
Abstract
Tumor microenvironment is essential for tumor cell proliferation, angiogenesis, invasion and metastasis through its provision of survival signals, secretion of growth and pro-angiogenic factors, and direct adhesion molecule interactions. This review examines its importance in the induction of an angiogenic response in tumors and in multiple myeloma. The encouraging results of pre-clinical and clinical trials in which tumors have been treated by targeting the tumor microenvironment are also discussed.
Similar content being viewed by others


Tumor growth and metastasis are angiogenesis dependent
The current wisdom is that both solid and hematological tumors are endowed with angiogenic capability and their growth, invasion and metastasis are angiogenesis dependent [1]. Judah Folkman, a highly rated pioneer and researcher in this field wrote: “Once tumor take has occurred, early increase in tumor cell population must be preceded by an increase in new capillaries that converge upon the tumor” [2]. Solid and hematological tumors are endowed with angiogenic capability and their growth, invasion and metastasis are angiogenesis dependent.
Angiogenesis is important for supplying oxygen, nutrients, growth factors, hormones, and proteolytic enzymes which control the coagulation and fibrinolytic systems, as well as dissemination of tumor cells to distal sites. Angiogenesis is controlled by the balance between molecules that have positive and negative regulatory activity. This concept led to the notion of the “angiogenic switch”, which depends on an increased production of one or more positive regulators of angiogenesis [3]. Endothelial cell turnover in the healthy adult organism is low, the quiescence being maintained by the dominant influence of endogenous angiogenesis inhibitors over angiogenic stimuli. In pathological conditions, angiogenesis may be triggered not only by the overexpression of pro-angiogenic factors, but also by the down-regulation of inhibitory factors.
About 30 angiogenic factors have been identified, including vascular endothelial growth factor (VEGF), fibroblast growth factor-2 (FGF-2), transforming growth factor alpha and beta (TGF-α and -β), platelet derived growth factor (PDGF), tumor necrosis factor alpha (TNF-α), angiogenin, interleukins (ILs), chemokines and angiopoietins (Angs). On the other hand, several anti-angiogenic factors have been described, such as angiostatin, endostatin and thrombospondin.
Neoplastic cell populations can only form a clinically observable tumor if the host produces a vascular network sufficient to sustain their growth. Furthermore, new blood vessels provide them with a gateway through which to enter the circulation and metastasize distant sites.
The importance of microenvironment in tumor growth
Studies on neoplastic transformation have focused on events that occur within transformed cells. They have addressed the microenvironment of tumor cells and documented its importance in supporting tumor progression. The pathogenesis of most cancers, in fact, includes complex and mutual interactions that affect the number and phenotype of the tumor cells and various normal stromal cells. The intricate tumor–microenvironmental interactions are increasingly recognized as critical features of several neoplasias.
Tumor cells are surrounded by an infiltrate of inflammatory cells: lymphocytes, neutrophils, macrophages and mast cells, which communicate via a complex network of intercellular signaling pathways, mediated by surface adhesion molecules, cytokines and their receptors. This infiltrate, particularly macrophages, may contribute to tumor angiogenesis, and there are many reports of associations between macrophage infiltration, vascularity and prognosis. Tumor-associated macrophages accumulate in poorly vascularized hypoxic or necrotic areas [4], and respond to experimental hypoxia by increasing the release of VEGF and FGF-2 and a broad range of other factors, such as TNF-α, urokinase and matrix netalloproteases (MMPs) [5]. Moreover, activated macrophages synthesize and release inducible nitric oxide synthase (NOS), which increases blood flow and promotes angiogenesis [6].
The importance of microenvironment in tumor metastasis
It has long been accepted that most malignant tumors show an organ-specific pattern of metastasis. For example, colon carcinomas metastasize usually to liver and lung but rarely to bone, skin or brain and almost never to kidneys, intestine or muscle. In contrast, other tumor entities, such as breast carcinomas, frequently form metastases in most of these organs. This specific formation of secondary tumors at distant sites appears to require a number of steps which must be successfully completed by metastazing tumor cells [7].
Various explanations have been proposed for the site selectivity of blood–bone metastases, including tumor cell surface characteristics [8, 9], response to organ derived chemotactic factors [10], adhesion between tumor cells and the target organ components [11, 12] and response to specific host tissue growth factors [13]. The relative importance of pre-existing tumor subpopulations with specific metastatic properties and the organ environment characteristics in determining metastatic homing have been debated [14–16].
An alternative explanation for the different sites of tumor growth involves interactions between the metastatic cells and the organ environment, possibly in terms of specific binding to endothelial cells and responses to local growth factors. Endothelial cells in the vasculature of different organs express different cell surface receptors and growth factors that influence the phenotype of the corresponding metastases. Greene and Harvey [17] first suggested that the organ distribution patterns of metastatic foci were dependent on the formation of sufficient adhesive bonds between arrested tumor cells and endothelial cells, and they hypothesized that these interactions were similar to lymphocyte/endothelial cells at the sites of inflammation.
The development of organ-derived microvascular endothelial cell cultures has allowed more specific studies on the preferential homing of tumor cells. Auerbach and co-workers [18, 19] found that different tumors showed differences in their adhesive propensity and preference for different endothelial cells, and in a few cases preferential adhesion was observed to the endothelial cells derived from the organ of origin and the target organ.
The “seed and soil” theory
In 1889, the English surgeon Stephen Paget published his “seed and soil” explanation of the non-random pattern of metastasis, and was the first to suggest that interactions between tumor cells and host cells in the microenvironment are critical in regulating tumorigenesis [20]. Certain favored tumor cells (the “seed”), he said, had a special affinity for the growth-enhancing milieu within specific organs (the “soil”), and hence metastasis only occurred when the “seed” and the “soil” were compatible.
The importance of several components of the “soil” in regulating tumor growth has since been emphasised: (1) the extracellular matrix (ECM); (2) stromal cells and their growth factors and inhibitors; (3) microvessels and angiogenic factors; (4) inflammatory cells. There is now substantial evidence that tumor growth and progression depend on the cross talk between malignant cells and their adjacent stromal compartment.
Angiogenesis in multiple myeloma
Multiple myeloma (MM) is a B-cell neoplasm characterized by clonal expansion of malignant plasma cells in the bone marrow, where they proliferate and acquire resistance to apoptosis and eventually lead to osteolysis, renal dysfunction and anemia. It is still incurable with a median survival of approximately 4 years.
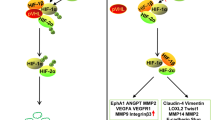
Bone marrow angiogenesis plays an important role in the pathogenesis and progression of MM, as in other hematological malignancies [21]. Growth is halted and a dormancy state is induced in the avascular phase (such as monoclonal gammopathy of undetermined significance (MGUS), or non-active MM) [22], whereas with clonal expansion and epigenetic modifications (hypoxia, shear stress) of the microenvironment tumor plasma cell subsets switch to an angiogenic phenotype that generates the “vascular phase” (active MM), and involves changes in the local balance between pro- and anti-angiogenic factors.
The cause of induction of the vascular phase is the subject of current investigation [23]. Several studies show overexpression and secretion of VEGF by the clonal plasma cells. VEGF stimulates proliferation and chemotaxis in both endothelial cells (EC) via VEGF receptor-2 (VEGFR-2) and stromal cells via VEGFR-1. These cells are rapidly phosphorylated by the interaction with VEGF, and signal via extracellular signal-related kinase-2 (ERK-2) [24].
A murine model for MM indicates that the switch is preceded by the expression of mRNA for VEGF and secretion of the protein by plasma cells, and by a shift from CD45 positive to CD45 negative plasma cells that are the VEGF producers [25]. Accordingly, CD45 expression (as the CD45 positive percentage) by a patient’s bone marrow plasma cells is inversely correlated with the degree of bone marrow angiogenesis [26]. Other studies demonstrate that the expression levels of VEGF, FGF-2 and their receptors overlap between MGUS, smoldering MM (SMM) and newly diagnosed MM (NMM). However, 63% of MGUS samples inhibit angiogenesis in vitro compared to 43% SMM and 4% NMM. Hence the angiogenic switch from MGUS to NMM is partly refefable to an increasing tumor burden rather than increased expression of VEGF and/or FGF-2 and by a loss of the MGUS anti-angiogenic activity [27].
The importance of microenvironment in regulating MM angiogenesis
Since MM mainly progresses in the bone marrow, signals from this microenvironment play a critical role in maintaining plasma cell growth, migration and survival. The pathogenesis of most cancers includes complex and mutual interactions that affect the number and phenotype of the tumor cells and host stromal cells [28]. Reciprocal positive and negative interactions between plasma cells and bone marrow stromal cells (BMSC), namely hematopoietic stem cells, fibroblasts, osteoblasts/osteoclasts, chondroclasts, EC, EC progenitor cells, T lymphocytes, macrophages and mast cells, are mediated by an array of cytokines, receptors, and adhesion molecules. The MM microenvironment is formed by clonal plasma cells, ECM proteins and BMSC, which are intimately involved in all biological stages of intramedullary growth [29]. Interactions between these components determine the proliferation, migration and survival of plasma cells, as well as their acquisition of drug resistance and the development of diseases [30–32]. Receptors expressed by plasma cells, such as αVβ3 integrin, are crucial for their relationships with each other (homotypic interrelationships) and with ECM proteins [33]. Very late activating antigen-4 (VLA-4), leukocyte function-associated antigen (LFA-1), mucin-1 antigen (MUC-1) expressed by plasma cells, and vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1) expressed by BMSC mediate their heterotypic and homotypic interactions. All these interactions result in enhanced expression and release of cytokines and growth factors needed for the plasma cell survival [34].
Plasma cells in the bone marrow microenvironment secrete cytokines, such as TNF-α [35], TGF-β, VEGF, FGF-2 [36], hepatocyte growth factor/scatter factor (HGF/SF) [37], Ang-1 and MMPs [36, 38]. Moreover, binding of plasma cells to BMSC triggers transcription and secretion by the latter of cytokines, such as IL-6 [39], insulin-like growth factor (IGF-1) [40] and VEGF [41] and CXCL12/stromal cell derived factor-1α (SDF-1α) [42], that mediate cell growth (IL-6, IGF-1, VEGF), survival (IL-6, IGF-1), drug resistance (IL-6, IGF-1, VEGF), migration (IGF-1, VEGF, MMPs, SDF-1α) and angiogenesis (VEGF) in the bone marrow. MM endothelial cells (MMEC) differ from umbilical vein EC (HUVEC). They produce growth and invasive factors for plasma cells, including VEGF, FGF-2, MMP-2, and MMP-9 [43].
Bone marrow MMEC express more mRNA and secrete larger amounts of the CXC-chemokines CXCL8/IL-8, CXCL11/interferon-inducible T-cell alpha chemoattractant (I-TAC), CXCL12/SDF-1α and CCL2/monocyte chemotactic protein-1 (MCP-1) than HUVEC, and paired plama cells express cognate receptors to a variable extents, which suggests that paracrine loops between MMEC and plasma cells involving CXC-chemokines and their receptors are operative in MM patients and mediate plasma cell proliferation and chemotaxis [42].
This tumor–host interplay highlights a reciprocal relationship that sustains and promotes the progression of MM by inducing pathological developments, such as angiogenesis and osteolysis [29].
How the pro-angiogenic activity of the cytokines in bone marrow microenvironment may be counteracted
Since the cytokine network between plasma cells and BMSC in the bone marrow milieu promotes plasma cell growth, survival and migration, and plasma cells in the bone marrow are resistant to conventional agent treatment, targeting this network constitute a rationale to the treatment of MM.
Inhibition of NF-kB activity by specific IkB kinase inhibitor down-regulates IL-6 secretion in BMSC and related plasma cell growth [44]. Furthermore, inhibition of either p38MAPK or TFG-β by specific inhibitors down-regulates IL-6 secretion in BMSC [45, 46]. Targeting inhibition of Ras/Raf/MEK/ERK signaling by using the farnesyltransferase inhibitors (FTI) SCH66336 and R115777 abrogates plasma cell growth [47]. The proteasome inhibitor bortezomib (formerly PS-341) can overcome the protective effect of IL-6 against dex-induced apoptosis in MM by inducing caspase-8, -9, -3 activation, which results in caspase-dependent gp130 cleavage [48, 49]. Moreover, bortezomib inhibits DNA repair and may restore the sensitivity of MM cells to DNA damaging chemotherapeutic agents, suggesting that its combination with conventional chemotherapy may augment clinical effectiveness and overcome resistance in patients with relapsed or refractory MM [50].
VEGF and VEGFR inhibitors may offer therapeutic promise. Thalidomide directly inhibits the growth and survival of MM cells and/or BMSC, modulates adhesive interactions between them, and alters the secretion and bioactivity of cytokines they release into the bone marrow milieu [51]. Treatment with two classes of thalidomide analogs, namely selected cytokine inhibitory drugs (Sel-CiDs) and immunomodulatory drugs (IMiDs), may alter MM adhesion to BMSC and fibronectin, and abrogate the up-regulation of IL-6 and VEGF induced by tumor cell binding. Ongoing studies are evaluating the efficacy of a humanized monoclonal antibody against VEGF, bevacizumab (Avastatin) in patients with relapsed or refractory MM (with or without thalidomide) [52].
PTK 787/ZK222584 is an oral tyrosine-kinase inhibitor that also binds VEGFR-1. Hence it acts directly on MM plasma cells and inhibits the autocrine VEGF/VEGFR-1-induced plasma cell growth and migration and the paracrine (IL-6-mediated) growth. PTK/ZK (1.25 mg/day) is currently being evaluated in clinical phase I trials [53]. The indazolypyrimidine GW654652 inhibits all three VEGF receptors with similar potency and the VEGF-triggered migration activity and proliferation of MM cell lines, including those sensitive and resistant to conventional therapy [54]. GW654652 also acts in the bone marrow milieu, since it inhibits both IL-6 and VEGF secretion, as well as proliferation of plasma cells induced by their binding to BMSC; it is anti-angiogenic too since blocks HUVEC proliferation [54].
The therapeutic action of bortezomib-induced inhibition of the proteasome in MM is probably a result of direct cytotoxicity and effects on the bone marrow milieu [49]. The anti-angiogenic effect of bortezomib is another potential mechanism of its anti-MM activity [55, 56]. Moreover, bortezomib down-regulates caveolin-1 expression and inhibits caveolin-1 tyrosine phosphorylation, which are required for VEGF-mediated MM cell migration on fibronectin, and blocks VEGF-induced tyrosine phosphorylation of caveolin-1 in HUVEC, thereby inhibiting ERK-dependent EC proliferation [52].
High local VEGF concentration in the MM bone marrow milieu suppresses the anti-proliferative effect of several chemotherapeutic agents, hence promoting multidrug resistance [57]. Combination of these agents along with drugs that block VEGF signaling may enhance anti-MM efficacy by normalizing and sensitizing the tumor vasculature and improving oxygenation and delivery of such agents to tumor cells and EC.
Inhibition of wild-type and constitutively activated FGFR-3 autophosphorylation in human MM cell lines by the FGFR-specific tyrosine kinase inhibitors SU5402, SU10991, PD173074 or CHIR258 is associated with decreased viability and tumor cell growth arrest, both in vitro and in vivo in a murine model [58–60].
IGF-1 receptor (IGF-1R) inhibition whether by neutralizing anti-IGF-1R specific monoclonal antibodies, antagonistic peptides, or selective IGF-1R kinase inhibitors prevents MM cell proliferation by blocking the Ras/Raf/MAPK and Pl3K/AKT-1 pathways; induces phosphorylation of proapoptotic FKHR; down-regulates intracellular anti-apoptotic proteins, and also increases telomerase activity [40, 61–65].
References
Ribatti D, Vacca A, Dammacco F (1999) The role of vascular phase in solid tumor growth: a historical review. Neoplasia 1:293–302
Folkman J (1984) What is the role of endothelial cells in angiogenesis? Lab Invest 51:601–604
Ribatti D, Nico B, Crivellato E, Roccaro AM, Vacca A (2007) The history of angiogenic switch concept. Leukemia 21:44–52
Leek RD, Lander RJ, Harris AL, Lewis CE (1999) Necrosis correlates with high vascular density and focal macrophages infiltration in invasive carcinoma of the breast. Br J Cancer 79:991–995
Bingle L, Brown NJ, Lewis CE (2002) The role of tumor associated macrophages in tumor progression: implications for new anticancer therapies. J Pathol 196:254–265
Jenkins DC, Charles IG, Thompson LL, Moss DW, Holmes LS, Baylis SA, Rhodes P, Westmore K, Emson PC, Moncada S (1995) Role of nitric oxide in tumor growth. Proc Natl Acad Sci USA 92:4392–4396
Chambers AF, Groom AC, Mac Donald IC (2002) Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer 2:563–572
Reading CL, Hutchins JF (1985) Carbohydrate structure in tumor immunity. Cancer Metastasis Rev 4:221–260
Turner GA (1982) Surface properties of the metastatic cell. Invasion Metastasis 2:197–216
Hujanen ES, Terranova VP (1985) Migration of tumor cells to organ-derived chemoattractants. Cancer Res 45:3517–3521
Nicolson GL (1988a) Cancer metastasis: tumor cell and host organ properties important in metastasis to specific secondary sites. Biochim Biophys Acta 948:175–224
Nicolson GL (1988b) Organ specificity of tumor metastasis: role of preferential adhesion, invasion and growth of malignant cells at specific secondary sites. Cancer Metastasis Rev 7:143–188
Nicolson GL, Dulski KM (1986) Organ specificity of metastatic tumor colonization is related to organ-selective growth properties of malignant cells. Int J Cancer 38:289–294
Fidler IJ (1986) Rationale and methods for the use of nude mice to study the biology and therapy of human cancer metastasis. Cancer Metastasis Rev 5:29–49
Talmadge JE, Fidler IJ (1982) Cancer metastasis is selective or random depending on the parent tumour population. Nature 297:593–594
Weiss L (1979) Dynamic aspects of cancer cell populations in metastasis. Am J Pathol 97:601–608
Greene HS, Harvey EK (1964) The relationship between the dissemination of tumor cells and the distribution of metastases. Cancer Res 24:799–811
Auerbach R, Alby L, Morrissey LW, Tu M, Joseph J (1985) Expression of organ-specific antigens on capillary endothelial cells. Microvasc Res 29:401–411
Auerbach R, Lu WC, Pardon E, Gumkowski F, Kaminska G, Kaminski M (1987) Specificity of adhesion between murine tumor cells and capillary endothelium: an in vitro correlate of preferential metastasis in vivo. Cancer Res 47:1492–1496
Paget S (1889) The distribution of secondary growths in cancer of the breast. Lancet 1:571–573
Ribatti D, Vacca A, Dammacco F, English D (2003) Angiogenesis and anti-angiogenesis in hematological malignancies. J Hematother Stem Cell Res 12:11–22
Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, Dammacco F (1994) Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol 87:503–508
Vacca A, Ribatti D (2006) Bone marrow angiogenesis in multiple myeloma. Leukemia 20:193–199
Vacca A, Ria R, Ribatti D, Semeraro F, Djonov V, Di Raimondo F, Dammacco F (2003b) A paracrine loop in the vascular endothelial growth factor pathway triggers tumor angiogenesis and growth in multiple myeloma. Haematologica 88:176–185
Asosingh K, De Raeve H, Menu E, Van Riet I, Van Mark E, Van Camp B, Vanderkerken K (2004) Angiogenic swithc during 512MM murine myeloma tumorigenesis: role of CD45 heterogeneity. Blood 103:3131–3177
Kumar S, Rajkumar SV, Kimlinger T, Greipp PR, Witzig TE (2005) CD45 expression by bone marrow plasma cells in multiple myeloma: clinical and biological correlations. Leukemia 19:1466–1470
Kumar S, Witzig TE, Timm M, Huagg J, Welik L, Kimlinger TK, Greipp PR, Rajkumar SV (2004) Bone marrow angiogenic ability and expression of angiogenic cytokines in myeloma: evidence favoring loss of marrow angiogenesis inhibitory activity with disease progression. Blood 104:1559–1165
Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, Porter D, Hu M, Chin L, Richardson A, Schnitt S, Sellers WR, Polyak K (2004) Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell 6:17–32
Kuehl WM, Bersagel PL (2002) Multiple myeloma: evolving genetic events and host interactions. Nature Rev Cancer 2:175–187
Damiano JS, Cress AE, Hazlehurst LA, Shtil AA, Dalton WS (1999) Cell adhesion mediated drug resistance (CAM-DR): role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 93:1658–1667
Hazlehurst LA, Damiano JS, Buyuksal I, Pledger WJ, Dalton WS (2000) Adhesion to fibronectin via beta 1 integrin regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 19:4319–4327
Hazlehurst LA, Enkemann SA, Beam CA, Argilagos RF, Painter J, Shain KH, Saporta S, Boulware D, Moscinski L, Alsina M, Dalton WS (2003) Genotypic and phenotypic comparisons of de novo and acquired melphalan resistance in an isogenic multiple myeloma cell line model. Cancer Res 63:7900–7906
Vacca A, Ria R, Presta M, Ribatti D, Iurlaro M, Merchionne F, Tanghetti E, Dammacco F (2001) Alpha (v) beta (3) integrin engagement modulates cell adhesion, proliferation, and protease secretion in human lymphoid tumor cells. Exp Hematol 29:993–1003
Hideshima T, Podar K, Chauhan D, Anderson KC (2005) Cytokines and signal transduction. Best Pract Res Clin Haematol 18:509–524
Hideshima T, Nakamura N, Chauhan D, Anderson HC (2001) The role of tumor necrosis factor alpha in the pathophysiology of human multiple myeloma: therapeutic applications. Oncogene 20:5991–6000
Vacca A, Ribatti D, Presta M, Minischetti M, Iurlaro M, Ria R, Albini A, Bussolino F, Dammacco F (1999) Bone marrow neovascularization, plasma cell angiogenic potential, and matrix metalloproteinase-2 secretion parallel progression of human multiple myeloma. Blood 93:3064–3073
Borset M, Hjorth-Hansen H, Seidel C, Sundan A, Waage A (1996a) Hepatocyte growth factor and its receptor c-met in multiple myeloma. Blood 88:3998–4004
Barillé S, Aknhoundi C, Colette M, Mellerin MP, Rapp MJ, Harousseau JL, Bataille R, Amiot M (1997) Metalloproteinases in multiple myeloma: production of matrix metalloproteinase-9 (MMP-9), activation of proMMP-2, and induction of MMP-1 by myeloma cells. Blood 90:1649–1655
Dankbar B, Padro T, Leo R, Feldmann B, Koropff M, Mesters RM, Serve H, Berdel WE, Kienast J (2000) Vascular endothelial growth factor and interleukin-6 in paracrine tumor-stromal cell interactions in multiple myeloma. Blood 95:2630–2636
Ferlin M, Noraz N, Hertogh C, Brochier J, Taylor N, Klein B (2000) Insulin-like growth factor induces the survival and proliferation of myeloma cells through an interleukin-6 independent transduction pathway. Br J Haematol 111:626–634
Gupta D, Treon SP, Shima Y, Hideshima T, Podar K, Tai YT, Lin B, Lentzsch S, Davies FE, Chauhan D, Schlossman RL, Richardson P, Ralph P, Wu L, Payvandi F, Muller G, Stirling DI, Anderson KC (2001) Adherence of multiple myeloma cells to bone marrow stromal cells up-regulates vascular endothelial growth factor secretion: therapeutic applications. Leukemia 15:1950–1961
Pellegrino A, Ria R, Di Pietro G, Cirulli T, Surico G, Pennisi A, Morabito F, Ribatti D, Vacca A (2005) Bone marrow endothelial cells in multiple myeloma secrete CXC-chemokines that mediates interactions with plasma cells. Br J Haematol 129:248–256
Vacca A, Ria R, Semeraro F, Merchionne F, Coluccia M, Boccarelli A, Scavelli C, Nico B, Gernone A, Battelli F, Tablio A, Guidolin D, Petrucci MT, Ribatti D, Dammacco F (2003) Endothelial cells in the bone marrow of patients with multiple myeloma. Blood 102:3340–3348
Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC (2002) NF-kappa B as a therapeutic target in mutiple myeloma. J Biol Chem 277:16639–16647
Hayashi T, Hideshima T, Nguyen AN, Munoz O, Podar K, Hamasaki M, Ishitsuka K, Yasui H, Richardson P, Chakravarty S, Murphy A, Chauhan D, Higgins LS, Anderson KC (2004) Transforming growth factor beta receptor I kinase inhibitor down-regulates cytokine secretion and multiple myeloma cell growth in the bone marrow microenvironment. Clin Cancer Res 10:7540–7546
Hideshima T, Podar K, Chauhan D, Ishitsuka K, Mitsiades C, Tai YT, Hamasaki M, Raje N, Hideshima H, Schreiner G, Nguyen AN, Navas T, Munshi NC, Richardson PG, Higgins LS, Anderson KC (2004) p38 MAPK inhibition enhances PS-341 (bortezomib)-induced cytotoxicity against multiple myeloma cells. Oncogene 23:8766–8776
Ochiai N, Uchida R, Fuchida S, Okano A, Okamoto M, Ashihara E, Inaba T, Fujita N, Matsubara H. Shimazaki C (2003) Effects of farnesyl transferase inhibitor R115777 on the growth of fresh and cloned myeloma cells in vitro. Blood 102:3349–3353
Hideshima T, Richardson P, Chauhan D, Palombella WJ, Elliott PJ, Adams J, Anderson KC (2001b) The proteasome inhibitor PS-341 inhibits growth, induces apotosis, and overcomes drug resistance in multiple myeloma cells. Cancer Res 61:3071–3073
Hideshima T, Chauhan D, Hayashi T, Akiyama M, Mitsiades N, Mitsiades C, Podar K, Munsji N, Richardson PG, Anderson KC (2003) Proteasome inhibitor PS-341 abrogates IL-6 triggering signaling cascade via caspase-dependent down-regulation of gp130 in multiple myeloma. Oncogene 22:8386–8393
Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Akiyama M, Hideshima T, Chauhan D, Joseph M, Libermann TA, Garcia-Echeverria C, Pearson MA, Hofmann F, Anderson KC. Kung AL (2004) Inhibition of the insulin-like growth factor receptor-1 tyrosine kinase activity as a therapeutic strategy for multiple myeloma, other hematologic malignancies, and solid tumors. Cancer Cell 5:221–230
Ribatti D, Vacca A (2005) Therapeutic renaissance of thalidomide in the treatment of hematological malignancies. Leukemia 18:1525–1531
Podar K, Anderson KC (2005) The pathophysiological role of VEGF in hematologic malignancies: therapeutic implications. Blood 105:1383–1395
Bruno B Giaccone L, Rotta M, Anderson K, Boccadoro M (2005) Novel targeted drugs for the treatment of multiple myeloma: from bench to bedside. Leukemia 19:1729–1738
Podar K, Catley LP, Tai YT, Shringarpure R, Carvalho P, Hayashi T, Burger R, Schlossman RL, Richardson PG, Pandite LN, Kumar R, Hideshima T, Chauhan D, Anderson KC (2004) GW654652, the pan-inhibitor of VEGF receptors, blocks the growth and migration of multiple myeloma cells in the bone marrow microenvironment. Blood 103:3474–3479
Le Blanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J, Gupta D, Richardson PG, Munshi NC, Anderson KC (2002) Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res 62:4996–5000
Roccaro AM, Hideshima T, Raje N, Kumar S, Ishitsuka K, Yasui H, Shiraishi N, Ribatti D, Nico B, Vacca A, Dammacco F, Richardson PG, Anderson KC (2006) Bortezomib mediates antiangiogenesis in multiple myeloma via direct and indirect effects on endothelial cells. Cancer Res 66:184–191
Tran J, Master Z, Yu JL, Rak J, Dumont DJ, Kerbel RS (2002) A role of survivin in chemoresistance of endothelial cells mediated by VEGF. Proc Natl Acad Sci USA 99:4349–4354
Grand EK, Chase AJ, Heath C, Rahemtulla A, Cross NC (2004) Targeting FGFR3 in mutiple myeloma: inhibition of t (4; 14)-positive cells by SU5402 and PD173074. Leukemia 18:962–966
Trudel S, Ely S, Farooqi Y, Affer M, Robbiani DF, Chesi M, Bergsagel PL (2004) Inhibition of fibroblast growth factor receptor 3 induces differentiation and apoptosis in t (4; 14) myeloma. Blood 103:3521–3528
Trudel S, Li ZH, Wei E, Weismann M, Chang H, Chen C, Reece D, Heise C, Stewart AK (2005) CHIR-258, a novel multitargeted tyrosine kinase inhibitor for the potential treatment of t (4; 14) multiple myeloma. Blood 105:2941–2948
Akiyama M, Hideshima T,Hayashi T, Tai YT, Mitsiades CS, Mitsiades N, Chauhan D, Richardson P, Munschi NC, Anderson KC (2002) Cytokines modulate telomerase activity in human multiple myeloma cell line. Cancer Res 62:3876–3882
Ge NI, Rudikoff S (2000) Insulin-like growth factor I as a dual effector of multiple myeloma cell growth. Blood 96:2856–2861
Mitsiades CS, Mitsiades N, Poulaki V, Schlossman R, Akiyama M, Chauhan D, Hideshima T, Treon SP, Munshi NC, Richardson PG, Anderson KC (2002) Activation of NF-kappa B and up-regulation of intracellular anti-apoptotic proteins via the IGF-1/Akt signaling in human multiple myeloma cells: therapeutic implications. Oncogene 21:5673–5683
Quiang YW, Kopantzev E, Rudikoff S (2002) Insulinlike growth factor I signaling in multiple myeloma: downstream elements, functional correlate, and pathway cross-talk. Blood 99:4138–4146
Tu Y, Gardner A, Lichtenstein A (2000) The phosphatidylinostol 3 kinase/AKT kinase pathway in multiple myeloma plasma cells: roles in cytokine-dependent survival and proliferative responses. Cancer Res 60:6763–6770
Acknowledgments
This study was supported by Associazione Italiana per la Ricerca sul Cancro (AIRC, National and Regional Funds), Milan, the Ministry for Education, the Universities and Research (FIRB 2001, PRIN 2005, Progetto Carso 72/2), Rome, and Fondazione Italiana per la Lotta al Neuroblastoma, Genoa, Italy.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ribatti, D., Vacca, A. The role of microenvironment in tumor angiogenesis. Genes Nutr 3, 29–34 (2008). https://doi.org/10.1007/s12263-008-0076-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12263-008-0076-3