
Summary
Antibody–drug conjugates (ADCs) are a relatively new class of highly potent molecules which combine the targeting properties of monoclonal antibodies with the cell destructive properties of cytotoxic agents in order to reduce systemic exposure and toxicity of the latter. Gemtuzumab–ozogamicin was the first-in-class drug approved by the US Food and Drug Administration (FDA) in 2000, but later approval was withdrawn. In the meantime, the number of these types of drugs available for clinical use is rapidly evolving. This review gives a brief overview of currently approved ADCs, with special consideration of pharmaceutical aspects
Similar content being viewed by others


Introduction
Classic chemotherapy is based on the theory that cytotoxic agents, interfering in cell division at different timepoints, would destroy cancer cells, whereas healthy cells would not be harmed due to their lower division rate. Nevertheless, chemotherapy causes well-known toxicities and the search for targeted therapies with specific efficacy on tumor cells is not yet completed satisfactorily.
Already the Nobel laureate Paul Ehrlich (1854–1915) developed the idea of what he called “magic bullet” [1]: a therapeutic agent, which would be able to identify its target, without harming the body itself. He also anticipated the concept of attaching a toxin (e.g., arsenic) to an antibody to improve therapeutic specificity [2]. Based on this understanding, we are nowadays able to design a multitude of highly complex molecular structures.
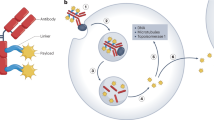
Antibody–drug conjugates (ADCs) are molecules consisting of three components (Fig. 1), all of which are relevant for pharmacological properties of the drug.

Schematic representation of antibody–drug conjugates (ADCs) and their mechanism of action. Clinical efficacy of ADCs is determined by fine-tuning combination of tumor antigen, targeting antibody, cytotoxic payload and conjugation strategy (a). ADC binds to tumor target cell surface antigens (b) leading to trigger a specific receptor mediated internalization (c). The internalized ADCs are decomposed to release cytotoxic payloads inside the tumor cell either through its linkage/linker sensitivity to protease, acidic, reductive agents or by lysosomal process, leading to cell death (d). Ab antibody. From: [24]. This is an open access article under the CC BY license (http://creativecommons.org/licenses/by/4.0/). This figure is not included under the Creative Commons CC BY license of this publication.
Antibody
The antibody is responsible for the delivery of the cytotoxic agent to the tumor cells. It needs target specificity and high target-binding affinity. Furthermore, low immunogenicity is important [3].
Linker
The linker has the essential function to maintain the conjugate in an inactive, nontoxic state while circulating in the blood; it unleashes the cytotoxic drug upon internalization in the tumor cells. Its chemical properties determine how and when this release occurs and therefore determine whether the so-called bystander effect might occur, an unwanted toxic effect on surrounding healthy cells [4].
Noncleavable linkers provide higher stability and do not unleash the cytotoxic agent at off-target sites and therefore reduce toxicity. The only approved ADC with an uncleavable linker is trastuzumab–emtansine (T-DM1) [5].
Most of the clinically approved ADCs have cleavable linkers [6] which use the inherent properties of tumor cells to release the cytotoxine: β‑glucuronidase richness in tumor necrotic regions, acidic tumor microenvironment, lysosomal proteases expressed by tumor cells, lower pH, elevated intracellular concentration of glutathione [7].
All available drugs (Table 1) are formulated as a lyophilized powder, which needs reconstitution and further dilution upon infusion. In clinical routine, those compounding procedures are performed by pharmaceutical professionals; thus, interdisciplinary communication between treating physicians and responsible pharmacists is essential, especially due to short in-use shelf-life once the drug is reconstituted.
Chemical characteristics of the linkers are not only responsible for the in vivo activity of the compound, but also for its physicochemical shelf-life. Whereas trastuzumab–emtansine [8] has a chemically stable, noncleavable linker and therefore long in-use stability (e.g., 24 h), drugs with a cleavable linker are more prone to chemical instability. Examples include inotuzumab–ozogamicin [9] and sacituzumab–govitecan (SG) [10]—both of which are stable for only 4 h, once reconstituted. Degradation via hydrolysis, resulting in both increased toxicity and/or reduced efficacy, is the most likely effect assumable for low in-use stability time. This might also be the reason why most of the products require protection from light from the beginning of reconstitution until end of application (e.g., trastuzumab–deruxtecan [11], gemtuzumab–ozogamicin [12]).
Payload
The cytotoxic payload becomes activated upon release from the ADC inside the cytoplasm of tumor cells. Essential properties are a small molecular weight and a long half-life.
Three principles of action are used in currently approved drugs:
-
Microtubule-disrupting agents: auristatin, maytansinoids.
-
DNA-alkylating agents: calicheamicins, pseudomonas exotoxin, pyrrolobenzodiazepine.
-
Topoisomerase inhibitors: camptothecin derivatives.
Drug–drug interactions
Cytochrome P450 with all its subtypes, especially CYP3A, plays a significant role in the metabolism of drugs. With auristatin [13] and maytansine [14] being metabolized via CYP3A4, the expectation would be significant interaction between ADCs containing these payloads and strong inhibitors or inductors. Even though these effects may be seen in in vitro investigations, no clinically significant changes in plasma levels are observed. One possible explanation could be the small amount of payload circulating in plasma, which is in line with the mechanism of the linker releasing the payload only upon cellular internalization.
This also applies for SN-38, a substrate of UDP-glucuronyltransferase (UGT1A) [15] which also underlies inducing and inhibiting alterations, for instance, through phenytoin and ciclosporin, respectively.
However, in order to provide subjects with best possible precautions, pharmaceutical counselling or drug-drug interaction (DDI) checks should be performed before any change of systemic oncological treatment [16].
Regarding toxicity management, it is worth noting that the cytotoxic payload of the conjugate might have a different safety profile than its equivalent used as classical chemotherapy. As an example, the emetogenic risk of the camptothecin derivates shall be mentioned: whereas irinotecan (including its active metabolite SN-38) and topotecan are usually classified as having low-to-moderate risk, sacituzumab–govitecan and trastuzumab–deruxtecan are of moderate-to-high risk [17].
To summarize, with newly developed antibodies, linkers and payloads, ADCs represent an interesting extension of medical oncological treatment options, which is an advancement of medicine toward Ehrlich’s vision of magic bullets in clinical practice.
References
Bäumler E. Auf der Suche nach der Zauberkugel. 1st ed. : Econ; 1963.
Strebhardt K, Ullrich A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat Rev Cancer. 2008;8(6):473–80.
Khongorzul P, Ling CJ, Khan FU, et al. Antibody-drug conjugates: a comprehensive review. Mol Cancer Res. 2020;18(1):3–19.
Staudacher AH, Brown MP. Antibody drug conjugates and bystander killing: is antigen-dependent internalisation required? Br J Cancer. 2017;5;117(12):1736–42.
Tsuchikama K, An Z. Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell. 2018;9(1):33–46.
Lyon R. Drawing lessons from the clinical development of antibody-drug conjugates. Drug Discov Today Technol. 2018;30:105–9.
Bargh JD, Isidro-Llobet A, Parker JS, Spring DR. Cleavable linkers in antibody-drug conjugates. Drug Discov Today Technol. 2018;30:105–9.
SMPC Kadcyla. 2018. https://www.ema.europa.eu/en/documents/product-information/kadcyla-epar-product-information_en.pdf. Accessed 06 Oct 2021.
SMPC Besponsa. 2017. https://www.ema.europa.eu/en/documents/product-information/besponsa-epar-product-information_en.pdf. Accessed 06 Oct 2021.
SMPC Trodelvy. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761115s000lbl.pdf. Accessed 06 Oct 2021.
SMPC Enhertu. 2021. https://www.ema.europa.eu/en/documents/product-information/enhertu-epar-product-information_en.pdf. Accessed 06 Oct 2021.
SMPC Mylotarg. 2018. https://www.ema.europa.eu/en/documents/product-information/mylotarg-epar-product-information_en.pdf. Accessed 06 Oct 2021.
Han TH, Gopal AK, Ramchandren R, et al. CYP3A-mediated drug-drug interaction potential and excretion of brentuximab vedotin, an antibody-drug conjugate, in patients with CD30-positive hematologic malignancies. J Clin Pharmacol. 2013;53(8):866–77.
Davis JA, Rock DA, Wienkers LC, et al. In vitro characterization of the drug-drug interaction potential of catabolites of antibody-maytansinoid conjugates. Drug Metab Dispos. 2012;40(10):1927–34.
Ma MK, McLeod HL. Lessons learned from the irinotecan metabolic pathway. Curr Med Chem. 2003;10(1):41–9.
van Leeuwen RWF, Jansman FGA, van den Bemt PMLA. Drug-drug interactions in patients treated for cancer: a prospective study on clinical interventions. Ann Oncol. 2015;26(5):992–7.
National Comprehensive Cancer Network NCCN. Clinical Practice Guidelines in Oncology—Antiemesis, Version 1.2021. 2020.
SMPC Adcetris. 2020. https://www.ema.europa.eu/en/documents/product-information/adcetris-epar-product-information_en.pdf. Accessed 06 Oct 2021.
SMPC Polivy. 2020. https://www.ema.europa.eu/en/documents/product-information/polivy-epar-product-information_en.pdf. Accessed 06 Oct 2021.
SMPC Blenrep. 2020. https://www.ema.europa.eu/en/documents/product-information/blenrep-epar-product-information_en.pdf. Accessed 06 Oct 2021.
SMPC Padcev. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761137s000lbl.pdf. Accessed 06 Oct 2021.
SMPC Lumoxiti. 2021. https://www.ema.europa.eu/en/documents/product-information/lumoxiti-epar-product-information_en.pdf. Accessed 06 Oct 2021.
SMPC Zynlonta. 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf. Accessed 06 Oct 2021.
Nejadmoghaddam MR, Minai-Teheran A, Ghahremanzadeh R, et al. Antibody-drug conjugates: possibilities and challenges. Avicenna J Med Biotechnol. 2019;11(1):3–23.
Funding
Open access funding provided by Medical University of Vienna.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
M.-B. Aretin declares that she has no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aretin, MB. Antibody–drug conjugates—the magic bullet?. memo 15, 125–128 (2022). https://doi.org/10.1007/s12254-021-00780-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12254-021-00780-8