
Abstract
The recombinant analogue of human glucagon-like peptide-2 (GLP-2) teduglutide (Gattex®, Revestive®) is a novel therapy for short bowel syndrome (SBS). GLP-2 is a naturally occurring hormone that regulates the growth, proliferation and maintenance of cells lining the gastrointestinal tract. Subcutaneous teduglutide is the first long-term medical therapy approved for the treatment of adult patients with SBS who are dependent on parenteral support (parenteral nutrition and/or intravenous fluids). In a pivotal, double-blind, multicentre, phase III study in adult patients with SBS who were dependent on parenteral support, a significantly higher proportion of teduglutide 0.05 mg/kg/day recipients than placebo recipients achieved at least a 20 % reduction from baseline in weekly parenteral support volume at week 20 and maintained at week 24 (primary endpoint). The overall mean reduction in weekly parenteral support volume from baseline was greater in patients who received teduglutide compared with those who received placebo. Additionally, more teduglutide-treated patients achieved at least a one-day reduction in parenteral support than those receiving placebo. Subcutaneous teduglutide had an acceptable tolerability profile; the most frequently reported adverse events were of gastrointestinal origin, consistent with the underlying disease condition and the known mechanism of action of teduglutide.
Similar content being viewed by others

First targeted long-term therapy approved in the USA and EU for the treatment of adult patients with short bowel syndrome, who are dependent on parenteral support |
Increases villus height and crypt depth of the intestinal epithelium resulting in enhanced absorptive capacity |
Facilitated the reduction in parenteral support volume and provided days off parenteral support in patients with short bowel syndrome |
Acceptable tolerability profile; the most frequently reported adverse events were of gastrointestinal origin |
1 Introduction
Short bowel syndrome (SBS), a serious and highly disabling condition, results from either loss of portions of intestine or loss of critical intestinal function, whereby the amount of remaining functional gut is too short to allow for adequate absorption of nutrients and fluids [1–3]. SBS can be a consequence of surgical resection, congenital defects, trauma or disease-associated loss of absorption [1–3]. In adults, the most common cause of SBS is extensive surgical resection (e.g. for vascular disease, Crohn’s disease, trauma or cancer) [1]. Although the symptom complex varies among patients, SBS is characterised by diarrhoea, steatorrhoea, abdominal pain, electrolyte disturbances, dehydration and malnutrition [2, 3]. These symptoms are due to loss of absorptive surface, site-specific transport processes, site-specific endocrine cells and gastrointestinal (GI) hormones, and/or ileocecal valve [1–3].
During the first couple of years following surgery, the remnant intestine has the ability to adapt in order to compensate for the reduction of absorptive surface area [1, 2, 4]. Structural adaptation involves increases in villus height, crypt cell depth and enterocyte proliferation, while functional adaptation refers primarily to a slowing of the transit time of the bowel, thus allowing for an increased time for absorption to occur [1, 2, 4]. The process of adaptation has been linked to a host of trophic hormones and peptides [1, 2, 4]. Glucagon-like peptide-2 (GLP-2), a hormone that regulates the growth, proliferation and maintenance of cells lining the GI tract, may play a major role in modulating bowel adaptation and nutrient absorption in humans [5]. The adaptive process is a highly variable process, yielding different levels of symptom control in each patient [1, 2].
The effective management of patients with SBS requires a comprehensive multidisciplinary approach [2, 3, 6]. Furthermore, treatment should be individualized because of the considerable differences in remnant bowel anatomy and function, psychosocial traits and personal lifestyles between patients [2, 3, 6]. The overall treatment goal is intestinal rehabilitation, a process of maximizing the digestive and absorptive capacity of the remnant GI tract [7]. Medical management of SBS may be accomplished pharmacologically (antidiarrheal agents [e.g. loperamide, codeine], antisecretory agents [e.g. H2-receptor antagonists, proton-pump inhibitors, somatotropin receptor antagonists] and supplementation of vitamins, minerals and intravenous fluids), through dietary manipulations, parenteral nutrition or intestinal transplantation [6–8].
In the absence of sufficient intestinal adaptation, many patients remain dependent on long-term parenteral nutrition and/or intravenous fluids (parenteral support) to maintain fluid, electrolyte, trace element, vitamin and nutrient balances [6]. In 1992, it was estimated there were ≈10,000 patients with SBS in North America who were dependent on home parenteral nutrition [9]. Moreover, long-term parenteral support is invasive, associated with numerous complications (e.g. catheter-related infections, biliary complications, venous occlusions and hepatic dysfunction) and deleterious effects on health-related quality of life [1, 7].
Growth factors and other trophic hormones have been used to enhance intestinal adaptation [10]. Recombinant human growth hormone was the first medication approved by the US FDA specifically for use in patients with SBS in conjunction with specialized nutritional support [1]. However, the use of growth hormone has been limited, largely owing to concerns with regard to efficacy and the fact that only short-term use was approved, with somatropin and l-glutamine approved for use up to 4 and 16 weeks, respectively, in the USA [8, 11, 12].
Until recently, there were no targeted long-term medical treatments aimed at intestinal rehabilitation to promote absorption of fluid and nutrients [6, 8, 11]. Teduglutide (Gattex®, Revestive®) is a novel intestinotrophic agent [13, 14]. It is a recombinant human GLP-2 analogue with a single amino acid substitution that substantially extends its half-life compared with native GLP-2, allowing it to be administered once daily by subcutaneous injection [13–16]. It is the first long-term medical therapy to be approved as a treatment for adults with SBS who are dependent on parenteral support in the USA [13] and EU [14].
Data sources:
Medical literature (including published and unpublished data) on teduglutide in patients with short bowel syndrome was identified by searching databases including MEDLINE (from 1946) and EMBASE (from 1996) [searches last updated 6 May 2013], bibliographies from published literature, clinical trial registries/databases and websites (including those of regional regulatory agencies and the manufacturer). Additional information (including contributory unpublished data) was also requested from the company developing the drug.
Search terms: Teduglutide, short bowel syndrome.
Study selection: Studies in patients with short bowel syndrome who received teduglutide. Inclusion of studies was based mainly on the methods section of the trials. When available, large, well-designed, comparative trials with appropriate statistical methodology were preferred. Relevant pharmacodynamic and pharmacokinetic data are also included.
Keywords: Teduglutide, small bowel syndrome, pharmacodynamics, pharmacokinetics, therapeutic use, tolerability.
2 Pharmacodynamic Properties
Data on the pharmacodynamic properties of subcutaneous teduglutide are derived from studies in healthy subjects [17–19] or patients with SBS [20–22]. Additional data were obtained from the EU summary of product characteristics (SPC) [14] and the US prescribing information [13].
2.1 Mechanism of Action
Teduglutide is an analogue of naturally occurring human GLP-2 (a peptide secreted by L cells of the distal intestine in response to luminal nutrients) that is manufactured in Escherichia coli using recombinant technology [13, 14]. Teduglutide has been shown to preserve mucosal integrity by promoting repair and normal growth of the intestine through an increase of villus height and crypt depth [13, 14]. GLP-2 is known to increase intestinal and portal blood flow, inhibit gastric acid secretion and decrease intestinal motility [23]. Teduglutide binds to the GLP-2 receptors located in intestinal subpopulations of enteroendocrine cells, subepithelial myofibroblasts, and enteric neurons of the submucosal and myenteric plexus [23]. Activation of these receptors results in the local release of multiple mediators, including insulin-like growth factor-1, nitric oxide and keratinocyte growth factor [23].
2.2 Effects on Gastrointestinal Absorption
Results of a 21-day, open-label, multicentre, pharmacodynamic study (n = 17) suggested that subcutaneous teduglutide 0.03, 0.10 or 0.15 mg/kg/day could benefit parenteral support-dependent patients with SBS in several ways (see Table 1) [20].
Compared with baseline, 21 days of teduglutide treatment increased intestinal wet weight absorption and reduced faecal wet weight, as measured by 72-h metabolic balance studies (see Table 1) [20]. In addition to the decrease in ostomy output, urine output increased by approximately 500 mL/day from baseline levels, demonstrating increased intestinal absorption of fluids and nutrients while parenteral support volume and oral intake were maintained at stable levels throughout the 21-day study [20]. Subgroup analyses revealed similar improvements in patients with end jejunostomy and in patients with ≥50 % colon continuity [20].
Teduglutide was associated with some improvements in energy and macronutrient absorption [20]. A significant decrease in faecal energy excretion was observed (Table 1) [20]. However, this reduction in energy excretion did not translate into an overall significant effect with respect to absolute or relative energy absorption, possibly because of variability in dietary energy intake [20]. No significant effect on intestinal fat or nitrogen absorption was demonstrated [20]. Consistent effects on energy balance were observed following 24 weeks of teduglutide treatment in a 72-h metabolic balance sub-study as part of a placebo-controlled, phase III trial in patients with SBS (study 004; see Sect. 4.1 for trial details) [21].
Absorption of some electrolytes improved during teduglutide treatment, with corresponding decreases in excretion of faecal and stomal fluid in the same study [20]. For example, faecal potassium levels were reduced by 12 mmol/day after 21 days of treatment (p < 0.05 vs. baseline), while faecal sodium levels were reduced by 25 mmol/day. Urinary sodium excretion was increased by 53 mmol/day (p < 0.001 vs. baseline) [20].
In SBS patients with distal bowel resections, malabsorption is not only caused by a reduction in absorptive area, but also by the disruption of the ileal brake feedback mechanism regulated by hormones such as GLP-1 and -2 and peptide YY [6, 24]. The lack of this meal-stimulated hormonal feedback leads to gastric hypersecretion, rapid gastric and intestinal transit, and poor intestinal adaptation [24]. The physiological feedback associated with the ileal brake mechanism appears to be restored during treatment with teduglutide in patients with an end jejunostomy [20].
In addition to 72-h metabolic balance studies, the phase II trial examined the histological changes in bowel biopsies obtained from eight patients with end-jejunostomy [20]. Significant structural adaptations in the intestinal mucosa of the jejunum were seen in seven of the eight teduglutide recipients [20]. For instance, significant increases from baseline in villus height (38 %; p = 0.030), crypt depth (22 %; p = 0.010) and mitotic index (115 %; p = 0.010) were observed after 21 days of teduglutide treatment [20]. These findings were confirmed after 24 weeks of treatment with teduglutide 0.05 mg/kg/day in study 004 [21, 25].
Of note, the improvements in intestinal absorption, decreases in faecal excretion and histological changes observed after treatment with teduglutide had reversed at follow up on days 39–42 after study completion (i.e. 18–21 days drug-free) [20].
2.3 Effects on Body Composition
Lean body mass, measured by dual energy x-ray absorptiometry, was increased by 0.82 kg in teduglutide 0.05 mg/kg/day recipients and decreased by 0.52 kg in placebo recipients after 24 weeks of treatment in study 004, suggesting that intestinal energy and/or fluid absorption was improved with teduglutide [21]. Fat mass was reduced by 1.6 and 3.8 % in teduglutide and placebo recipients, respectively. A numerical increase in total body bone mineral content was observed in teduglutide recipients versus placebo recipients (+0.009 vs. −0.016 kg) [21]. The effect of teduglutide on bodyweight is discussed in Sect. 4.
2.4 Effects on Plasma Citrulline
Plasma citrulline, an amino acid produced by enterocytes of the intestinal mucosa, is regarded as a biomarker of the functional enterocyte mass (i.e. a decrease in citrulline levels reflects a decrease in enterocyte mass) [26]. In general, plasma levels of citrulline were increased from baseline following teduglutide administration in patients with SBS [21, 22]. In two placebo-controlled phase III studies (004 and 020; see Sect. 4 for study details), plasma citrulline levels at week 24 were significantly (p ≤ 0.0001) higher than baseline levels in patients receiving teduglutide 0.05 mg/kg/day, whereas plasma citrulline levels were not significantly changed in placebo recipients [21, 22].
According to additional data from the extension phase of study 004, plasma citrulline was significantly (p = 0.0001) increased by 68 % after a total of 52 weeks of teduglutide treatment [27]. Four weeks after treatment withdrawal, plasma citrulline levels decreased by 20 %; however, levels did remain higher than baseline levels (i.e. at the start of the placebo-controlled trial) [27].
2.5 Other Effects
Subcutaneous teduglutide 4 mg once daily for 10 days had no significant effect on gastric emptying, as measured by paracetamol (acetaminophen) pharmacokinetics in a placebo-controlled trial in 36 healthy volunteers [18, 19]. There were no significant differences in the area under the serum concentration-time curve (AUC) from time zero to the last measurable concentration, AUC from time zero to infinity (AUC∞) or peak serum concentrations (Cmax) of paracetamol between teduglutide and placebo recipients on day 0 or day 10 [18, 19].
Subcutaneous teduglutide 4 mg/day had no clinically significant effect on serum insulin, glucagon or glucose levels in healthy volunteers [18].
Therapeutic and supratherapeutic dosages of teduglutide did not have a significant effect on cardiac repolarization, according to results of a ‘thorough corrected QT (QTc) interval study’ in 72 volunteers [17]. Furthermore, teduglutide had no relevant effect on other ECG parameters (e.g. QRS interval), T or U wave morphology, or heart rate [17].
As teduglutide is a recombinant therapeutic protein, there is potential for formation of antibodies to teduglutide and to E. coli protein (residual host cell protein from the manufacture) [13, 14]. In clinical trials, non-neutralizing antibodies to teduglutide, as well as antibodies to E. coli protein were detected in teduglutide recipients (see Sect. 5) [13, 14, 22]. The development of anti-teduglutide antibodies has not been associated with any clinically relevant effects on the efficacy or tolerability or pharmacokinetics of teduglutide during up to 1.5 years of treatment [13, 14].
Based upon the pharmacodynamic effect of teduglutide, there is a potential for increased absorption of concomitant oral medications such as benzodiazepines and psychotropic agents [13]. In clinical trials, altered mental status during concomitant treatment with teduglutide and benzodiazepines has been observed (see Sect. 5.3) [13]. Therefore, it is recommended that patients receiving concurrent therapy with oral drugs (e.g. benzodiazepines, phenothiazines) requiring titration or with a narrow therapeutic index may require dose adjustment of their concurrent therapy while receiving teduglutide [13].
3 Pharmacokinetic Properties
The pharmacokinetics of subcutaneous teduglutide have been investigated in healthy volunteers [15, 16], patients with SBS [16] and in subjects with renal impairment [28]. Additional data are available from the US prescribing information [13]. Dosages and patient populations are specified where available.
3.1 Absorption and Distribution
The pharmacokinetics of subcutaneous teduglutide (across the dosage range of 2.5–80 mg once daily) were best described by a one-compartment model with a site-specific first-order constant of absorption in the abdomen, arm and thigh, as assessed in a population pharmacokinetic modelling study using data from several trials in healthy volunteers and patients with SBS [16]. Teduglutide displays linear pharmacokinetics, with exposure to the drug increasing in proportion to the dose over the dose range of 10 to 80 mg [15]. There was minimal accumulation of teduglutide following repeated subcutaneous administration in healthy volunteers [15].
Teduglutide was rapidly absorbed following subcutaneous administration, with the Cmax of teduglutide attained approximately 3–5 h after single and multiple doses in healthy volunteers [13]. In patients with SBS, the median Cmax of teduglutide was 36 ng/mL and the median AUC∞ was 0.15 μg·h/mL following a 0.05 mg/kg subcutaneous dose of teduglutide [13].
Following a single dose of teduglutide 0.12 mg/kg administered subcutaneously into the abdomen, the mean absolute bioavailability of teduglutide was approximately 87 % in healthy volunteers [15]. The relative bioavailabilities of teduglutide when administered into the thigh and arm, compared with abdominal administration, were 86.5 and 89.2 %, respectively [15].
The volume of distribution of teduglutide is 103 mL/kg in healthy volunteers, which is similar to blood volume [11, 13], suggesting that teduglutide was not widely distributed throughout the body and was primarily retained in the intravascular compartment.
3.2 Metabolism and Excretion
The metabolic pathway of teduglutide has not been investigated in humans [13]. Since teduglutide is a peptide, it is not likely to be metabolized by common drug metabolizing enzymes (e.g. cytochrome P450 [CYP] isozymes, uridine diphosphate glucuronyltransferase, glutathione-S-transferase) [13]. It is likely that teduglutide is metabolized via hydrolytic degradation, like native GLP-2, to form small peptides and amino acids [13].
The terminal elimination half-life of teduglutide was ≈2 h in healthy subjects and ≈1.3 h in patients with SBS [13]. Following intravenous administration, the plasma clearance of teduglutide was ≈123 mL/h/kg, which is similar to the glomerular filtration rate, suggesting that teduglutide is primarily eliminated via the kidneys [13].
3.3 Special Patient Populations and Drug Interactions
Age and gender had no clinically relevant effect on the pharmacokinetics of teduglutide [13]. Bodyweight was identified as a covariate; in general, an increase in bodyweight was associated with increases in the volume of distribution and the elimination half-life of teduglutide [16].
Decreasing renal function was found to be associated with increased teduglutide exposure [13]. Relative to individuals with normal renal function, the AUC∞ and Cmax values of teduglutide were greater (up to 2.6- and 2.1-fold, respectively) in individuals with progressive renal impairment up to and including end-stage renal disease (see Sect. 6 for dosing recommendations) [13].
The pharmacokinetics of teduglutide were not altered to any clinically significant extent in individuals with moderate hepatic impairment [13]. The pharmacokinetics of teduglutide have not been assessed in severe hepatic impairment [11].
No formal drug-drug interaction studies have been conducted in patients with SBS receiving teduglutide [13]. In vitro studies revealed no significant inhibition or induction of tested CYP isozymes at a teduglutide concentration of 2 μg/mL [11]. Furthermore, teduglutide was neither a substrate nor an inhibitor of P-glycoprotein at a concentrations above 2 μg/mL [11]. Thus, it is considered that the potential for CYP or P-glycoprotein-related pharmacokinetic drug interactions with teduglutide is low.
4 Therapeutic Efficacy
The clinical efficacy of teduglutide in the treatment of adult patients with SBS was evaluated in two 24-week, randomized, double-blind, placebo-controlled, multicentre, phase III studies: study 004 (n = 83) [21] and study 020 (STEPS [Study of Teduglutide Effectiveness in Parenteral nutrition-dependent SBS subjects]; n = 86) [22]. Long-term efficacy and safety of teduglutide has also been demonstrated in two extension studies: study 005 (n = 52) [27] and 021 (n = 88; available as a poster) [29].
Key trial details and patient characteristics at baseline are shown in Table 2. Briefly, adults (aged ≥18 years) with SBS secondary to intestinal surgery and at least 12 continuous months of parenteral support dependency were eligible [21, 22]. Parenteral support dependency was defined as requiring parenteral nutrition and/or IV fluids at least three times per week to meet caloric, fluid or electrolyte needs [21, 22].
Both studies consisted of two stages [21, 22]. The first stage included a screening visit and optimization (up to 8 weeks) and stabilization (4 to 8 weeks) periods [21, 22]. During the optimization period, the goal was to establish a urine output of 1.0–2.0 L/day [21, 22]. During the stabilization period, parenteral support usage was to match prescribed parenteral support, and oral fluid intake and urine volume could not deviate more than 25 % from the optimized levels [21, 22]. Although the osmolality and oral intake were not strictly controlled, patients were asked to try to keep the timing, quantity and quality of beverages as constant as possible during the 48 h collection periods so that increased intestinal fluid absorption would be reflected in increased urinary output [21, 22]. For the second stage, patients underwent randomization and a 24-week treatment period [21, 22].
4.1 Study 004
Eligible patients were randomized to receive subcutaneous teduglutide 0.05 mg/kg/day (n = 35), teduglutide 0.10 mg/kg/day (n = 32) or placebo (n = 16) once daily in study 004 [21]. A strict parenteral support weaning algorithm was followed during treatment that allowed for no more than a 10 % reduction in parenteral support volume at 4-week intervals [21].
The primary efficacy endpoint was initially a dichotomous responder criterion (percentage of patients who demonstrated a ≥20 % reduction from baseline in weekly parenteral support volume at weeks 20 and 24); however, an expanded primary endpoint was later introduced to compare teduglutide with placebo in terms of a graded response score criterion assessed in the intent-to-treat (ITT) population. The graded response score criterion accounted for intensity and duration of response at the end of the 24-week period (from 20–100 % reduction in weekly parenteral volume and the responses from weeks 16 to 20 and weeks 20 and 24) [21]. The statistical analysis plan for the primary endpoint specified a step-down procedure that required teduglutide 0.10 mg/kg/day to be statistically significantly greater than placebo before evaluation of the 0.05 mg/kg/day dosage [21].
Teduglutide 0.10 mg/kg/day was no more effective than placebo for reducing parenteral support requirements in adults with SBS who were dependent on parenteral support as indicated by the primary efficacy endpoint analysis (a graded categorical score) which revealed no statistically significant differences between the teduglutide 0.10 mg/kg/day and placebo groups at 24 weeks [21]. The authors suggest that limitations in the study design, the parenteral support weaning algorithm and an imbalance between the treatment groups at baseline in the parenteral support requirements (parenteral volume and parenteral energy requirements at baseline were 1816 mL/day and 5296 kJ/day, respectively, in the teduglutide 0.10 mg/kg/day group compared with 1531 mL/day and 3385 kJ/day, respectively, in the placebo group) may account for the lack of a significant difference between the teduglutide 0.10 mg/kg/day group and the placebo group for the primary endpoint [21]. Therefore, further efficacy findings for this dosage are not, presented here; this section focuses instead on exploratory findings for the 0.05 mg/kg/day dosage, which is the recommended dosage subsequently approved by the regulatory authorities in the EU [14] and USA [13] (see Sect. 6).
Analysis of the primary endpoint revealed that teduglutide 0.05 mg/kg/day was associated with significantly (p = 0.007) improvements in the graded response score compared with placebo [21]. However, these results are considered hypothesis generating only, because the pre-specified statistical analysis plan for the primary endpoint stated that no further statistical testing was to be done if the teduglutide 0.10 mg/kg/day dosage was not found to be significant [21].
The proportion of patients who were responders was also significantly higher in the teduglutide 0.05 mg/kg/day arm compared with the placebo arm (46 vs. 6 %; p = 0.005) [21].
At week 24, parenteral support volume was reduced from baseline by 2.5 L/week in patients receiving teduglutide (at both dose levels) compared with a reduction of 0.9 L/week in placebo recipients [21]. Furthermore, two patients receiving teduglutide 0.05 mg/kg/day were completely weaned off parenteral support at 24 weeks [21]. At baseline, one patient had been receiving 5.4 L of parenteral support per week for 25 years and the other patient had been receiving 3.5 L of parenteral support per week for 6.5 years [21].
Teduglutide 0.05 mg/kg/day significantly (p < 0.05) increased bodyweight, compared with placebo, at weeks 4, 8 and 16 in patients with SBS who were dependent on parenteral support [21]. Non-significant increases were observed at the other time points [21]. After 24 weeks, mean increases in bodyweight were 1.2 kg in teduglutide recipients and 0.2 kg in placebo recipients, these changes were driven by improvements in lean body mass [21].
4.2 Study 020
The results of study 004 [21] supported the rationale for the use of the 0.05 mg/kg/day dosage of teduglutide in the 020 study, which is the pivotal clinical trial of teduglutide [22]. Eligible patients were randomized to receive subcutaneous teduglutide 0.05 mg/kg/day (n = 43) or placebo (n = 43) once daily [22]. A strict parenteral support weaning algorithm was followed during treatment that allowed for parenteral support volume reduction of 10–30 % [22].
The primary endpoint was the responder rate, defined as the percentage of patients who attained a 20–100 % reduction from baseline in weekly parenteral support volume at week 20 with this response maintained at week 24 [22].
In adult patients with SBS who were dependent on parenteral support, subcutaneous teduglutide 0.05 mg/kg/day was effective in reducing parenteral support fluid requirements, as indicated by a significantly higher responder rates (primary endpoint) in the teduglutide group than in the placebo group (63 vs. 30 %; p = 0.002) [22].
Analyses of the secondary efficacy measures and exploratory endpoints generally supported the benefit of teduglutide [22]. For example, patients receiving teduglutide achieved a greater absolute reduction from baseline in parenteral support volume compared with patients receiving placebo at all visits, beginning at week 4 (1.1 vs. 0.5 L/week) through to week 24 (4.4 vs. 2.3 L/week secondary endpoint), with statistically significant (p ≤ 0.05) differences observed from week 8 until study end at week 24 [22].
In terms of exploratory analyses, the proportion of patients who achieved a reduction from baseline of 20–100 % in prescribed weekly parenteral support was also significantly higher with teduglutide than placebo at week 24 (77 vs. 46 %; p = 0.01) [22]. Furthermore, 54 % of teduglutide-treated subjects experienced at least one additional day off parenteral support at 24 weeks, compared with 23 % of placebo recipients (p = 0.005) [22]. Corresponding proportions of patients who gained more than two additional days off parenteral support were 21 and 8 % at 24 weeks [22]. However, no patients were completely weaned from parenteral support at week 24 during study 020 [22].
Teduglutide was also associated with significantly greater reductions in the fluid composite effect, a putative measure of intestinal fluid absorption (defined as defined as summation of the increase in urine production, reduction in parenteral nutrition and/or intravenous fluid volume and reduction in oral fluid intake [L/week]) than placebo [22]. At week 24, the fluid composite effect was reduced from baseline by 5.4 L/week in teduglutide recipients, compared with a reduction of 1.1 L/week in placebo recipients (p < 0.0006) [22]. Of note, oral fluid intake was significantly higher in placebo recipients than teduglutide recipients at weeks 12, 20 and 24 (all p < 0.05) [22].
Over the 24-week treatment period, teduglutide recipients experienced a mean increase in bodyweight of 1.0 kg, while placebo recipients had a mean decrease in bodyweight of 0.6 kg [22].
4.2.1 Health-Related Quality of Life
Health-related quality of life (QOL) was an exploratory outcome measure of study 020, which was investigated a subset of the per-protocol population (n = 35 teduglutide and 35 placebo recipients) [30]. In this analysis, the short bowel syndrome-quality of life scale (SBS-QOL), a newly validated scale comprising of 17 items (divided into two subscales comprising 11 and 6 items), was used to assess the burden of SBS on patients’ health-related QOL [30]. The total score ranged from 0 (perfect) to 170 (worst), with individual items rated on a 100 mm visual analogue scale [30].
According to the primary HR-QOL analysis, three factors significantly influenced on the change in QOL from baseline to week 24 in patients with SBS who were dependent on parenteral support [30]. These three factors were parenteral support volume reduction (p = 0.0194), baseline SBS-QOL score (p = 0.0009) and the interaction between treatment and parenteral support volume reduction (p = 0.0290); in contrast, the factor ‘treatment’ was found to have no significant influence [30].
Teduglutide significantly improved SBS-QOL total (69.0 vs. 79.7 points at baseline; p = 0.0038), subscale one (48.4 vs. 53.1 points; p = 0.0109) and subscale two (24.2 vs. 26.7 points; p = 0.0225) scores from baseline to week 24 [30]. No significant changes were observed in the placebo group. However, the changes in SBS-QOL observed in the teduglutide group were not significant compared with placebo [30].
Teduglutide significantly (p < 0.05) reduced the scores of 9 of the 17 individual items (including diarrhoea/stomal output, gastrointestinal symptoms, sleep and everyday activities) of the SBS-QOL scale from baseline at week 24 [30]. Only diarrhoea/stomal output was significantly reduced in the placebo group (p = 0.001) [30].
4.3 Long-Term Extension Studies
Because SBS is a chronic disease, it is important to understand whether patients experience continued benefit with teduglutide therapy during long-term use. Patients from the two placebo-controlled trials could enrol in extension trials. Study 005 was a 28-week, randomized, double-blind, multicentre extension of study 004 [27], while study 021 (STEPS-2) is a 2-year open-label extension of the pivotal 020 trial [29]. This section briefly discusses results for the 0.05 mg/kg/day dosage from these trials; some data available as abstracts [29, 31].
4.3.1 Study 005
Study 005 enrolled 65 of 71 eligible patients, i.e., those who had completed study 004. Patients who had originally received teduglutide continued receiving teduglutide 0.05 (n = 25) or 0.10 mg/kg/day (n = 27) in study 005, while those who had previously received placebo were randomized to receive 0.05 (n = 6) or 0.10 mg/kg/day (n = 7) for 28 weeks [31]. Data are reported for the 52 patients (teduglutide 0.05 [n = 25] or 0.10 mg/kg/day [n = 27]) who received a total of 52 weeks of teduglutide therapy during the combined treatment period of the placebo-controlled and the extension studies [27].
Improvements in parenteral support requirements were generally maintained during long-term treatment (i.e. 52 weeks) with teduglutide 0.05 mg/kg/day in patients with SBS who were dependent on parenteral support [27]. At the end of the combined 52 week treatment period, 68 % of patients in the teduglutide group were responders (reductions of ≥20 % of baseline parenteral support volume [i.e. start of study 004]) [27].
Following 52 weeks of teduglutide treatment, parenteral support volume was reduced from baseline (in initial placebo-controlled trial) by 4.9 L/week (52 %), intravenous energy intake was reduced by 3511.09 kcal/week and 68 % of patients had experienced at least one additional day off parenteral support [27]. Moreover, 75 % of patients who were responders in study 004 (i.e. at 24 weeks), while receiving teduglutide, remained responders at 52 weeks [27]. Two teduglutide recipients who were weaned off parenteral support during study 004 (see Sect. 4.1) remained completely weaned off parenteral support at the end of the 28-week extension [27, 31]. Furthermore, one additional patient was weaned off parenteral support during the extension study [27].
Four weeks after treatment withdrawal, mean parenteral support volume was increased from 4.0 L/week at the end of the 52-week treatment period to 5.5 L/week in teduglutide recipients [27].
4.3.2 Study 021
The extension study (STEPS-2) included 88 patients, of whom 37 patients had previously received teduglutide, 39 patients had received placebo and 12 patients had been optimized and stabilized during study 020 but not treated or included in that trial [29]. There were no significant differences between groups for the mean parenteral support volume required or mean number of days per week of support volume required at baseline [29]. All patients received subcutaneous teduglutide 0.05 mg/kg/day [29].
According to interim results of study 021, treatment with teduglutide 0.05 mg/kg/day was associated with a durable response, as reflected by continued improvements in and the maintenance of reductions in parenteral support needs in patients with SBS who were dependent on parenteral support [29].
At last follow-up (31 May 2012), 74 of the 88 patients enrolled in the extension trial had received 12 months of teduglutide therapy in the extension study [29]. Of these patients, 68 % achieved a 20–100 % reduction in parenteral support volume from baseline (i.e. start of initial placebo-controlled phase [study 020]) [29]. Of the 37 patients originally randomised to teduglutide in study 020, 88 % achieved clinically meaningful reductions from baseline in parenteral support volume after a total of 18 months (6 months of treatment in study 020 and 12 months in study 021) of teduglutide treatment [29].
During the extension study, additional clinically meaningful reductions in parenteral support volumes were observed over those seen in study 020 [29]. In patients who had received teduglutide throughout (total 18 months’ treatment), parenteral support volumes reduced from 7.6 L/week at the start of the extension study to 5.8 L/week at 18 months (baseline at the start of study 020 was 12.9 L/week) [29]. A reduction in parenteral support volume was also observed in patients who switched from placebo to teduglutide therapy in the extension study, with support volumes reducing from 10.5 L/week at the start of the extension study to 8.4 L/week at 18 months (baseline 13.2 L/week) [29]. Similar improvements were also seen in patients who had not been included in study 020 and who had started teduglutide therapy during the extension study [29]. Furthermore, following 12 months of teduglutide treatment in the extension study, 38 of 74 patients (51 %) experienced at least one additional day off parenteral support [29].
In addition, some patients achieved complete independence from parenteral support during the first year of the extension study [29]. As of 16 October 2012, complete independence from parenteral support was achieved in 12 (14 %) of 88 patients after 28–114 weeks of treatment with teduglutide 0.05 mg/kg/day [29]. At baseline, parenteral support requirement in these patients ranged between 3.5 and 13.4 L/week and they had been receiving parenteral support for 2–19 years. Although demographics and disease characteristics varied widely among the completely weaned patients, more of them had colon-in-continuity (≈67 % of patients) and lower baseline parenteral support requirements (i.e. <7 L/week; ≈67 %) [29].
5 Tolerability
Discussion in this section focuses on tolerability data from the two phase III trials (study 004 [safety population n = 83] and 020 [safety population n = 85]; see Sect. 4 for study details) [21, 22]. These data are supplemented with information from the US prescribing information [13] and the SPC [14]. While study 004 included a teduglutide 0.10 mg/kg/day arm, discussion in this section focuses on the recommended teduglutide dosage of 0.05 mg/kg/day, unless stated otherwise.
5.1 Short-Term Tolerability
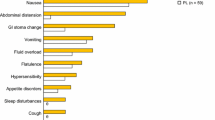
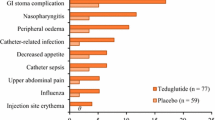
Subcutaneous teduglutide was generally well tolerated in adult patients with SBS who were dependent on parenteral support [21, 22]. The most frequently reported adverse events were usually GI-related (e.g. abdominal pain, nausea), consistent with the underlying disease condition and the known mechanism of action of teduglutide (Fig. 1) [13].

Tolerability of teduglutide 0.05 mg/kg/day in patients with short bowel syndrome who were dependent on parenteral support. Incidence (≥5 % of patients and more frequently than placebo) of clinical adverse events occurring in the 24-week randomized, double-blind, placebo-controlled, multicentre, 004 and 020 studies [13]. GI gastrointestinal, PL placebo, TED teduglutide, URTI upper respiratory tract infection. θ indicates incidence of 0 %
Treatment-emergent adverse events occurred in 94 % of teduglutide and 94 % of placebo recipients in study 004 [21] and in 83 and 79 % of patients, respectively, in study 020 [22]. The majority (≈80 %) of treatment-emergent adverse events were considered not to be related to treatment, and were of mild to moderate severity [14].
Serious adverse events occurred in 37 % of teduglutide and in 31 % of placebo recipients in study 004 [21] and in 36 and 28 % of patients, respectively, in study 020 [22]. In study 004, haemorrhoidal bleeding (n = 1), coma (n = 1), dysgeusia (n = 1) and hypersomnia (n = 1) were reported as serious adverse events in teduglutide group, while catheter sepsis (n = 1) was reported in the placebo group [21]. In study 020, two serious adverse events (acute cholecystitis and small intestinal stenosis) in the teduglutide group were deemed to be treatment-related [22].
Withdrawal rates because of treatment-related adverse events in teduglutide and placebo recipients were 17 and 6 %, respectively, in study 004; one event in a was judged as serious the placebo recipient [21]. In study 020, the respective rates of treatment withdrawal were 5 and 7 %; no events were deemed as serious [22].
No deaths were reported during study 004 or 020 [21, 22]. However, one patient died from a bleeding ulcer during the screening phase of study 004 [21].
Non-neutralizing anti-teduglutide antibodies were detected in six patients who received teduglutide during study 020 [22]. Additionally, the anti-teduglutide antibodies were cross-reactive to native GLP-2 in five of the six (83 %) patients who developed anti-teduglutide antibodies [13]. According to the SPC, 30 % of patients developed anti-teduglutide antibodies and 40 % of patients developed antibodies against E. coli protein during treatment with teduglutide for up to 1 year [14]. There was no correlation between presence of antibodies with short-term (up to 1.5 years) efficacy, tolerability or pharmacokinetic parameters [13].
5.2 Long-Term Tolerability
According to preliminary results of pooled data from the placebo-controlled trials and their respective extension studies (total n = 173), long-term treatment with teduglutide 0.05 or 0.10 mg/kg/day was generally well tolerated in patients with SBS who were dependent on parenteral support [32]. Treatment-emergent adverse events were reported in 95 % of patients in the teduglutide (0.05 or 0.10 mg/kg/day) groups [32]. Serious adverse events and adverse events leading to discontinuation were reported in 56 and 17 % of patients who had received teduglutide 0.05 or 0.10 mg/kg/day, respectively [32]. Abdominal pain followed by GI stoma complications and nausea were the most common adverse events leading to discontinuation [32]. Two deaths were reported during long-term therapy with teduglutide [32].
The most commonly reported adverse events in teduglutide (0.05 or 0.10 mg/kg/day) recipients were GI-related (abdominal pain, nausea) [32]. Generally, adverse events occurred early during therapy (e.g. during the first 6 months of treatment) and their incidence decreased over time [32].
In study 021, the immunogenicity incidence rate increased over time, with anti-teduglutide antibodies being detected in 27 % of teduglutide recipients at 12 months and in 38 % of teduglutide recipients at 18 months [13].
5.3 Adverse Events of Special Interest
Based on the pharmacologic activity and findings in animals, teduglutide has the potential to cause hyperplastic changes, including small intestinal and/or colonic neoplasia (see Sect. 6 for recommendations) [13]. Where reported [21, 25], no dysplastic transformation was observed during histopathological evaluation of the intestinal tissue samples from patients participating in study 004. However, three patients were diagnosed with malignancy during study 021 (STEPS-2), all of whom were male and had received teduglutide in study 020 [29, 32]. One subject had a history of abdominal radiation for Hodgkin’s disease two decades prior to receiving teduglutide and prior liver lesion on computed tomography scan, and was diagnosed with metastatic adenocarcinoma of unconfirmed origin after 11 months of exposure to teduglutide [32]. Two subjects had extensive smoking histories, and were diagnosed with lung cancers (non-small cell and squamous) after 3 and 12 months of teduglutide exposure, respectively [32].
Overall, six patients were diagnosed with intestinal polyps during the clinical trials [32]. Of these patients, one placebo recipient and one teduglutide recipient was diagnosed during the placebo-controlled trials (inflammatory stomal and hyperplastic sigmoidal polyp after 3 and 5 months, respectively) [32]. Four polyp cases were reported in teduglutide 0.05 or 0.10 mg/kg/day recipients in the extension studies (two villous adenomas, one hyperplastic polyp and one small duodenal polyp) [13, 32].
Six teduglutide (0.05 or 10 mg/kg/day)-treated patients during the placebo-controlled trials and six teduglutide-treated patients during the extension studies reported one or more episodes of intestinal obstruction/stenosis [13, 32]. A recurrence of obstruction was reported in two of the six patients during the extension studies [32].
Three patients who had received teduglutide during the placebo-controlled trials were diagnosed with cholecystitis, all of whom had a prior history of gallbladder disease [32]. No cases of cholecystitis were reported in the placebo group [13]. There were reports of acute cholecystitis (n = 3), new-onset cholelithiasis (n = 2) and cholestasis secondary to an obstructed biliary stent (n = 1) with teduglutide in the extension studies [32]. In teduglutide recipients, one case of pancreatic pseudocyst was reported during the placebo-controlled trials and one case each of chronic pancreatitis and acute pancreatitis was reported during the extension studies [32].
Fluid overload occurred in 9 of 77 teduglutide-treated patients (12 %) during the placebo-controlled trials. Of these, two cases were in patients with congestive heart failure (one serious) [32].
During the placebo-controlled trials, one patient, who was also receiving the benzodiazepine prazepam, experienced dramatic deterioration in mental status progressing to coma during her first week of teduglutide treatment [32]. Her benzodiazepine level was found to be >300 μg/L. She recovered following withdrawal of teduglutide and prazepam [32]. See Sect. 2.5 for details of potential drug interactions and recommendations.
6 Dosage and Administration
In adult patients with SBS who are dependent on parenteral support, the recommended dosage of teduglutide is 0.05 mg per kg of bodyweight, administered once daily via subcutaneous injection [13, 14]. Teduglutide injection site should be alternated between one of the four abdominal quadrants, and in some cases the thigh can also be used (e.g. when use of the abdomen is hampered by pain, scarring or hardening of the tissue) [13, 14].
In patients with moderate or severe renal impairment and in those with end-stage renal disease, it is recommended that the teduglutide dosage be reduced by 50 % (see Sect. 3) [13, 14]. No dosage adjustment is necessary for patients with mild and moderate hepatic impairment [13, 14].
Due to the risk of neoplastic growth associated with teduglutide (see also Sect. 5.3), a colonoscopy of the entire colon with removal of polyps should be conducted prior to initiating therapy and also recommended after one year of treatment [13, 14]. Subsequent colonoscopies should be conducted as needed, but no less frequently than every five years. In patients at increased risk for malignancy, the clinical decision to use teduglutide should be considered only if the benefits outweigh the risks. Teduglutide therapy should be discontinued in patients with active gastrointestinal malignancy (GI tract, hepatobiliary, pancreatic). In patients with active non-gastrointestinal malignancy, the clinical decision to continue teduglutide should be made based on benefit:risk considerations [13, 14].
Local manufacturer’s prescribing information should be consulted for further information on treatment regimens, warnings, precautions, and contraindications, as well as the use of teduglutide in special patient populations.
7 Current Status of Teduglutide in Short Bowel Syndrome
Teduglutide is approved in the USA [13] and EU [14] for the treatment of adult patients with SBS who are dependent on parenteral support.
In the pivotal phase III, randomized, double-blind, placebo-controlled study 020 (STEPS), subcutaneous teduglutide 0.05 mg/kg given once daily was effective in the treatment of patients with SBS who required parenteral support as assessed by responder rates (primary endpoint) (Sect. 4.2). A benefit of therapy (as assessed by a graded response score) was also observed with the 0.05 mg/kg/day dosage of teduglutide in the similarly designed study 004 (Sect 4.1). However, no statistically significant difference was observed between teduglutide 0.10 mg/kg/day recipients and placebo recipients in this study.
Subcutaneous teduglutide 0.05 mg/kg/day had an acceptable tolerability profile (Sect. 5); the most frequently reported adverse events were generally of GI origin (e.g. abdominal pain, nausea), consistent with the underlying disease condition and the known mechanism of action of teduglutide. Adverse events of special interest were malignancy; colorectal polyps; GI stenosis/obstruction; gallbladder, biliary, and pancreatic disease; fluid overload; and increased risk of absorption of concomitant oral medications.
The long-term efficacy and tolerability of teduglutide in patients with SBS is currently being investigated in study 021 (STEPS-2) an 2-year open-label extension of study 020, with preliminary interim results suggesting that teduglutide recipients experience additional clinically meaningful reductions in parenteral support during long-term treatment (Sect. 4.3.2). Furthermore, complete independence from parenteral support was achieved in 12 patients, between 28 and 114 weeks after starting teduglutide, in the extension study. The tolerability profile of teduglutide during long-term treatment was similar to what was similar to that observed during short-term treatment. Generally, adverse events occurred early (i.e. during the first 6 months of treatment) and the incidence decreased over time. An extension trial of study 021 [33] and a planned SBS registry (follow-up of 10 years) [34] will provide further information on the long-term efficacy and tolerability of teduglutide. Additionally, a risk evaluation and mitigation strategy (REMS) is in place to ensure that the benefits of teduglutide outweigh the risk [35].
There are no published guidelines regarding the recommended approach for weaning a patient off parental support. However, an article by Seidner et al. [7] discusses the clinical considerations and best practice recommendations for intestinal rehabilitation, focusing on: optimization of fluids, electrolytes and nutrients; integration of teduglutide therapy; and approaches to parenteral nutrition and fluid weaning.
Although demographics and disease characteristics varied widely among the completely weaned patients in study 021, most patients had lower baseline parenteral support requirements (i.e. <7 L/week). Teduglutide may be of most benefit in patients who are borderline between being dependent or independent of parenteral support and it appears likely that these patients will be the ones who are able to completely discontinue parenteral support during teduglutide therapy [36]. However, it has been suggested that even one night free from parenteral support has the potential to improve QOL for many patients [36].
Further clinical trials investigating the optimal timing of teduglutide treatment following onset of SBS and the effects on patients after the discontinuation of teduglutide would be beneficial. In the meantime, the acceptable benefit: risk ratio of teduglutide in study 020 combined with the limited treatment options and seriousness of the condition make teduglutide a welcome addition for the management of SBS.
References
Buchman AL, Scolapio J, Fryer J. AGA technical review on short bowel syndrome and intestinal transplantation. Gastroenterology. 2003;124(4):1111–34.
O’Keefe SJ, Buchman AL, Fishbein TM, et al. Short bowel syndrome and intestinal failure: consensus definitions and overview. Clin Gastroenterol Hepatol. 2006;4(1):6–10.
Misiakos EP, Macheras A, Kapetanakis T, et al. Short bowel syndrome: current medical and surgical trends. J Clin Gastroenterol. 2007;41(1):5–18.
Drozdowski LA, Clandinin MT, Thomson AB. Morphological, kinetic, membrane biochemical and genetic aspects of intestinal enteroplasticity. World J Gastroenterol. 2009;15(7):774–87.
Jeppesen PB, Hartmann B, Thulesen J, et al. Glucagon-like peptide 2 improves nutrient absorption and nutritional status in short-bowel patients with no colon. Gastroenterology. 2001;120(4):806–15.
Nightingale J, Woodward JM. Guidelines for management of patients with a short bowel. Gut. 2006;55(Suppl 4):iv1–12.
Seidner DL, Schwartz LK, Winkler MF, et al. Increased intestinal absorption in the era of teduglutide and its impact on management strategies in patients with short bowel syndrome-associated intestinal failure. JPEN J Parenter Enteral Nutr. 2013;37(2):201–11.
Steiger E. Guidelines for pharmacotherapy, nutritional management, and weaning parenteral nutrition in adult patients with short bowel syndrome: introduction. J Clin Gastroenterol. 2006;40(Suppl 2):S73–4.
Oley Foundation. North American home parenteral and enteral nutrition patient registry annual report. 1994. http://www.oley.org/documents/Patient_Registry_AnnualReportFinal.pdf. Accessed 8 May 2013.
McMellen ME, Wakeman D, Longshore SW, et al. Growth factors: possible roles for clinical management of the short bowel syndrome. Semin Pediatr Surg. 2010;19(1):35–43.
Food and Drug Administration. FDA briefing information for the 16 October 2012 meeting of the GastroIntestinal Drugs Advisory Committee. 2012. http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/GastrointestinalDrugsAdvisoryCommittee/UCM323504.pdf. Accessed 8 May 2013.
Scolapio JS. Short bowel syndrome: recent clinical outcomes with growth hormone. Gastroenterology. 2006;130(2 Suppl 1):S122–6.
NPS Pharmaceuticals. Gattex (teduglutide): US prescribing information. 2012. http://www.npsp.com/file_depot/0-10000000/0-10000/262/folder/2023/Gattex_PI-IFU_FINAL_2012-12-21.pdf. Accessed 8 May 2013.
European Medicines Agency. Revestive (teduglutide): summary of product characteristics. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002345/WC500132926.pdf. Accessed 8 May 2013.
Marier JF, Beliveau M, Mouksassi MS, et al. Pharmacokinetics, safety, and tolerability of teduglutide, a glucagon-like peptide-2 (GLP-2) analog, following multiple ascending subcutaneous administrations in healthy subjects. J Clin Pharmacol. 2008;48(11):1289–99.
Marier J-F, Mouksassi M-S, Gosselin NH, et al. Population pharmacokinetics of teduglutide following repeated subcutaneous administrations in healthy participants and in patients with short bowel syndrome and Crohn’s disease. J Clin Pharmacol. 2010;50(1):36–49.
Hartmann M, Timmer W, Schultz A, et al. A thorough QT study of teduglutide in healthy subjects. Clin Pharmacol Drug Dev. 2012;1(2):57–66.
Berg J, Kim E, Li Y, et al. A randomized, double-blind, placebo-controlled, multiple-dose, parallel-group study to assess the effects of teduglutide on gastric emptying in healthy subjects [abstract no. Sa1959]. Digestive Disease Week 2012 (DDW); 20–22 May 2012; San Diego.
Berg JK, Kim EH, Li B, et al. A randomized, double-blind, placebo-controlled study to assess the effects of teduglutide on gastric emptying in healthy subjects [abstract no. 1520397]. American Society for Parenteral and Enteral Nutrition (ASPEN) Clinical Nutrition Week 2013 (CNW13); 9–12 Feb 2013; Phoenix.
Jeppesen PB, Sanguinetti EL, Buchman A, et al. Teduglutide (ALX-0600), a dipeptidyl peptidase IV resistant glucagon-like peptide 2 analogue, improves intestinal function in short bowel syndrome patients. Gut. 2005;54(9):1224–31.
Jeppesen PB, Gilroy R, Pertkiewicz M, et al. Randomised placebo-controlled trial of teduglutide in reducing parenteral nutrition and/or intravenous fluid requirements in patients with short bowel syndrome. Gut. 2011;60(7):902–14.
Jeppesen PB, Pertkiewicz M, Messing B, et al. Teduglutide reduces need for parenteral support among patients with short bowel syndrome with intestinal failure. Gastroenterology. 2012;143(6):1473–81.
Wallis K, Walters JRF, Gabe S. Short bowel syndrome: the role of GLP-2 on improving outcome. Curr Opin Clin Nutr Metab Care. 2009;12(5):526–32.
Van Citters GW, Lin HC. The ileal brake: a fifteen-year progress report. Curr Gastroenterol Rep. 1999;1(5):404–9.
Tappenden KA, Edelman J, Joelsson B. Teduglutide enhances structural adaptation of the small intestinal mucosa in patients with short bowel syndrome. J Clin Gastroenterol. 2013. doi:10.1097/MCG.0b013e3182828f57
Papadia C, Sherwood RA, Kalantzis C, et al. Plasma citrulline concentration: a reliable marker of small bowel absorptive capacity independent of intestinal inflammation. Am J Gastroenterol. 2007;102(7):1474–82.
O’Keefe SJ, Jeppesen PB, Gilroy R, et al. Safety and efficacy of teduglutide after 52 weeks of treatment in patients with short bowel syndrome intestinal failure. Clin Gastroenterol Hepatol. 2013. doi:10.1016/j.cgh.2012.12.029.
Nave R, Halabi A, Herzog R, et al. Pharmacokinetics of teduglutide in subjects with renal impairment. Eur J Clin Pharmacol. 2012. doi:10.1007/s00228-012-1455-7.
Schwartz L, Fujioka K, Jeppesen PB, et al. Continued improvement and maintenance of parenteral nutrition and/or intravenous fluid support volume reduction in patients with long-term teduglutide treatment: results of an ongoing long-term open-label study [abstract no. 1520168 plus poster]. American Society for Parenteral and Enteral Nutrition (ASPEN) Clinical Nutrition Week 2013 (CNW13); 9–12 Feb 2013; Phoenix.
Jeppesen PB, Pertkiewicz M, Forbes A, et al. Quality of life in patients with short bowel syndrome treated with the new glucagon-like peptide-2 analogue teduglutide - analyses from a randomised, placebo-controlled study. Clin Nutr. 2013. doi:10.1016/j.clnu.2013.03.016.
Gilroy R, Allard J, Jeppesen PB, et al. Treatment out to 1 year with a GLP-2 analog, teduglutide, safely reduces parenteral nutrition (PN) needs in PN-dependent short bowel syndrome patients [abstract no. 273]. Am J Gastroenterol. 2008;103(s1):S105.
Fujioka K, Seidner DL, Delmaestro E, et al. Long-term safety and tolerability of teduglutide in patients with intestinal failure associated with short bowel syndrome: combined data from phase III trials [abstract no. 1520553 plus poster]. American Society for Parenteral and Enteral Nutrition (ASPEN) Clinical Nutrition Week 2013 (CNW13); 9–12 Feb 2013; Phoenix.
NPS Pharmaceuticals. A one-year, open-label study with teduglutide for subjects who completed study CL0600-021 (STEPS 3) (ClinicalTrials.gov identifier NCT01560403) US National Institutes of Health, ClinicalTrials.gov. 2012. http://www.clinicaltrials.gov/ct2/show/NCT01560403. Accessed 8 May 2013.
European Medicines Agency. Revestive: EPAR-Public assessment report. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Public_assessment_report/human/002345/WC500132928.pdf. Accessed 8 May 2013.
NPS Pharmaceuticals. Risk evaluation and mitigation strategy (REMS). 2013. http://www.gattexrems.com/. Accessed 8 May 2013.
Buchman AL. Teduglutide and short bowel syndrome: every night without parenteral fluids is a good night. Gastroenterology. 2012;143(6):1416–20.
Disclosure
The preparation of this review was not supported by any external funding. During the peer review process, the manufacturer of the agent under review was offered an opportunity to comment on this article. Changes resulting from comments received were made by the author on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Additional information
The manuscript was reviewed by: L. Matarese Division of Gastroenterology, Hepatology and Nutrition, East Carolina University, Greenville, North Carolina, USA; E. P. Misiakos Attikon University Hospital, Athens, Greece; A. Shatnawei Department of Gastroenterology and Hepatology, Digestive Disease Institute, The Cleveland Clinic, Cleveland, Ohio, USA; D. L. Sigalet Department of Surgery, Alberta Children’s Hospital & University of Calgary, Calgary, Alberta, Canada; E. Steiger Nutrition Support and Vascular Access Department, Cleveland Clinic Foundation, Cleveland, Ohio, USA; J. S. Thompson Department of Surgery, University Nebraska Medical Center, Omaha, Nebraska, USA; J. Woodward Department of Gastroenterology, Addenbrooke’s Hospital, Cambridge, UK.
Rights and permissions
About this article
Cite this article
Burness, C.B., McCormack, P.L. Teduglutide: A Review of its Use in the Treatment of Patients with Short Bowel Syndrome. Drugs 73, 935–947 (2013). https://doi.org/10.1007/s40265-013-0070-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-013-0070-y