
Summary
Purpose. This phase II, open-label, multicenter study assessed the oral, multitargeted, tyrosine kinase inhibitor sunitinib in patients with advanced gastric or gastroesophageal junction adenocarcinoma who had received prior chemotherapy. Experimental design. Patients received sunitinib 50 mg/day on Schedule 4/2 (4 weeks on treatment, followed by 2 weeks off treatment). The primary endpoint was objective response rate; secondary endpoints included clinical benefit rate, duration of response, progression-free survival (PFS), overall survival (OS), pharmacokinetics, pharmacodynamics, safety and tolerability, and quality of life. Results. Of 78 patients enrolled, most had gastric adenocarcinoma (93.6%) and metastatic disease (93.6%). All were evaluable for safety and efficacy. Two patients (2.6%) had partial responses and 25 patients (32.1%) had a best response of stable disease for ≥6 weeks. Median PFS was 2.3 months (95% confidence interval [CI], 1.6–2.6 months) and median OS was 6.8 months (95% CI, 4.4–9.6 months). Grade ≥3 thrombocytopenia and neutropenia were reported in 34.6% and 29.4% of patients, respectively, and the most common non-hematologic adverse events were fatigue, anorexia, nausea, diarrhea, and stomatitis. Pharmacokinetics of sunitinib and its active metabolite were consistent with previous reports. There were no marked associations between baseline soluble protein levels, or changes from baseline, and measures of clinical outcome. Conclusions. The progression-delaying effect and manageable toxicity observed with sunitinib in this study suggest that although single-agent sunitinib has insufficient clinical value as second-line treatment for advanced gastric cancer, its role in combination with chemotherapy merits further study.
Similar content being viewed by others


Introduction
Gastric cancer is the fourth most common cancer globally, with an estimated 934,000 new cases in 2002 [1]. Patients presenting or relapsing with metastatic disease have a poor prognosis, and with 700,000 deaths annually, gastric cancer is the second most common cause of death from cancer worldwide [1]. In Japan and Korea, mass screening has led to a shift towards diagnosis at earlier stages of the disease, and the 5-year survival rate is relatively high at 40–60% [2, 3]. Globally, 5-year survival is lower, at approximately 20% [2]. In clinical trial patients with advanced gastric cancer, reported median survival commonly ranges from 8 months to 11 months in the first-line treatment setting and approximately 5 months to 6 months in the second-line treatment setting [4–6].
Combination chemotherapy prolongs survival and improves quality of life in patients with gastric cancer, compared with best supportive care [7, 8]. Recently, a meta-analysis showed a small but significant survival benefit for combination chemotherapy versus single-agent chemotherapy, though at a cost of higher toxicity [8]. There is no globally accepted standard regimen for first-line treatment of advanced gastric cancer, though a 5-fluorouracil-based regimen in combination with a platinum analog is reported to be the most widely accepted regimen [9]. As yet, there are no data showing acceptable efficacy for gastric cancer in the second line setting. New treatment strategies are still needed to improve the survival of patients with advanced gastric cancer, both in the first-line treatment setting and in those patients whose disease has progressed during or after chemotherapy.
Tumor angiogenesis, growth, and metastasis can be inhibited by blocking receptor tyrosine kinases (RTKs), including vascular endothelial growth factor receptors (VEGFRs) and platelet-derived growth factor receptors (PDGFRs), which are both expressed or overexpressed in gastric cancer [10–12]. VEGF and PDGF-A expression have been linked to tumor progression and poor survival in gastric cancer [13, 14], and both VEGF and VEGFR expression have been correlated with increasing stage of disease [15]. Treatments that specifically interrupt RTK signalling pathways have been investigated in phase II studies in advanced gastric cancer, including a study of single-agent gefitinib [16, 17] and targeted therapies such as bevacizumab [18], cetuximab [19, 20], and erlotinib [21] in combination with chemotherapy. These targeted agents act through a single receptor pathway. However, many gastric tumors co-express several RTKs [10] and drugs targeting multiple RTKs involved in angiogenesis may deliver additional benefits relative to single receptor target inhibition.
Sunitinib malate (SUTENT®; Pfizer Inc., New York, NY) is an oral, multitargeted tyrosine kinase inhibitor of VEGFR-1, -2, and -3, PDGFR-α and -β, and several other related RTKs [22–24]. In a murine xenograft model of gastric carcinoma, sunitinib exhibited antiangiogenic and antitumor activity at a dose of 40 mg/kg/day (Pfizer Inc. Data on file). At a dose of 50 mg/day given on Schedule 4/2 (4 weeks on treatment followed by 2 weeks off treatment), sunitinib has demonstrated superior efficacy to previous standard treatments and acceptable tolerability in gastrointestinal stromal tumors refractory or intolerant to imatinib, and advanced renal cell carcinoma [25, 26]. This phase II trial investigated the use of single-agent sunitinib in patients with previously-treated, advanced gastric carcinoma.
Materials and methods
Patients
Patients eligible for inclusion were males and females aged ≥18 years with histologically or cytologically confirmed diagnosis of gastric adenocarcinoma or gastroesophageal junction adenocarcinoma (i.e. adenocarcinoma with >50% extension in the stomach) that was not amenable to surgery, radiation, or combined modality therapy with curative intent, and who had disease progression or recurrence after treatment with one prior chemotherapy regimen for advanced or metastatic disease (last dose ≥4 weeks before study entry).
Patients who had received prior adjuvant therapy were eligible if relapse occurred >6 months after completing adjuvant treatment and had received one regimen for relapsed disease. Those who had received prior palliative radiotherapy to metastatic lesions were also eligible, if at least one measurable lesion had not been irradiated. Patients were excluded if they had: major surgery or radiation therapy <4 weeks before starting study treatment; grade 3 hemorrhage (based on the National Cancer Institute [NCI] Common Terminology Criteria for Adverse Events [CTCAE]) <4 weeks before starting study treatment; presence of clinically relevant ascites (requiring paracentesis) and/or grade ≥2 weight loss; active inflammatory bowel disease, partial or complete bowel obstruction, or chronic diarrhea; known brain metastases, spinal cord compression, or carcinomatous meningitis; uncontrolled hypertension; clinically significant cardiovascular disease (severe/unstable angina, myocardial infarction, coronary artery bypass graft, symptomatic congestive heart failure), pulmonary embolism, or cerebrovascular accident within 12 months prior to study drug administration; ongoing cardiac dysrhythmias (NCI CTCAE grade ≥2), atrial fibrillation, or prolongation of the QTc interval; or any other severe acute or chronic medical or psychiatric condition making the patient inappropriate for entry into the study in the judgment of the investigator.
All patients had: measurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST); Eastern Co-operative Oncology Group (ECOG) performance status of 0 or 1; adequate hepatic, renal, and hematologic function; and life expectancy of ≥3 months; and were required to provide written, informed consent.
Study design and treatment
In this phase II, open-label, 2-stage, multicenter study, patients received oral sunitinib 50 mg/day on Schedule 4/2 (4 weeks on treatment, followed by 2 weeks off treatment) in repeated 6-week cycles, until disease progression, unacceptable toxicity, or withdrawal of consent. Dose reduction to 37.5 mg/day and then to 25 mg/day was allowed, and therapy could be interrupted or delayed for up to 4 weeks according to individual tolerability.
The primary objective of this study was to determine the antitumor activity of single-agent sunitinib in this population. The primary endpoint was the overall objective response rate (ORR), defined as the percentage of all patients who experienced a confirmed complete response (CR) or partial response (PR), as defined by RECIST [27]. Secondary endpoints included duration of response (in those with an objective response of CR or PR); clinical benefit rate (CBR, defined as the percentage of patients with CR, PR, or stable disease [SD] ≥24 weeks); progression-free survival (PFS); time to progression (TTP); OS; one-year survival rate; safety and tolerability; health-related quality of life (HRQoL); and measurement of trough sunitinib and SU12662 (the major active metabolite of sunitinib) plasma levels, as well as levels of plasma biomarkers (VEGF, soluble (s) VEGFR2, sVEGFR3, and sKIT). This study was approved by the institutional review board of each participating center and was performed in accordance with the International Conference on Harmonization Good Clinical Practice guidelines, as well as applicable local laws and regulatory requirements.
Assessments
Tumor response was assessed according to RECIST version 1.0, with a minor modification such that lesions assessed using spiral computed tomography (CT) scan qualified as measurable if they were twice the reconstruction interval used (up to 8 mm) and at least 10 mm at baseline. Tumor response was assessed: on day 28 of every cycle; whenever disease progression was suspected; to confirm a CR or PR (at least 4 weeks after initial documentation of response); and at the end of study treatment or withdrawal from the study. Tumors were imaged using CT scan or magnetic resonance imaging.
Safety was assessed at regular intervals by monitoring and recording adverse events and by measuring hematology and clinical chemistries. Additional safety assessments included 12-lead electrocardiograms, vital signs, physical examination, and ECOG performance status. Adverse events were graded using NCI CTCAE, version 3.0.
Blood samples were taken for pharmacokinetic analysis of sunitinib and SU12662 prior to sunitinib treatment on study day 1, on days 14 and 28 of the first treatment cycle, on days 1 and 28 of cycles 2 and 3, and on day 28 of cycle 5. Sunitinib and SU12662 concentrations were analyzed using a validated, sensitive, and specific isocratic liquid chromatographic tandem mass spectrometric method, as previously described [28]. Blood samples for biomarker assessment were taken prior to sunitinib treatment on study day 1, on days 14 and 28 of the first treatment cycle, on days 1 and 28 of cycle 2, and on day 28 of cycle 5.
Patient-reported outcomes were assessed using the validated, self-administered European Organisation for Research and Treatment of Cancer (EORTC) Quality of Life Questionnaire QLQ-C30, and the stomach cancer-specific questionnaire QLQ-STO22 [29, 30]. The questionnaires were completed on the first day of each cycle during a patient’s clinic visit prior to other clinical activities including the administration of the study drugs, and at the end of treatment or withdrawal from the study.
Statistical considerations
This study followed a 2-stage Simon design. If ≤1 objective response (CR or PR) was observed in the first 38 eligible patients, then enrollment to the study would end. If ≥2 of these patients achieved a CR or PR, then the study was planned to proceed to Stage 2 by enrolling 25 additional patients. Based on Simon’s 2-stage design, this study had 85% power to reject the null hypothesis of a 5% response rate (considered not clinically meaningful) when the true response rate for sunitinib was ≥15% (considered favorable in this patient population). With a significance level (α) of 5%, 63 eligible patients were required, and at the end of the study, the null hypothesis would be rejected if ≥7 objective tumor responses were observed.
The study population for all analyses was defined as the number of patients enrolled in the study who received at least one dose of sunitinib, and (for analysis of ORR, duration of response, CBR, TTP, and PFS) had measurable disease at baseline. The number (%) of patients who achieved an objective response was summarized along with the corresponding 95% exact confidence interval (CI). Time-to-event variables, 1-year survival rate, and a 2-sided 95% CI were estimated and summarized using the Kaplan-Meier method.
Results
Patient characteristics and study treatment
In total, 78 patients were enrolled in the study (Fig. 1), of whom 73 (93.6%) had a diagnosis of gastric adenocarcinoma, and 5 (6.4%) had adenocarcinoma of the gastroesophageal junction. A total of 73 patients (93.6%) had metastatic disease. Baseline characteristics are summarized in Table 1.

Patient disposition. ITT, intention-to-treat. RECIST, Response Evaluation Criteria in Solid Tumors
The median duration of treatment was 1.6 months (range, 0.1–15.4), and the median number of cycles started was 2 (range, 1–17). Fourteen patients (17.9%) required at least one dose reduction to 37.5 mg/day, mainly due to hematologic adverse events; three of these patients had ≥2 dose reductions. Median relative dose intensity was 93.5%. The relative dose intensity was highest during cycles 1 and 2 (96.4% and 100%, respectively) and ranged from 50.0% to 96.4% during cycles 3–17. Sixteen patients (20.5%) required one or more doses of sunitinib to be delayed, with 12 dose delays lasting for ≥1 week, 6 for ≥2 weeks, and 1 for ≥3 weeks. Reasons for study discontinuation were lack of efficacy (n = 55), adverse events (n = 11), death (n = 8), and withdrawal of consent (n = 2).
During follow-up, among 69 patients for whom data were available, 39 received post-study chemotherapy; the most common regimens were single-agent taxanes, FOLFIRI or FOLFOX, or cisplatin-based combinations. Japanese and Korean patients were most likely to receive later lines of chemotherapy (approximately 75% of enrolled patients) but no significant differences were noted in the types of chemotherapy delivered. Five patients received radiotherapy during the follow-up period, and one underwent surgical resection of metastatic ovarian cancer.
Efficacy
All 78 patients had measurable disease at baseline and were included in the efficacy analyses. Two patients achieved confirmed investigator-determined PR, with a response duration of 20 weeks in one patient and at least 6 weeks (before study discontinuation) in the other patient. Both patients achieving a PR were enrolled in Stage 1 of the study, hence the study proceeded to Stage 2. However, with no further responses seen during Stage 2, the primary endpoint of the study was not met, with an ORR of 2.6%. Twenty-five patients (32.1%) had stable disease (SD) for ≥6 weeks, including four patients (5.1%) experiencing SD lasting ≥24 weeks. The clinical benefit rate was 7.7%. Forty-two patients (53.8%) experienced disease progression; the remaining nine patients (11.5%) had missing evaluations or were not evaluable.
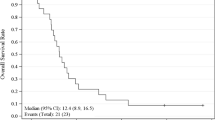
By intent-to-treat analysis (n = 78), median TTP was 2.3 months (95% CI, 1.7–2.6 months), median PFS was 2.3 months (95% CI, 1.6–2.6 months; Fig. 2a), and median OS was 6.8 months (95% CI, 4.4–9.7 months; Fig. 2b). The probability of 1-year survival was 24.2% (95% CI, 14.4–34.1%).

Kaplan-Meier curve of a progression-free survival and b overall survival following treatment with sunitinib 50 mg/day on Schedule 4/2
Pharmacokinetics and pharmacodynamics
Steady-state observed trough concentrations (Ctrough) were dose-corrected to the starting dose (i.e. reference dose) where appropriate, to adjust for individual dose changes during the study. Mean, dose-corrected, plasma Ctrough on day 28 (steady state) of cycles 1, 2, 3, and 5 ranged from 62.2 ng/mL to 65.6 ng/mL for sunitinib, 26.0 ng/mL to 33.7 ng/mL for its active metabolite SU12662, and 90.7 ng/mL to 97.9 ng/mL for total drug (sunitinib + SU12662), respectively. The mean dose-corrected Ctrough box plot of the total drug concentration versus cycle/day is displayed in Fig. 3. No unexpected accumulation of sunitinib and SU12662 was observed throughout the study.

Total drug (sunitinib + SU12662) dose-corrected (reference dose: 50 mg) plasma trough concentration versus cycle/day box plot. Box boundaries denote 25th and 75th percentiles; lines within the box show the median value and expected range of the median. Whiskers indicate the minimum and maximum data values; where outliers are present (asterisks), whiskers extend to a maximum of 1.5 times the interquartile range
Baseline soluble protein (biomarker) levels or changes from baseline at each time point were analyzed in patients stratified by tumor response category (clinical benefit [PR or SD ≥24 weeks] versus progressive disease). Significant associations with clinical benefit were only observed between high sKIT ratio to baseline at cycle 1 day 28 (P = 0.0081), and between low VEGF-C ratio at cycle 2 day 1 (P = 0.0326), though the number of patients with clinical benefit was relatively small (n = 6). Analysis of patients stratified according to whether they were above or below median time-to-event endpoints for PFS or TTP found no significant differences in any of the soluble proteins studied; there was a modest association between elevated baseline plasma VEGF-C levels and above-median OS (P = 0.0241).
Safety
All 78 patients received at least one dose of sunitinib and were included in the safety analyses (Table 2). The most commonly reported treatment-emergent, all-causality, non-hematologic adverse events were fatigue, anorexia, nausea, diarrhea, and stomatitis (Table 2). Most non-hematologic adverse events were grade 1 or 2. Grade 3 or 4 events included fatigue (10.3%), anorexia, hand–foot syndrome, hyperbilirubinemia (6.4% each), and abdominal pain (5.1%). The most common hematologic toxicities were thrombocytopenia (61.5% of patients; 34.6% grade 3 or 4, and one patient with a grade 5 event) and neutropenia (52.6% of patients, 29.4% grade 3 or 4). Thirteen patients (16.7%) experienced grade 3 or 4 anemia. There were no cases of neutropenic fever. Of non-hematologic laboratory adverse events, blood alkaline phosphatase was increased in 10.3% of the study population and occurred at grade 3, the maximum grade reported, in only two patients. Increases in gamma glutamyl transferase were infrequent (2.6%) and of grade 2 severity.
Twenty-four patients (30.8%) permanently discontinued study treatment due to an adverse event; in 14 patients, the adverse events were judged by the investigators to be treatment related. Non-fatal, treatment-related adverse events leading to discontinuation were grade 3 fatigue (n = 2) and grades 2 and 4 mucositis, grade 3 nausea, grade 1 ascites, grade 4 thrombocytopenia, grade 3 hand–foot syndrome, grade 4 abdominal pain plus grade 1 anorexia, and combined grade 2 thrombocytopenia and grade 1 nausea, stomatitis, fatigue, skin erosion and hand–foot syndrome (n = 1 each). Non-treatment-related discontinuations due to adverse events were attributed by investigators to the disease under study (n = 8) or other illness (n = 2; stomach cancer perforation and infection, respectively). Nine patients (11.5%) had a dose reduction due to treatment-emergent adverse events, all of which were treatment-related.
Eleven patients (14.1%) died during the reporting period (during treatment or within 28 days after the last dose of study drug), with eight of these patients having death as the reason for discontinuation of the study. Four of the 11 deaths were considered to be treatment-related adverse events (three during treatment and one within 28 days after the last dose of sunitinib) and seven due to adverse events unrelated to treatment (six due to disease progression; one due to hypotension, depressed level of consciousness and hypopnea). The deaths considered to be treatment-related were due to thrombocytopenia and pulmonary embolism; brain herniation (preceded by upper gastrointestinal bleeding at day 14); cardiac arrest; and brainstem hemorrhage occurring 21 days after the last dose of study drug, respectively.
HRQoL
QLQ-C30 questionnaires were completed by 64 patients at baseline (cycle 1, day 1); completion rates were generally high during treatment but upon withdrawal from the study the completion rate fell to 69.2%. From a mean baseline global health status/HRQoL of 62.3, HRQoL was maintained by sunitinib treatment during the first three cycles of this study, though the domains of diarrhea and reflux symptoms were noticeably worse compared to baseline. Beyond cycle 3, HRQoL data were available for <10 patients per cycle due to study discontinuations.
At patients’ last evaluation (end of treatment or withdrawal from the study), noticeable changes (deterioration) were observed in most scales and measures of the EORTC QLQ-C30 and QLQ-STO22 compared to the baseline. The domains for perceived financial difficulties, body image, and hair loss did not change noticeably.
Discussion
In this study, sunitinib showed preliminary activity in the second-line treatment setting in patients with advanced gastric adenocarcinoma or gastroesophageal junction adenocarcinoma. Following two objective responses in Stage 1, both stages of the study were enrolled, but overall the study did not meet its primary endpoint, with only two patients achieving a PR by RECIST for an overall RECIST-defined ORR of 2.6%. However, the clinical benefit rate was 7.7% and one-third of patients experienced a best response of SD. The median OS duration of 6.8 months, and the median PFS and TTP of 2.3 months with single-agent sunitinib in this study are comparable with those reported in the second-line treatment setting in similar phase II trials of single-agent chemotherapy, such as docetaxel [31, 32], paclitaxel [33, 34], irinotecan [35], or mitomycin C [36], as well as various chemotherapy combinations [4–6]. This level of efficacy is clearly insufficient to support further study of sunitinib as a single-agent in this population, although these data support the proof of concept that sunitinib does affect the late clinical course of gastric cancer.
Recently, the use of trastuzumab in combination with chemotherapy was found to significantly prolong survival when given as first-line treatment for patients with HER2-positive gastric or gastroesophageal cancer [37]—this is the first time that a regimen including a targeted agent has been shown to provide a survival benefit in patients with advanced gastric cancer. It can be hypothesized that the progression-delaying effects observed with sunitinib in our trial might be enhanced if sunitinib is given in combination with chemotherapy, and this is being investigated in the first-line treatment setting in phase I trials at present.
In general, the type and frequency of reported adverse events were consistent with those previously reported with sunitinib when administered as a single agent [25, 26, 38, 39]. Adverse events were generally manageable, as dose schedule modifications (mainly dosing delays) were required in less than half of the patients, though the incidence of permanent discontinuations due to treatment-related adverse events was 18%. This included four (5.1%) treatment-related deaths (thrombocytopenia/pulmonary embolism, brain herniation preceded by upper gastrointestinal bleeding, cardiac arrest, and one patient who died 21 days after the last dose of study drug from brainstem hemorrhage). The predominant non-fatal, treatment-related adverse events leading to discontinuation were fatigue and mucositis. Most non-hematologic adverse events were Grade 1 or 2 in severity. The most common Grade 3 or 4 non-hematologic events included fatigue, anorexia, hand–foot syndrome, hyperbilirubinemia, and abdominal pain, each reported in ≤10% of patients. However, the incidence and severity of hematologic adverse events during sunitinib treatment was higher in this population than in gastrointestinal stromal tumor (GIST) and metastatic renal cell carcinoma (mRCC) patients [25, 26]. Grade 3 or 4 neutropenia or thrombocytopenia was reported in approximately one-third of patients, but only one case of hemorrhagic thrombocytopenia was reported, and there were no cases of neutropenic fever. The majority of adverse events were managed by standard medical intervention and sunitinib dosing interruption, with or without dose reduction.
Analysis of the HRQoL endpoints measuring gastric cancer-related symptoms, general cancer-related symptoms, overall health status and quality of life shows that these scores were largely maintained during the first three cycles of sunitinib treatment. Given that patients discontinued sunitinib treatment due to insufficient clinical response, the subsequent worsening in health status was more likely due to disease progression than to drug toxicity in this single-arm study of limited sample size.
Based on the dose-corrected Ctrough data, the pharmacokinetics of sunitinib and its metabolite in advanced/metastatic gastric cancer patients were consistent with previous experience with sunitinib at 50 mg/day on Schedule 4/2 in patients with advanced solid tumors [40]. No unexpected accumulation of sunitinib or its metabolite was observed throughout the study. Assessment of soluble protein levels versus measures of clinical outcome only showed associations between clinical benefit and high sKIT ratio to baseline at cycle 1 day 28 (P = 0.0081), and between clinical benefit and low VEGF-C ratio at cycle 2 day 1 (P = 0.0326). However, there were a limited number of patients with clinical benefit to include in these analyses. In general, the patterns of pharmacodynamic changes in soluble protein levels observed were similar to those seen in previously reported sunitinib studies [41].
In exploring the potential of sunitinib in gastric cancer, several hypotheses can be proposed that may have had an impact on the limited efficacy observed in this trial. Firstly, in the absence of known predictive biomarkers, it was not possible in this trial to select a subset of gastric cancer patients who might be more likely to respond to sunitinib, which is in contrast to the ability to preselect HER2-positive patients likely to be sensitive to trastuzumab in the ToGA trial [37]. Further understanding of who may benefit from treatment could help to refine the target population for future studies. It is also notable that ORR assessed using RECIST was selected as the primary endpoint of this study. However, observations with targeted agents in other tumor types, for example imatinib in GIST [42] and sunitinib in RCC [43], suggest that one-dimensional RECIST measurements can miss important information about changes in tumor density and metabolic response. This raises the question as to whether ORR is the most suitable endpoint for assessing sunitinib in gastric cancer.
Dose selection and schedule could also be explored further. On the intermittent schedule used in this and other studies of sunitinib, pharmacodynamic modulation of several plasma proteins associated with angiogenesis was reversible during the off-treatment period [38, 43–45]. This raises the question of whether continuous administration of sunitinib might be of benefit, to ensure continuous suppression of angiogenesis. Ultimately, these hypotheses would all require testing in a trial setting.
In summary, the preliminary activity and manageable toxicity observed with sunitinib in this study suggest that although single-agent sunitinib has insufficient clinical value as second-line treatment for advanced gastric cancer, its role in combination with chemotherapy is worthy of further study. The results of ongoing phase I trials in the first-line treatment setting will provide more insight into the use of multiple-RTK inhibitors such as sunitinib in the treatment of advanced gastric cancer.
References
Parkin DM, Bray F, Ferlay J, Pisani P (2005) Global cancer statistics, 2002. CA Cancer J Clin 55:74–108. doi:10.3322/canjclin.55.2.74
Inoue M, Tsugane S (2005) Epidemiology of gastric cancer in Japan. Postgrad Med J 81:419–424. doi:10.1136/pgmj.2004.029330
Jung KW, Yim SH, Kong HJ, Hwang SY, Won YJ, Lee JK, Shin HR (2007) Cancer survival in Korea 1993–2002: a population-based study. J Korean Med Sci 22 Suppl:S5–S10.:S5–S10. doi: 10.3346/jkms.2007.22.S.S5
Scartozzi M, Galizia E, Verdecchia L, Berardi R, Antognoli S, Chiorrini S, Cascinu S (2007) Chemotherapy for advanced gastric cancer: across the years for a standard of care. Expert Opin Pharmacother 8:797–808. doi:10.1517/14656566.8.6.797
Wilson D, Hiller L, Geh JI (2005) Review of second-line chemotherapy for advanced gastric adenocarcinoma. Clin Oncol (R Coll Radiol) 17:81–90. doi:10.1016/j.clon.2004.10.006
Wohrer SS, Raderer M, Hejna M (2004) Palliative chemotherapy for advanced gastric cancer. Ann Oncol 15:1585–1595. doi:10.1093/annonc/mdh422
Glimelius B, Ekstrom K, Hoffman K, Graf W, Sjoden PO, Haglund U, Svensson C, Enander LK, Linne T, Sellstrom H, Heuman R (1997) Randomized comparison between chemotherapy plus best supportive care with best supportive care in advanced gastric cancer. Ann Oncol 8:163–168
Wagner AD, Grothe W, Haerting J, Kleber G, Grothey A, Fleig WE (2006) Chemotherapy in advanced gastric cancer: a systematic review and meta-analysis based on aggregate data. J Clin Oncol 24:2903–2909. doi:10.1200/JCO.2005.05.0245
Ohtsu A (2008) Chemotherapy for metastatic gastric cancer: past, present, and future. J Gastroenterol 43:256–264. doi:10.1007/s00535-008-2177-6
Drescher D, Moehler M, Gockel I, Frerichs K, Muller A, Dunschede F, Borschitz T, Biesterfeld S, Holtmann M, Wehler T, Teufel A, Herzer K, Fischer T, Berger MR, Junginger T, Galle PR, Schimanski CC (2007) Coexpression of receptor-tyrosine-kinases in gastric adenocarcinoma—a rationale for a molecular targeting strategy? World J Gastroenterol 13:3605–3609
Zhang H, Wu J, Meng L, Shou CC (2002) Expression of vascular endothelial growth factor and its receptors KDR and Flt-1 in gastric cancer cells. World J Gastroenterol 8:994–998
Becker JC, Muller-Tidow C, Serve H, Domschke W, Pohle T (2006) Role of receptor tyrosine kinases in gastric cancer: new targets for a selective therapy. World J Gastroenterol 12:3297–3305
Katano M, Nakamura M, Fujimoto K, Miyazaki K, Morisaki T (1998) Prognostic value of platelet-derived growth factor-A (PDGF-A) in gastric carcinoma. Ann Surg 227:365–371
Maeda K, Chung YS, Ogawa Y, Takatsuka S, Kang SM, Ogawa M, Sawada T, Sowa M (1996) Prognostic value of vascular endothelial growth factor expression in gastric carcinoma. Cancer 77:858–863
Takahashi Y, Cleary KR, Mai M, Kitadai Y, Bucana CD, Ellis LM (1996) Significance of vessel count and vascular endothelial growth factor and its receptor (KDR) in intestinal-type gastric cancer. Clin Cancer Res 2:1679–1684
Doi T, Koizumi W, Siena S, Cascinu S, Ohtsu A, Michael M, Takiuchi H, Swaisland H, Gallagher N, Van Cutsem E (2003) Efficacy, tolerability and pharmacokinetics of gefitinib (ZD1839) in pretreated patients with metastatic gastric cancer. Proc Am Soc Clin Oncol 22:Abstract 1036.
Rojo F, Tabernero J, Albanell J, Van CE, Ohtsu A, Doi T, Koizumi W, Shirao K, Takiuchi H, Cajal S, Baselga J (2006) Pharmacodynamic studies of gefitinib in tumor biopsy specimens from patients with advanced gastric carcinoma. J Clin Oncol 24:4309–4316. doi:10.1200/JCO.2005.04.2424
Shah MA, Ramanathan RK, Ilson DH, Levnor A, D’Adamo D, O’Reilly E, Tse A, Trocola R, Schwartz L, Capanu M, Schwartz GK, Kelsen DP (2006) Multicenter phase II study of irinotecan, cisplatin, and bevacizumab in patients with metastatic gastric or gastroesophageal junction adenocarcinoma. J Clin Oncol 24:5201–5206. doi:10.1200/JCO.2006.08.0887
Pinto C, Di FF, Siena S, Cascinu S, Rojas Llimpe FL, Ceccarelli C, Mutri V, Giannetta L, Giaquinta S, Funaioli C, Berardi R, Longobardi C, Piana E, Martoni AA (2007) Phase II study of cetuximab in combination with FOLFIRI in patients with untreated advanced gastric or gastroesophageal junction adenocarcinoma (FOLCETUX study). Ann Oncol 18:510–517. doi:10.1093/annonc/mdl459
Han SW, Oh DY, Im SA, Park SR, Lee KW, Song HS, Lee NS, Lee KH, Choi IS, Lee MH, Kim MA, Kim WH, Bang YJ, Kim TY (2009) Phase II study and biomarker analysis of cetuximab combined with modified FOLFOX6 in advanced gastric cancer. Br J Cancer 100:298–304. doi:10.1038/sj.bjc.6604861
Dragovich T, McCoy S, Fenoglio-Preiser CM, Wang J, Benedetti JK, Baker AF, Hackett CB, Urba SG, Zaner KS, Blanke CD, Abbruzzese JL (2006) Phase II trial of erlotinib in gastroesophageal junction and gastric adenocarcinomas: SWOG 0127. J Clin Oncol 24:4922–4927. doi:10.1200/JCO.2006.07.1316
Abrams TJ, Lee LB, Murray LJ, Pryer NK, Cherrington JM (2003) SU11248 inhibits KIT and platelet-derived growth factor receptor beta in preclinical models of human small cell lung cancer. Mol Cancer Ther 2:471–478
Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, Chan E, Moss KG, Haznedar JO, Sukbuntherng J, Blake RA, Sun L, Tang C, Miller T, Shirazian S, McMahon G, Cherrington JM (2003) In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res 9:327–337
O’Farrell AM, Abrams TJ, Yuen HA, Ngai TJ, Louie SG, Yee KW, Wong LM, Hong W, Lee LB, Town A, Smolich BD, Manning WC, Murray LJ, Heinrich MC, Cherrington JM (2003) SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 101:3597–3605. doi:10.1182/blood-2002-07-2307
Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich MC, Morgan JA, Desai J, Fletcher CD, George S, Bello CL, Huang X, Baum CM, Casali PG (2006) Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet 368:1329–1338. doi:10.1016/S0140-6736(06)69446-4
Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, Chen I, Bycott PW, Baum CM, Figlin RA (2007) Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med 356:115–124
Therasse P, Arbuck SG, Eisenhauer EA, Wanders J, Kaplan RS, Rubinstein L, Verweij J, Van GM, van Oosterom AT, Christian MC, Gwyther SG (2000) New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 92:205–216. doi:10.1093/jnci/92.3.205
Bello CL, Sherman L, Zhou J, Verkh L, Smeraglia J, Mount J, Klamerus KJ (2006) Effect of food on the pharmacokinetics of sunitinib malate (SU11248), a multi-targeted receptor tyrosine kinase inhibitor: results from a phase I study in healthy subjects. Anticancer Drugs 17:353–358
Aaronson NK, Ahmedzai S, Bergman B, Bullinger M, Cull A, Duez NJ, Filiberti A, Flechtner H, Fleishman SB, de Haes JC (1993) The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst 85:365–376. doi:10.1093/jnci/85.5.365
Blazeby JM, Conroy T, Bottomley A, Vickery C, Arraras J, Sezer O, Moore J, Koller M, Turhal NS, Stuart R, Van CE, D’haese S, Coens C (2004) Clinical and psychometric validation of a questionnaire module, the EORTC QLQ-STO 22, to assess quality of life in patients with gastric cancer. Eur J Cancer 40:2260–2268. doi:10.1016/j.ejca.2007.07.005
Giuliani F, Gebbia V, De Vita F, Maiello E, Di Bisceglie M, Catalano G, Gebbia N, Colucci G (2003) Docetaxel as salvage therapy in advanced gastric cancer: a phase II study of the Gruppo Oncologico Italia Meridionale (G.O.I.M.). Anticancer Res 23:4219–4222.
Jo JC, Lee JL, Ryu MH, Sym SJ, Lee SS, Chang HM, Kim TW, Lee JS, Kang YK (2007) Docetaxel monotherapy as a second-line treatment after failure of fluoropyrimidine and platinum in advanced gastric cancer: experience of 154 patients with prognostic factor analysis. Jpn J Clin Oncol 37:936–941. doi:10.1093/jjco/hym123
Hironaka S, Zenda S, Boku N, Fukutomi A, Yoshino T, Onozawa Y (2006) Weekly paclitaxel as second-line chemotherapy for advanced or recurrent gastric cancer. Gastric Cancer 9:14–18. doi:10.1007/s10120-005-0351-6
Kodera Y, Ito S, Mochizuki Y, Fujitake S, Koshikawa K, Kanyama Y, Matsui T, Kojima H, Takase T, Ohashi N, Fujiwara M, Sakamoto J, Akimasa N (2007) A phase II study of weekly paclitaxel as second-line chemotherapy for advanced gastric Cancer (CCOG0302 study). Anticancer Res 27:2667–2671
Chun JH, Kim HK, Lee JS, Choi JY, Lee HG, Yoon SM, Choi IJ, Ryu KW, Kim YW, Bae JM (2004) Weekly irinotecan in patients with metastatic gastric cancer failing cisplatin-based chemotherapy. Jpn J Clin Oncol 34:8–13. doi:10.1093/jjco/hyh006
Hartmann JT, Quietzsch D, Daikeler T, Kollmannsberger C, Mayer F, Kanz L, Bokemeyer C (1999) Mitomycin C continuous infusion as salvage chemotherapy in pretreated patients with advanced gastric cancer. Anticancer Drugs 10:729–733
Van Cutsem E, Kang Y, Chung H, Shen L, Sawaki A, Lordick F, Hill J, Lehle M, Feyereislova A, Bang Y (2009) Efficacy results from the ToGA trial: A phase III study of trastuzumab added to standard chemotherapy (CT) in first-line human epidermal growth factor receptor 2 (HER2)-positive advanced gastric cancer (GC). J Clin Oncol 27:abstr LBA4509.
Burstein HJ, Elias AD, Rugo HS, Cobleigh MA, Wolff AC, Eisenberg PD, Lehman M, Adams BJ, Bello CL, DePrimo SE, Baum CM, Miller KD (2008) Phase II study of sunitinib malate, an oral multitargeted tyrosine kinase inhibitor, in patients with metastatic breast cancer previously treated with an anthracycline and a taxane. J Clin Oncol 26:1810–1816. doi:10.1200/JCO.2007.14.5375
Socinski MA, Novello S, Brahmer JR, Rosell R, Sanchez JM, Belani CP, Govindan R, Atkins JN, Gillenwater HH, Pallares C, Tye L, Selaru P, Chao RC, Scagliotti GV (2008) Multicenter, phase II trial of sunitinib in previously treated, advanced non-small-cell lung cancer. J Clin Oncol 26:650–656. doi:10.1200/JCO.2007.13.9303
Faivre S, Delbaldo C, Vera K, Robert C, Lozahic S, Lassau N, Bello C, Deprimo S, Brega N, Massimini G, Armand JP, Scigalla P, Raymond E (2006) Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J Clin Oncol 24:25–35. doi:10.1200/JCO.2005.02.2194
DePrimo SE, Bello C (2007) Surrogate biomarkers in evaluating response to anti-angiogenic agents: focus on sunitinib. Ann Oncol 18 Suppl 10:x11–9.:x11–x19. doi:10.1093/annonc/mdm409
Choi H, Charnsangavej C, Faria SC, Macapinlac HA, Burgess MA, Patel SR, Chen LL, Podoloff DA, Benjamin RS (2007) Correlation of computed tomography and positron emission tomography in patients with metastatic gastrointestinal stromal tumor treated at a single institution with imatinib mesylate: proposal of new computed tomography response criteria. J Clin Oncol 25:1753–1759. doi:10.1200/JCO.2006.07.3049
Motzer RJ, Michaelson MD, Redman BG, Hudes GR, Wilding G, Figlin RA, Ginsberg MS, Kim ST, Baum CM, DePrimo SE, Li JZ, Bello CL, Theuer CP, George DJ, Rini BI (2006) Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol 24:16–24. doi:10.1200/JCO.2005.02.2574
Norden-Zfoni A, Desai J, Manola J, Beaudry P, Force J, Maki R, Folkman J, Bello C, Baum C, DePrimo SE, Shalinsky DR, Demetri GD, Heymach JV (2007) Blood-based biomarkers of SU11248 activity and clinical outcome in patients with metastatic imatinib-resistant gastrointestinal stromal tumor. Clin Cancer Res 13:2643–2650. doi:10.1158/1078-0432.CCR-06-0919
Yoon SY, Kim SY, Cho YH, Chung HW, So Y, Lee HM (2009) Hepatic metastases of gastric adenocarcinoma showing metabolic remission on FDG-PET despite an increase in size on CT. Cancer Res Treat 41:100–103. doi:10.4143/crt.2009.41.2.100
Acknowledgments
Thanks to the global network of investigators who participated in this trial:
CHINA: Rongcheng Luo, Shukui Qin, Lin Shen, Yan Sun. HONG KONG: Wai-Tong Ng, Winnie Yeo. ITALY: Stefano Cascinu, Alberto Sobrero. JAPAN: Narikazu Boku, Toshihiko Doi, Kuniaki Shirao. PORTUGAL: José Evaristo Sanches. REPUBLIC OF KOREA: Yung-Jue Bang, Hyun-Cheol Chung, Won Ki Kang, Yoon-Koo Kang. TAIWAN: Jen-Shi Chen, Kun-Huei Yeh.
Editorial assistance was provided by Jenni Macdougall of ACUMED® (Tytherington, UK) and was funded by Pfizer Inc. The authors also acknowledge data analysis from Charles Harmon (an employee of Atrium Inc., and a paid consultant to Pfizer Inc.) and Zhixiao Wang (Outcomes Research, Pfizer Oncology, New York, NY, USA), and input and review of the manuscript from Richard Chao, Darrel Cohen, and Kristen Letrent (Pfizer Oncology, La Jolla, CA, USA, and New York, NY, USA).
This trial was sponsored by Pfizer Inc.
Conflict of interest
Y-J Bang has received commercial research support, honoraria from speakers bureau, and compensation for consultation from Pfizer.
Y-K Kang has received honoraria from speakers bureau from Pfizer.
HC Chung has received research funding from Pfizer for the clinical trial.
T Doi has received compensation for consultation for Pfizer’s Sutent GIST program.
JM Tursi, MJ Lechuga, DR Lu, and A Ruiz-Garcia are employees of Pfizer.
JM Tursi, MJ Lechuga, and DR Lu own stock in Pfizer.
A Ruiz-Garcia owns restricted stocks in Pfizer.
A Sobrero participated as chairman to advisory boards for Pfizer in 2009 and 2008.
WK Kang, N Boku, JS Chen, Y Sun, L Shen, SK Qin, and WT Ng have nothing to disclose.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Bang, YJ., Kang, YK., Kang, W.K. et al. Phase II study of sunitinib as second-line treatment for advanced gastric cancer. Invest New Drugs 29, 1449–1458 (2011). https://doi.org/10.1007/s10637-010-9438-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-010-9438-y