that dealt with how to incorporate non-validated (by the original WHO data) data into FRAXTM still did not have enough data to add CKD to the clinical risk menu to calculate fracture risk. Nevertheless, clinical recognition of the higher fracture risk for fracture that is observed in severe (stage 4–5) CKD is useful since it emphasizes the additional risk that CKD–MBD derangements in bone metabolism add to management decisions intended to reduce risk. Therefore, it is critical that the managing physicians faced with patients who fracture and have CKD make the correct diagnosis. Management decisions differ if the patient has osteoporosis as opposed to the metabolic bone diseases that encompass the ranges of renal osteodystrophy or CKD–MBD as define by the Kidney Disease Improving Global Outcome working group.
Dual energy X-ray absorptiometry (DXA) may underestimate the fracture risk in stage 4–5 CKD,40,55 though more recent data suggests that 2 dimensional DXA is as robust as 3 dimensional skeletal measurements to predict risk in severe CKD.58–60
Biochemical markers of bone turnover in CKD
The human being remodels (turns over) bone at both cortical as well as cancellous sites. Bone remodeling is regulated by both systemic (e.g., parathyroid hormone, phosphorus, 1,25 vitamin D, circulating sclerostin and, perhaps FGF-23) as well as local bone microenviroment factors (Rank-Ligand, osteoprotogerin, sclerostin, insulin growth factors, ephrin B2/eph B4), all lumped into a single label, ‘clastokines’.61–69 One major purpose of remodeling is to repair the microdamage that occurs in the skeleton with daily mechanical stress on bone. In clinical practice, there are a number of biochemical markers of bone turnover that can be measured in the serum.70,71 Bone resorption and bone formation markers can be measured.72–77 Data suggest that specific serum BTM levels including serum PTH may help to discriminate among the heterogeneous forms of renal bone disease;13–15,78–81 and are valuable as well to assess systemic bone turnover in post-menopausal osteoporosis or the response to agents that inhibit bone turnover (anti-resorptive agents) or stimulate bone turnover (anabolic agents).
Two markers that are not cleared by the kidney: a resorption (or, more accurate, an osteoclast cellular number marker) is tartrate resistant acid phosphatase (TRAP5b); and, the formation (or more accurate, an osteoblast activity marker), bone-specific alkaline phosphatase. The osteoblast-derived marker propeptide type I collagen (PINP) is currently measured by two assays: one that measures the intact (monomer and trimer) form of PINP (Roche Diagnostics, Indianapolis, IN, USA), and one that measures only the trimer form of PINP (Immunodiagnostics, Phoenix, AZ, USA). The trimer is not cleared by the kidney, while the intact is cleared by the kidney. The only FDA-approved assay for PINP (a radioimmunoassay) measures the trimeric form (OrionTM). Currently, there are insufficient data to know if these differences influence PINP clearance enough to influence clinical utilization of the intact PINP in determining osteoblast activity. However, since from the pooled clinical teriparatide trial data, a rise of PINP of >10 μg·L−1, one to 3 months after the initiation of teriparatide (20 μg·day−1) is highly correlated with an improvement in BMD and/or bone microarchitecture.82 The only FDA-approved assay available in the United States at this time is the radioimmunoassay (the trimeric form) of PINP (Immunodiagnostics and marketed as the radioimmunoassay, OrionTM).
In the management of patients with stage 4–5 CKD and low T-scores or fragility fractures, adynamic bone disease is probably the most important bone disease to exclude since lowering bone turnover, by the use of antiresorptive drugs, may not be desired in patients, including diabetics, whose skeleton has developed into a low bone turnover state.1,2,83–89
Bone biopsy for quantitative purposes
Transiliac bone biopsy done with prior double tetracycline labeling is the most sensitive and specific means for discriminating among the various renal bone diseases;47,51–54 and by exclusion, making the diagnosis of osteoporosis.1,2 Transiliac bone biopsies are safe and have a very low morbidity when done by experienced operators. Tetracycline enters bone attached to calcium and because it fluoresces under a fluorescent microscope, is used as a means of quantifying certain dynamic parameters of bone turnover. The science underpinning quantitative histomorphometry is rooted in robust data sets defining normal and abnormal bone turnover.34,35,40 Whereas hyperparathyroid bone parameters display a histomorphometric spectrum according to the severity and longevity of the hyperparathyroid disorder, osteomalacia has a clear set of criteria required for its definition, and adynamic bone disease generally is considered to be a turnover disorder best defined by the absence of any single or double tetracycline labels (Figure 2).47 Though bone biopsy is definitive in the diagnosis of osteomalacia, the disease also always has a biochemical cause.40,41 Hence, if a patient has an unexplained elevated bone ALP level and has no other cause for it, osteomalacia may be the most probable cause. If there are no identifiable biochemical abnormalities suggesting a cause for osteomalacia, bone biopsy is a definitive means for making the diagnosis.

Renal adynamic bone disease.47
Treatment of osteoporosis in stage 1–3 CKD
Osteoporosis management and treatment should not differ in stage 1–3 CKD as it is in persons without CKD, as long as there are no biochemical markers suggestive of the presence of CKD–MBD.1,2 While greater detail on the use of bisphosphonates, denosumab and teriparatide will be described in the following section, mention should be made with regard to the use of calcitonin, estrogens, androgens and selective estrogen receptor modulators' role in the treatment of post-menopausal osteoporosis. Recently, an FDA advisory panel suggested that salmon calcitonin marketing for osteoporosis be discontinued due to data suggesting a possible increase in the risk of gastrointestinal cancers. Estrogens are still registered for the prevention of osteoporosis, though the FDA suggests that other bone active agents are preferred over estrogens due to the concerns that estrogen carries for increased risk of cardiovascular and breast cancer events. The selective estrogen receptor modulator raloxifene is FDA-registered for PMO as well as invasive breast cancer. Raloxifene has a documented benefit to reduce the risk of vertebral but not of non-vertebral or hip fracture risk. Androgens also are of benefit to the skeleton and muscle (sarcopenia) and should be considered in hypogonadal men particularly if they are symptomatic of hypogonadism. Androgen benefit to risk, however, should be carefully assessed in the CKD population due to the relationships between androgen exposure and increase risk of prostate cancer.
Treatment of osteoporosis in stage 4–5 CKD
Treatment of osteoporosis is an important consideration for patients who have suffered a fragility fracture due to the high mortality as well as morbidity associated with osteoporotic fractures, and even more so in the CKD population. Population data have confirmed the short-term as well as long-term mortality in both genders and with all fractures, including vertebral fractures.34 This mortality is even greater in patients with CKD probably associated with the overall greater mortality from all causes in severe CKD.
The limitations in FDA-approved pharmacological choices for osteoporosis is the lack of evidence for fracture risk reduction in patients with severe (stage 4–5) CKD, with the exception of a few post-hoc analyses in smaller sample sizes of the registered cohorts for PMO. Here, I focus on the most widely used treatments: the bisphosphonates and denosumab representing the anti-resorptive agents and teriparatide as the only available osteoanabolic agent.
Anti-resorptive agents
Anti-resorptive agents all inhibit bone resorption.90 The FDA-approved anti-resorptive agents include calcitonin, estrogens, selective estrogen receptor modulators, bisphosphonates and denosumab. Each anti-resorptive agent has each owns unique mechanism of action. Since bisphosphonates and densoumab are the most widely used anti-resorptive agents for osteoporosis, these two agents will be focused here.
Bisphosphonates are chemical analogues of naturally occurring pyrophosphates (P–O–P), degradation products of adenosine triphosphate metabolism. Pyrophosphates are rapidly metabolized by the ubiquitous presence of pyrophosphatases, while bisphosphonates (P–C–P) are not metabolized. Once entering the blood stream, bisphosphonates are rapidly taken up by bone, the only tissue that binds bisphosphonates.91,92 In bone, bisphosphonates inhibit bone resorption by two mechanisms: a physiochemical one stabilizing the calcium–phosphorus surface and a cellular one by inhibiting osteoclast activity.91,92 Bisphosphonates are cleared by the kidney both by filtration and active proximal tubular secretion.93,94 Bisphosphonates are retained in bone in the remodeling resorption cavity and the amount of bisphosphonate retained in probably a function of the baseline remodeling space, the chronic rate of bone turnover and the GFR. While oral bisphosphonates are poorly (<1% of a single dose) absorbed and 50% of that excreted unchanged by the kidney, intravenous bisphosphonate show a 100% bioavailability with still 50% of an intravenous dose excreted by the kidney. Oral bisphosphonates have never been shown to have any renal toxicity while intravenous bisphosphonates, especially zoledronic acid, may acutely reduce the GFR via a tubular lesion that mimics acute tubular necrosis.94,95 While intravenous ibandronate, the only other intravenous bisphosphonate registered for osteoporosis has not been shown in either clinical trials nor post-marketing reports to have a negative effect on the kidney, there have never been any head-to-head studies in normal, healthy subjects or in subjects with impaired GFR on renal effects between these two bisphosphonates.94,96 Even zoledronic acid, when administered slower than the registered label recommends (15 min), seems safe in clinical experience even in those patients with impaired GFR. Data from the zoledronic acid cancer trials provide an insight that the potential renal damage that can be seen with zoledronic acid is related to dose and rate of infusion.97,98 Zoledronic acid given as a dose of 8 mg monthly vs. 4 mg monthly and given over 5 min vs. 15 min induced a large proportion of acute renal failure not seen with the lower dose–slower infusion rate. These data would suggest that from a pharmacokinetic profile that renal damage might be related to the CMax (the peak concentration of the drug) rather than the area under the curve. Nevertheless, because of the renal clearance and lack of clinical trial data in subjects with GFR <30 mL·min−1, bisphosphonates carry either a warning or a contraindication in the label for use in patients with GFR <30–35 mL·min−1. In that regard, use of bisphosphonates is an off-label use in patients with stage 4–5 CKD, but if used, should be administered very slowly (60 min).
In two post-hoc analyses from the pooled risedronate registration studies and the alendronate fracture intervention trials, both of these oral bisphosphonates in their original registration formulations (5 mg·day−1 of risedronate and 10 mg·day−1 of alendronate) were used in approximately 600 patients per trial (300 treated and 300 placebo) to treat subjects with PMO with eGFR by Cockgroft-Gault equations between 15 and 30 mL·min−1. In both trials the two bisphosphonates reduced the incidence of either morphometric vertebral fractures or all clinical fractures significantly as compared to placebo over an average of 2.6 years duration without any change in renal function.99,100 The initial clinical trials developed for registration did not require or measure in any pre-planned design eGFR or GFR as inclusion/exclusion criteria but only serum creatinine concentration. It took the osteoporosis community longer than the nephrology community to grasp the data that a serum creatinine concentration may fall within a ‘normal’ laboratory reference range, yet the subject with low BMI may still have a significant reduction in GFR. In that regard, only the registration studies for zoledronic acid and densoumab used eGFR as randomization criteria.
Bisphosphonates as a class should be administered for 3–5 years and then discontinued in lower-risk subjects, e.g., the ‘bisphosphonate drug holiday’. The FDA advises this strategy based on the lack of efficacy of bisphosphonates on fracture risk reduction beyond 5 years as well as the appearance of bisphosphonate-associated atypical femur fractures with long duration of use, especially beyond 5 years.101 While these atypical fractures are rare, they have a high morbidity. In addition, the risk for these ASFF falls by at least 70% within the first year of bisphosphonate discontinuation, even though the pharmacology of bisphosphonates indicates that they are still being recycled into and out of bone.91,92,102 The biological answer to why risk of AFF declines despite continual reduction in bone turnover is not defined, but it is important for physicians to know that even though risk declines, it does not disappear. In that regard, patients should be made aware of a prodromal symptom that may precede the fracture displacement weeks before the break: a deep anterior thigh or groin pain that does not go away with rest or a supine position. In these cases, an X-ray of the femoral shaft may reveal the classic stress fracture and periosteal reaction. In high-risk patients (those with femoral neck T-scores—2.5 or lower) on bisphosphonates for 3–5 years, the benefit of continuation from the alendronate ‘Fosamax Long-Term Extension’ trial seems to far outweigh the risks.103 Recent reviews on bisphosphonate benefit to risk ratio provide helpful guidelines.104,105 For those patients at high risk who have discontinued bisphosphonates, the FDA advises switching to a different approved osteoporosis therapy with a different mechanism of action. While there are no data on duration of bisphosphonate use in more advanced CKD, it seems logical that because bone retention may be greater in CKD, that duration of use could be shorter than 3–5 years.
Finally, with regard to use of bisphosphonates, there is a growing amount of retrospective cohort data, suggesting that bisphosphonates may be associated with a reduction in all-cause mortality, including cardiovascular mortality.106 If there is such a link, the mechanism is unknown though hisphosphonates have been shown to alter cellular pathways in vascular endothelial cells that influence vascular calcification.107–109 These data are relevant to the issue of bone turnover and vascular calcification in CKD. Since there may be a link between low bone turnover and a greater risk for vascular calcification in severe CKD,110 pharmacological lowering bone turnover should only be done in this population if very low (e.g., adynamic) bone disease has been excluded.
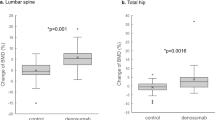
Denosumab (ProliaTM) is a fully human monoclonal antibody that binds to an osteoblast (and osteocyte)-derived glycoprotein, Rank-Ligand, inhibiting Rank-Ligand from binding to an osteoclast membrane receptor, RANK, and thereby inhibiting osteoclastogenesis and action.111,112 Denosumab was FDA-registered for PMO June 2010. Denosumab is a powerful and reversible inhibitor of bone turnover with a substantial reduction of BTM and induces a transient complete loss of the histomorphometric appearance of osteoclasts on biopsy.113 Denosumab clinical trial data show a complete reversibility of both BTM and BMD effect 6 months after administration of the 60 mg dose and a return to responsiveness with repeat administration.114 The registration trials show strong evidence for fracture risk reduction in PMO of all fractures (hip, non-vertebral and vertebral) with the registered dose of denosumab (60 mg SQ Q6 months).115 In the extension trials of denosumab that have now been reported out through 8 years, there is a continual increase in BMD.116,117
Since denosumab is metabolized (in the reticuloendotehlial system) and the biological effect wanes after 6 months, it seems but is not established as of this time, that denosumab must be continued indefinitely to have a benefit. One of the fundamental limitations of all osteoporosis clinical trials is that the placebo arms cannot be continued indefinitely, especially in high-risk patients so that after 3–5 years in most trials, the continual fracture benefit is always compared to the reduction in fracture risk seen with the original randomized population.
From a safety stand-point, denosumab seems to be safe with long-term exposure. In the original clinical trial, the only significantly different safety signal in the treated group as opposed to the placebo arm were reports of skin ‘cellulitis’, most of which cleared with topical or antibiotic therapy.115 Nevertheless, since denosumab is a ‘biologic’ and the Rank-Ligand-Rank system is ubiquitous throughout the body, general immune suppression must be a safety consideration, especially in patients with immune suppression, including the other biologics and post-transplantation. There is a paucity of data in these groups.
There is also the observation that in hemodialysis patients, denosumab may induce significant hypocalcemia.118 This hypocalcemic effect may be mitigated by ensuring adequate 25 hydroxy-vitamin D levels and calcium intake. Also, in these patients, the serum calcium concentration should not be below normal.
In addition, for the renal population, there are additional considerations regarding denosumab use. On quantitative bone histomorphometry in the original registration trials, there were significantly more subjects who had no single tetracycline labels in the treated as opposed to the placebo groups.113 While absent single tetracycline labels may be seen in <5% of healthy normal subjects, the preponderance of absent labels with denosumab suggests the absence of bone mineralization during the administration in a subset of the clinical trial subjects. Though BTM rebound to even greater than baseline within 6 months after discontinuation of denosumab, it is unknown whether mineralization returns to the level of the original registration cohort. In a separate non-registration study (acronym: ‘STAND’), double labels were seen in subjects exposed to denosumab after discontinuation.117 However, it is unknown if these subjects had absent labels at baseline. The point is that if suppression of remodeling is a concern in renal patients with adynamic bone disease, and until we have more definitive data, denosumab should be avoided in stage 4–5 CKD unless the managing physician knows that the patients does not have pre-existing adynamic bone disease. The challenge is compounded in that denosumab has no FDA lower GFR warning or contraindication due to the fact that densoumab is not cleared by the kidney. In addition, in a post-hoc analysis from the original denosumab registration trial, densoumab significantly increase BMD and reduced incident vertebral fractures in subjects whose GFR was as low as 15 mL·min−1.119 The issues of efficacy vs. safety in these populations are separate considerations. Finally, since vascular calcification is the major risk factor for death in the CKD population, there should be provided some data with regard to vascular calcification in the denosumab trials. This is important since serum osteoprotogerin levels rise with denosumab administration as a regulatory response once Rank pathways are inhibited. There are conflicting and opposing data with regard to the influence of osteoprotogerin on vascular calcification.120,121 In the densoumab registration trial, vascular calcification was assessed by lateral lumbar spine X-rays done in order to assess for incident vertebral fracture. Data recently published suggest that vascular calcification scores did not changes between treated vs. placebo groups over the 3 year duration of the trial.122 Larger prospective trials are being designed to examine this relationship between densoumab use and vascular calcification by a variety of more sensitive means to measure vascular calcification.
Anabolic agents
The only osteoanabolic agent FDA-registered for the treatment of osteoporosis in women and men, as well as glucocorticoid-induced osteoporosis, is recombinant human 1–34 PTH (teriparatide) (ForteoTM).123–126 Teriparatide stimulates the formation of new bone by cellular and regulatory pathways.127 The initial registration trial completed for FDA approval for PMO lasted 18 months, 16 months shorter than the FDA requires for the registration of treatment for osteoporosis.128 The teriparatide trial was cut short in part due to the appearance of osteogenic sarcoma in a Fischer strain of rat, an animal model that predominately models rather than remodels bone. The abbreviated study, nevertheless, showed significant reductions in vertebral and non-vertebral fracture.
The teriparatide registration trial, like other registration trials for osteoporosis, did not randomize subjects with known stage 4–5 CKD. However, like the previously mentioned post-hoc analysis for alendronate, risedronate and denosumab that had subsets of the randomized population with eGFR values down to 15 mL·min−1, the teriparatide trials had small sub-sets that had eGFR down to 30 mL·min−1.129 In these subsets, there were similar increases in BMD and PINP across tertiles of eGFR. Fracture numbers were too small to have power for statistical analysis across these three tertiles. There were no changes in renal function as assessed by changes in serum creatinine or serum calcium concentrations as a function of eGFR during the registration trial with the approved 20 µg·day−1 or the higher 40 µg·day−1 doses of teriparatide. While 24-h urine calcium excretion increased on average ∼50 mg·day−1 greater than placebo, there was no greater risk of clinical nephrolithiasis, though pre-existing kidney stones were an exclusionary criteria for trial randomization. Serum uric acid did rise significantly more than placebo, though the clinical consequences of this change in serum uric acid over the trial duration are unknown.
There are no data on the effect of teriparatide in subjects with stage 4–5 CKD, nor in subjects with bone biopsy proven adynamic renal bone disease. The use of teriparatide in stage 4–5 CKD is off label and its use in known adynamic bone disease is only predicated on the knowledge that an anabolic agent can increase bone turnover and improve bone microarchitecture and shows a strong correlation with increases in BMD and fracture risk reduction; and, is a disease where therapies are unknown.130–134 Hence, it is possible, though unproven, that teriparatide may have a beneficial role in idiopathic renal adynamic bone disease. Recent uncontrolled clinical observations do suggest that teriparatide increases bone formation in bisphosphonate-associated ASFF135 and, may heal fractures that have non-union. It is also unknown if teriparatide will have the same anabolic effect in subjects with pre-existing secondary hyperparathyroidism. Baseline PTH levels were only measured in a small subset of the teriparatide PMO registration trials, and were normal. Hence, it is unknown if sustained and uncorrected elevated PTH levels could mitigate the anabolic effect of teriparatide.
The future
There are new pharmacological agents in development that have completely different mechanism of actions than currently available agents; and, might offer unique investigative opportunities in patients with CKD. One agent is an anti-resorptive agent that, unlike previous anti-resorptive agents, does not decrease osteoblastic activity following inhibition of osteoclastic activity. This agent is a cathepsin K inhibitor, odanacatib. Cathepsin K is released by the osteoclast and induces bone resorption by dissolving bone collagen outside the osteoclast. The cathepsin K inhinitor, odanacatib, inhibits the activity of cathepsin K, leading to reduced bone resorption without affecting the cellular integrity of the osteoclast, which is altered by previously described anti-resorptive agents.136,137 Signals between the osteoclast and the osteoblast, usually directional in nature termed ‘coupling’, regulate cellular activity of each cell line. When the osteoclast anatomy is altered by bisphosphonates or densoumab, osteoblast activity is also reduced. This coupling is ‘un-coupled in the use of a cathepsin K inhibitor’. Thus, osteoblast activity is retained, a feature that may have specific advantages in disorders of low bone formation.
Low bone formation in CKD may be due, in part, to the increase in serum sclerostin observed in CKD.138 Sclerostin, an osteocyte derived protein regulates osteoblast activity and sclerostin binding to the osteoblast reduces osteoblast activity.139 There are two monoclonal antibodies to sclerostin in development for the treatment of PMO, romoozumab and blosozumab.140,141 Bone formation increases with both of these agents and their mechanism of actions may have a unique feature not seen with the approved anabolic agent, teriparatide: diminished increased osteoclastic activity (e.g., coupling from the osteoblast-side), while the monoclonal antibodies induce a greater increase in bone formation markers than bone resorption markers. Thus, the anabolic window may be widened with these anabolic agents. The same may be said from phase 2 data of the third anabolic agent in development, PTH-related peptide analog, abaloparatide.142 It is unclear why there may be a difference between teriparatide and these newer anabolic agents, but it might be due to a lower induction of osteoblast-derived Rank-Ligand with newer agents as compared to teriparatide. A lower production of Rank-Ligand would lead to a lower induction of osteoclast activity and a wider window of bone formation.
Conclusions
The management of patients with fragility fractures across the spectrum of CKD should not differ between persons without reductions in eGFR or persons with stage 1–3 CKD, at least as it pertains to patients with age-related reductions in GFR. This suggestion is predicated on the absence of information that could suggest the presence of CKD–MBD. In patients with stage 4–5 CKD and who have fragility fractures, the first management step is making the correct diagnosis. Diagnosis of osteoporosis in stage 4–5 CKD is an exclusionary one. Exclusion is best made by quantitative histomorphometry, a clinical science that is under-utilized. Biochemical markers of bone turnover, in particular serum PTH and tissue-specific alkaline phosphatase, may provide differentiation between biopsy proven adynamic, hyperparathyroid and/or osteomalacia. The exclusion, in particular, of renal adynamic bone disease is important since, even off-label use of anti-resorptive agents may not, in theory, be beneficial in persons with no bone turnover to begin with. There is a great need to gain knowledge and evidence for a beneficial or non-beneficial effect of registered therapies for post-menopausal, male or steroid-induced osteoporosis in very high-risk stage 4–5 CKD subjects who have sustained a low trauma fracture.
Anabolic agents that inhibit sclerostin production or sclrostin binding to osteoblasts might offer specific targeted treatment for conditions characterized by low bone formation and elevated sclerostin. These include teriparatide, romozumab, blosozumab and abaloparatide. While teriparatide has been FDA-approved for 12 years and has a known safety record when implemented and monitored appropriately, newer anabolic agents will need to also have a large benefit/risk ratio for newer agents that may have a broader anabolic than resorptive mechanism. However, the opportunities to treat patients with more targeted therapies are exciting.
References
Miller PD . Bone disease in CKD: a focus on osteoporosis diagnosis and management. Am J Kidney Dis 2014; 64: 290–304.
Miller PD . Chronic kidney disease and osteoporosis: evaluation and management. Bonekey Rep 2014; 3: 542.
Coresh J, Astor BC, Greene T, Eknoyan G, Levey AS . Prevalence of chronic kidney disease and decreased kidney function in the adult U.S. population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis 2013; 41: 1–12.
Levey AS, Coresh J, Balk E et al. National Kidney Foundation practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Ann Intern Med 2003; 139: 137–147.
Levey AS, Eckardt KU, Tsukamoto Y et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 2005; 67: 2089–2100.
Stevens LA, Levey AS . Chronic kidney disease in the elderly—how to assess risk. N Engl J Med 2005; 352: 2122–2124.
Stevens LA, Coresh J, Greene T, Levey AS . Assessing kidney function—measured and estimated glomerular filtration rate. N Engl J Med 2006; 354: 2473–2483.
National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Kidney Disease Outcomes Quality Initiative. Am J Kidney Dis 2002; 39: S1–S266.
Kirsztajn GM, Suassuna JH, Bastos MG . Dividing stage 3 of chronic kidney disease (CKD): 3A and 3B. Kidney Int 2009; 76: 462–463; author reply 463–464.
Jüppner H, Wolf M, Salusky IB . FGF-23: more than a regulator of renal phosphate handling? J Bone Miner Res 2010; 25: 2091–2097.
Silver J, Naveh-Many T . FGF-23 and secondary hyperparathyroidism in chronic kidney disease. Nat Rev Nephrol 2013; 9: 641–649.
Kidney Disease: Improving Global Outcomes (KDIGO) CKD–MBD Work Group. KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of chronic kidney disease–mineral and bone disorder (CKD–MBD). Kidney Int Suppl 2009; (113): S1–S130.
Coen G, Ballanti P, Bonucci E et al. Bone markers in the diagnosis of low turnover osteodystrophy in hemodialysis patients. Nephrol Dial Transplant 1998; 13: 2294–2302.
Couttenye MM, D’Haese PC, van Hoof VO et al. Low serum levels of alkaline phosphatase of bone origin: a good marker of adynamic bone disease in hemodialysis patients. Nephrol Dial Transplant 1996; 11: 1065–1072.
Garrett G, Sardiwal S, Lamb EJ, Goldsmith DJ . PTH—a particularly tricky hormone: why measure it at all in kidney patients? Clin J Am Soc Nephrol 2013; 8: 299–312.
NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA 2001; 285: 785–795.
Miller PD . Guidelines for the diagnosis of osteoporosis: T-scores vs fractures. Rev Endocr Metab Disord 2006; 7: 75–89.
Assessment of fracture risk and its application to screening for postmenopausal osteoporosis . Report of a WHO Study Group. World Health Organ Tech Rep Ser 1994; 843: 1–129.
Melton LJ 3rd. How many women have osteoporosis now? J Bone Miner Res 1995; 10: 175–177.
Baim S, Binkley N, Bilezikian JP et al. Official Positions of the International Society for Clinical Densitometry and executive summary of the 2007 ISCD Position Development Conference. J Clin Densitom 2008; 11: 75–91.
Bouxsein ML . Non-invasive measurements of bone strength: promise and peril. J Musculoskelet Neuronal Interact 2004; 4: 404–405.
Seeman E . Bone quality: the material and structural basis of bone strength. J Bone Miner Metab 2008; 26: 1–8.
Kanis JA, Johnell O, Oden A, Johansson H, McCloskey E . FRAX and the assessment of fracture probability in men and women from the UK. Osteoporos Int 2008; 19: 385–397.
Engelke K, Adams JE, Armbrecht G et al. Implementation and use of FRAX in clinical practice. J Clin Densitom 2013; 14: 226–236.
Leslie WD, Lix LM . Comparison between various fracture risk assessment tools. Osteoporos Int 2014; 25: 1–21.
Hans D, Goertzen AL, Krieg MA, Leslie WD . Bone microarchitecture assessed by TBS predicts osteoporotic fractures independent of bone density: the Manitoba study. J Bone Miner Res 2011; 26: 2762–2769.
Keaveny TM . Biomechanical computed tomography-noninvasive bone strength analysis using clinical computed tomography scans. Ann NY Acad Sci 2010; 1192: 57–65.
Krueger D, Fidler E, Libber J, Aubier-Rozier B, Hans D, Binkley N . Spine trabecular bone score subsequent to bone mineral density improves fracture discrimination in women. J Clin Densitom 2014; 17: 60–65.
Silva BC, Leslie WD, Resch H et al. Trabecular Bone Score: a noninvasive analytical method based upon the DXA image. J Bone Miner Res 2014; 29: 518–530.
Ensrud KE, Lui LY, Taylor BC et al. Renal function and risk of hip and vertebral fractures in older women. Arch Intern Med 2007; 167: 133–139.
Dukas L, Schacht E, Stähelin HB . In elderly men and women treated for osteoporosis a low creatinine clearance of < 65 ml/min is a risk factor for falls and fractures. Osteoporos Int 2005; 16: 1683–1690.
Fried LF, Biggs ML, Shlipak MG et al. Association of kidney function with incident hip fracture in older adults. J Am Soc Nephrol 2007; 18: 282–286.
Nickolas TL, Cremers S, Zhang A et al. Discriminants of prevalent fractures in chronic kidney disease. J Am Soc Nephrol 2011; 22: 1560–1572.
Nickolas TL, Leonard MB, Shane E . Chronic kidney disease and bone fracture: a growing concern. Kidney Int 2008; 74: 721–731.
Alem AM, Sherrard DJ, Gillen DL et al. Increased risk of hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58: 396–399.
Ball AM, Gillen DL, Sherrard D et al. Risk of hip fracture among dialysis and renal transplant recipients. JAMA 2002; 288: 3014–3018.
Jadoul M, Albert JM, Akiba T et al. Incidence and risk factors for hip or other bone fractures among hemodialysis patients in the Dialysis Outcomes and Practice Patterns Study. Kidney Int 2006; 70: 1358–1366.
Stehman-Breen CO, Sherrard DJ, Alem AM et al. Risk factors for hip fracture among patients with end-stage renal disease. Kidney Int 2000; 58: 2200–2205.
Ensrud KE . Fracture risk in CKD. Clin J Am Soc Nephrol 2013; 8: 1282–1283.
Jamal SA, West SL, Miller PD . Bone and kidney disease: diagnostic and therapeutic implications. Curr Rheumatol Rep 2012; 14: 217–223.
Miller PD . Unrecognized and underappreciated secondary causes of osteoporosis. Endocrinol Metab Clin North Am 2012; 41: 613–628.
Jüppner H, Wolf M, Salusky IB . FGF-23: more than a regulator of renal phosphate handling? J Bone Miner Res 2010; 25: 2091–2097.
Cejka D, Herberth J, Branscum AJ et al. Sclerostin and Dikkopf-1 in renal osteodystrophy. Clin J Am Soc Nephrol 2011; 6: 877–882.
Parfitt AM, Drezner MK, Glorieux FH et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 1987; 2: 595–610.
Dempster DW, Compston JE, Dreznar MK et al. Standarized nomenclature, symbols, and units for bone histomorphonetry: a 2012 update of the report of the ASBMR Histomorphometry Nomenclature Committee. J Bone Miner Res 2013; 28: 2–17.
Andress DL, Sherrard DJ . The osteodystrophy of chronic renal failure. In: Schrier RW, editor. Diseases of the kidney and urinary tract. Philadelphia, PA: Lippincott Williams and Wilkins, 2003: 2735–2768.
Miller PD . The role of bone biopsy in patients with chronic renal failure. Clin J Am Soc Nephrol 2008; 3(Suppl 3): S140–S150.
Frost HM . Tetracycline-based histological analysis of bone remodeling. Calcif Tissue Res 1969; 3: 211–237.
Hitt O, Jaworski ZF, Shimizu AG, Frost HM . Tissue-level bone formation rates in chronic renal failure, measured by means of tetracycline bone labeling. Can J Physiol Pharmacol 1970; 48: 824–828.
Parfitt AM . Renal bone disease: a new conceptual framework for the interpretation of bone histomorphometry. Curr Opin Nephrol Hypertens 2003; 12: 387–403.
Trueba D, Sawaya BP, Mawad H, Malluche HH . Bone biopsy: indications, techniques, and complications. Semin Dial 2003; 16: 341–345.
Miller PD, Jamal SA, Evenepoel P, Eastell R, Boonen S . Renal safety in patients treated with bisphosphonates for osteoporosis: a review. J Bone Miner Res 2013; 28: 2049–2059.
Gal-Moscovici A, Sprague SM . Osteoporosis and chronic kidney disease. Semin Dial 2007; 20: 423–430.
Lamb EJ, Vickery S, Ellis AR . Parathyroid hormone, kidney disease, evidence and guidelines. Ann Clin Biochem 2007; 44(Pt 1): 1–4.
Jamal SA, West SL, Miller PD . Fracture risk assessment in patients with chronic kidney disease. Osteoporos Int 2012; 23: 1191–1198.
Zangeneh F, Clarke BL, Hurley DL, Watts NB, Miller PD . Chronic kidney disease–mineral and bone disorder (CKD–MBDs): what the endocrinologist needs to know. Endocr Pract 2014; 20: 500–516.
Fang Y, Ginsburg C, Sugatani T, Monier-Faugere MC, Malluche H, Hruska KA . Early chronic kidney disease–mineral bone disorder stimulates vascular calcification. Kidney Int 2014; 85: 142–150.
Jamal SA, Hayden JA, Beyene J . Low bone mineral density and fractures in long-term hemodialysis patients: a meta-analysis. Am J Kidney Dis 2007; 49: 674–681.
Jamal S, Cheung AM, West S, Lok C . Bone mineral density by DXA and HR pQCT can discriminate fracture status in men and women with stages 3 to 5 chronic kidney disease. Osteoporos Int 2012; 23: 2805–2813.
West SL, Jamal SA . The interpretation and utility of bone mineral by dual energy X-ray absorptiometry in chronic kidney disease. Semin Dial 2014; 27: 569–571.
Boyce BF . Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts. J Bone Miner Res 2013; 28: 711–722.
Takyar FM, Tonna S, Ho PW et al. Ephrin B2/EphB4 inhibition in the osteoblast lineage modifies the anabolic response to parathyroid hormone. J Bone Miner Res. 2013; 28: 912–925.
Canalis E . Wnt signaling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol 2013; 9: 575–583.
Rhee Y, Lee EY, Lezcano V et al. Resorption controls bone anabolism driven by parathyroid hormone (PTH) receptor signaling in osteocytes. J Biol Chem 2013; 288: 29809–29820.
Boyce BF . Advances in the regulation of osteoblasts and osteoclasts functions. J Dent Res 2013; 92: 860–867.
Bonewald LF . The amazing osteocyte. J Bone Miner Res 2011; 26: 229–238.
Hernrisksen K, Neutzsky-Wulff AV, Bonewald LF, Kardsal MA . Local communication on and within the bone controls bone remodeling. Bone 2009; 44: 1026–1033.
Bonewald LF, Johnson ML . Osteocytes, mechanosensing and Wnt signaling. Bone 2008; 42: 606–615.
Teti A . Mechanisms of osteoclast-dependent bone formation. Bonekey Rep 2013; 2: 449.
Eastell R, Hannon RA . Biomarkers of bone health and osteoporosis risk. Proc Nutr Soc 2008; 67: 157–162.
Civitelli R, Armamento-Villareal R, Napoli N . Bone turnover markers: understanding their value in clinical trials and clinical practice. Osteoporos Int 2009; 20: 843–851.
McCloskey EV, Vasikaran S, Cooper C ; FRAX® Position Development Conference Members. Official positions for FRAX® clinical regarding biochemical markers from Joint Official Positions Development Conference of the International Society for Clinical Densitometry and International Osteoporosis Foundation on FRAX®. J Clin Densitom 2011; 14: 220–222.
Chavassieux PM, Delmas PD . Bone remodeling: biochemical markers or bone biopsy? J Bone Miner Res 2006; 21: 178–179.
Schafer AL, Vittinghoff E, Ramachandran R, Mahmoudi N, Bauer DC . Laboratory reproducibility of biochemical markers of bone turnover in clinical practice. Osteoporos Int 2010; 21: 439–445.
Vasikaran S, Eastell R, Bruyère O et al. Markers of bone turnover for the prediction of fracture risk and monitoring of osteoporosis treatment: a need for international reference standards. Osteoporos Int 2011; 22: 391–420.
Bauer D, Leary E, Silverman S et al. National Bone Health Alliance Bone Turnover Marker Project: current practices and the need for US harmonization, standardization, and common reference ranges. Osteoporos Int 2012; 23: 2425–2433.
Lemming DJ, Alexandersen P, Kardsal MA, Qvist P, Schaller S, Tankó LB . An update of biomarkers of bone turnover and their utility in biomedical research and clinical practice. Eur J Clin Pharmacol 2006; 62: 781–792.
Coen G, Ballanti P, Bonnucci E et al. Renal osteodystrophy in predialysis and hemodialysis patients: comparison of histologic patterns and diagnostic predictively of intact PTH. Nephron 2002; 91: 103–111.
Malluche HH, Manier-Faugere MC . Pth 1–84, Pth fragmants and bone turnover. Am J Kidney Dis 2003; 41: 1127.
Coen G, Ballanti P, Bonnucci E et al. Bone markers in the diagnosis of low turnover osteodystrophy in haemodialysis patients. Nephrol Dial Transplant 1998; 13: 2294–2302.
Couttenye MM, D’Haese PC, van Hoof VO, et al. Low serum levels of alkaline phosphatase of bone origin: a good marker of adynamic bone disease in haemodialysis patients. Nephrol Dial Transplant 1996; 11: 1065–1072.
Krege JH, Lane NE, Harris JM, Miller PD . PINP as a biological response marker during teriparatide treatment for osteoporosis. Osteoporos Int 2014; 25: 2159–2171.
Hofbauer LC, Brueck CC, Singh SK et al. Osteoporosis in patients with diabetes mellitus. J Bone Miner Res 2007; 22: 1317–1328.
Inzerillo AM, Epstein S . Osteoporosis and diabetes mellitus. Rev Endocr Metab Disord 2004; 5: 261–268.
Rosen CJ, Motyl KJ . No bones about it: insulin modulates skeletal remodeling. Cell 2010; 142: 198–200.
Coen G . Adynamic bone disease: an update and overview. J Nephrol 2005; 18: 117–122.
Farr JN, Drake MT, Amin S, Melton LJ 3rd, McCready LK, Khosla S . In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. J Bone Miner Res 2014; 29: 787–795.
Miller PD . Osteoporosis in patients with chronic kidney disease: diagnosis, evaluation and management. BoneKEy Rep 2014; 3: 542.
Miller PD . Fragility fractures in chronic kidney disease: an opinion-based approach. Cleve Clin J Med 2009; 76: 715–723.
Miller PD . Anti-resorptives in the management of osteoporosis. Best Pract Res Clin Endocrinol Metab 2008; 22: 849–868.
Russell RG, Watts NB, Ebetino FH, Rogers MJ . Mechanisms of action of bisphosphonates: similarities and differences and their potential influence on clinical efficacy. Osteoporos Int 2008; 19: 733–759.
Diab DL, Watts NB, Miller PD . Bisphosphonates: pharmacology and use in the treatment of osteoporosis. In: Marcus R, Feldman D, Dempster D, Lucket M, Cauley J, (ed.) Osteoporosis 2013. 4th ed. Waltham, MA: Academic Press, 2013: 1859–1872.
Lühe A, Künkele KP, Haiker M et al. Preclinical evidence for nitrogen-containing bisphosphonate inhibition of farnesyl diphosphate (FPP) synthase in the kidney: implications for renal safety. Toxicol In Vitro 2008; 22: 899–909.
Miller PD . The kidney and bisphosphonates. Bone 2011; 49: 77–81.
Markowitz GS, Fine PL, Stack JI et al. Toxic acute tubular necrosis following treatment with zoledronate (Zometa). Kidney Int 2003; 64: 281–289.
Miller PD, Ragi-Eis S, Mautalan C, Ramimeriz F, Jonkamski I . Effects of intravenous ibandronate injection on renal function in women with postmenopausal osteoporosis at high risk for renal disease—the DIVINE study. Bone 2011; 49: 1317–1322.
Rosen LS, Gordon D, Kaminski M et al. Long-term efficacy and safety of zoledronic acid compared with pamidronate disodium in the treatment of skeletal complications in patients with advanced multiple myeloma or breast carcinoma: a randomized, double-blind, multicenter, comparative trial. Cancer 2003; 98: 1735–1744.
Saad F, Gleason DM, Murray R et al. Zoledronic Acid Prostate Cancer Study Group. A randomized, placebo-controlled trial of zoledronic acid in patients with hormone-refractory metastatic prostate carcinoma. J Natl Cancer Inst 2002; 94: 1458–1468.
Jamal SA, Bauer DC, Ensrud KE et al. Alendronate treatment in women with normal to severely impaired renal function: an analysis of the fracture intervention trial. J Bone Miner Res 2007; 22: 503–508.
Miller PD, Roux C, Boonen S, Barton IP, Dunlap LE, Burgio DE . Safety and efficacy of risedronate in patients with age-related reduced renal function as estimated by the Cockcroft and Gault method: a pooled analysis of nine clinical trials. J Bone Miner Res 2005; 20: 2105–2115.
Whitaker M, Guo J, Kehoe T, Benson G . Bisphosphonates for osteoporosis—where do we go from here? N Engl J Med 2012; 366: 2048–2051.
Shane E, Burr D, Abrahamsen B et al. Atypical subtrochanteric and diaphyseal femoral fractures: second report of a task force of the American Society for Bone and Mineral Research. J Bone Miner Res 2014; 29: 1–23.
Black DM, Schwartz AV, Ensrud KE et al. FLEX Research Group. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): a randomized trial. JAMA 2006; 296: 2927–2938.
Khosla S, Bilezikian JP, Dempster DW et al. Benefits and risks of bisphosphonate therapy for osteoporosis. J Clin Endocrinol Metab 2012; 97: 2272–2282.
McClung M, Harris ST, Miller PD et al. Bisphosphonate therapy for osteoporosis: benefits, risks, and drug holiday. Am J Med 2013; 126: 13–20.
Sambrook PN, Cameron ID, Chen JS et al. Oral bisphosphonates are associated with reduced mortality in frail older people: a prospective five-year study. Osteoporos Int 2011; 22: 2551–2556.
Thompson B, Towler DA . Arterial calcification and bone physiology: role of the bone–vascular axis. Nat Rev Endocrinol 2012; 8: 529–543.
Peris P, Atkinson EJ, Gössl M et al. Effects of bisphosphonate treatment on circulating osteogenic endothelial progenitor cells in postmenopausal women. Mayo Clin Proc 2013; 88: 46–55.
Santos LL, Cavalcanti TB, Bandeira FA . Vascular effects of bisphosphonates—a systematic review. Clin Med Insights Endocrinol Diabetes 2012; 5: 47–54.
Hruska KA, Saab G, Mattew S, Lund R . Renal osteodystrophy, phosphate homeostasis, and vascular calcification. Semin Dial 2007; 20: 309–315.
Miller PD . Denosumab: anti-RANKL antibody. Curr Osteoporos Rep 2009; 7: 18–22.
Miller PD . A review of the efficacy and safety of denosumab in postmenopausal women with osteoporosis. Ther Adv Musculoskelet Dis 2011; 3: 271–282.
Reid IR, Miller PD, Brown JP et al. on behalf of the Denosumab Phase 3 Bone Histology Study Group. Effects of denosumab on bone histomorphometry: the FREEDOM and STAND studies. J Bone Miner Res 2010; 25: 2256–2265.
Miller PD, Bolognese MA, Lewiecki EM et al. for the AMG 162 Bone Loss Study Group. Effect of denosumab on bone density and turnover in postmenopausal women with low bone mass after long-term continued, discontinued, and restarting of therapy: a randomized blinded phase 2 clinical trial. Bone 2008; 43: 222–229.
Cummings SR, San Martin J, McClung MR et al. For the FREEDOM Trial. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 2009; 361: 756–765.
McClung MR, Lewiecki EM, Geller ML et al. Effects of denosumab on bone mineral density and biochemical markers of bone turnover: 8-year results of a phase 2 clinical trial. Osteoporos Int 2013; 24: 227–235.
Kendler DL, Roux C, Benhamou CL et al. Effects of denosumab on bone mineral density and bone turnover in postmenopausal women transitioning from alendronate therapy. J Bone Miner Res 2010; 25: 72–81.
Block GA, Bone HG, Fang L, Lee E, Padhi D . A single-dose study of denosumab in patients with various degrees of renal impairment. J Bone Miner Res 2012; 27: 1471–1479.
Jamal SA, Ljunggren O, Stehman-Breen C et al. Effects of denosumab on fracture and bone mineral density by level of kidney function. J Bone Miner Res 2011; 26: 1829–1835.
West SL, Lokb CE, Jamal SA . Osteoprotegerin and fractures in men and women with chronic kidney disease. J Bone Miner Metab 2014; 32: 428–433.
Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR . The effect of a single dose of osteoprotogerin in postmenopausal women. J Bone Miner Res 2001; 16: 348–360.
Egbuna OI, Cheung AM, Siddhanti S, Wang A, Daizadeh N, Anthony M . Treatment of osteoporosis by RANKL inhibition with denosumab in women at high cardiovascular risk and with renal impairment does not accelerate vascular calcification. J Bone Miner Res 2013; in press.
Miller PD, Bilezikian JP, Deal C, Harris ST, Ci RP . Clinical use of teriparatide in the real world: initial insights. Endocr Pract 2004; 10: 139–148.
Orwoll ES, Scheele WH, Paul S et al. The effect of teriparatide [human parathyroid hormone (1–34)] therapy on bone density in men with osteoporosis. J Bone Miner Res 2003; 18: 9–17.
Kurland ES, Cosman F, McMahon DJ, Rosen CJ, Lindsay R, Bilezikian JP . Parathyroid hormone as a therapy for idiopathic osteoporosis in men: effects on bone mineral density and bone markers. J Clin Endocrinol Metab 2000; 85: 3069–3076.
Saag KG, Shane E, Boonen S et al. Teriparatide or alendronate in glucocorticoid-induced osteoporosis. N Engl J Med 2007; 357: 2028–2039.
Bilezikian JP, Matsumoto T, Bellido T et al. Targeting bone remodeling for the treatment of osteoporosis: summary of the proceedings of an ASBMR workshop. J Bone Miner Res 2009; 24: 373–385.
Neer RM, Arnaud CD, Zanchetta JR et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med 2001; 344: 1434–1441.
Miller PD, Schwartz EN, Chen P, Misurski DA, Krege JH . Teriparatide in postmenopausal women with osteoporosis and mild or moderate renal impairment. Osteoporos Int 2007; 18: 59–68.
Dempster DW, Cosman F, Kurland ES et al. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. J Bone Miner Res 2001; 16: 1846–1853.
Chen P, Miller PD, Delmas PD, Misurski DA, Krege JK . Change in bone mineral density (BMD) and fracture risk reduction in teriparatide-treated women with osteoporosis. J Bone Miner Res 2005; 20(Suppl 1): S5.
Cannata-Andía JB, Rodriguez García M, Gómez Alonso C . Osteoporosis and adynamic bone in chronic kidney disease. J Nephrol 2013; 26: 73–80.
Frazão JM, Martins P . Adynamic bone disease: clinical and therapeutic implications. Curr Opin Nephrol Hypertens 2009; 18: 303–307.
Lindsay R, Scheele WH, Neer R et al. Sustained vertebral fracture risk reduction after withdrawal of teriparatide in postmenopausal women with osteoporosis. Arch Intern Med 2004; 164: 2024–2030.
Miller PD, McCarthy EF . Bisphosphonate-associated atypical sub-trochanteric femur fractures: paired bone biopsy quantitative histomorphometry before and after teriparatide administration. Semin Arthritis Rheum 2014; doi: 10.1016/j.semarthrit.2014.09.005. [Epub ahead of print]
Ng KW, Martin TJ . New therapeutics for osteoporosis. Curr Opin Pharmacol 2014; 16: 58–63.
Pennypacker BL, Chen CM, Zheng H et al. Inhibition of cathepsin K increases modeling-based bone formation, and improves cortical dimension and strength in adult ovariectomized monkeys. J Bone Miner Res 2014; 29: 1847–1858.
Pelletier S, Dubourg L, Carlier MC, Hadj-Aissa A, Fouque D . The relation between renal function and serum sclerostin in adult patients with CKD. Clin J Am Soc Nephrol 2013; 8: 819–823.
Bonewald LF . The amazing osteocyte. J Bone Miner Res 2011; 26: 229–238.
McClung MR, Grauer A . Romosozumab in postmenopausal women with osteopenia. N Engl J Med 2014; 370: 1664–1665.
Recker R, Benson C, Matsumoto T et al. A randomized, double-blind phase 2 clinical trial of blosozumab, a sclerostin antibody, in postmenopausal women with low bone mineral density. J Bone Miner Res 2014; doi: 10.1002/jbmr.2351. [Epub ahead of print]
Polyzos SA, Makras P, Efstathiadou Z, Anastasilakis AD . Investigational parathyroid hormone receptor analogs for the treatment of osteoporosis. Expert Opin Investig Drugs 2014; doi: 10.1517/13543784.2015.973021. [Epub ahead of print]
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution NonCommercial-NoDerivs 3.0 Unported License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article

Cite this article
Miller, P. Chronic kidney disease and the skeleton. Bone Res 2, 14044 (2014). https://doi.org/10.1038/boneres.2014.44
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/boneres.2014.44
This article is cited by
-
Nephrologisches Management und Medikamentendosierung bei Rheumapatienten mit Niereninsuffizienz
rheuma plus (2023)
-
Nephrologisches Management und Medikamentendosierung bei Rheumapatienten mit Niereninsuffizienz
Wiener klinisches Magazin (2023)
-
Nephrologisches Management und Medikamentendosierung bei Rheumapatienten mit Niereninsuffizienz
Zeitschrift für Rheumatologie (2022)
-
Skeletal Muscle Complications in Chronic Kidney Disease
Current Osteoporosis Reports (2022)
-
Kidney function and its association to imminent, short- and long-term fracture risk—a longitudinal study in older women
Osteoporosis International (2020)
