
Abstract
The pathogenesis of Coronavirus disease 2019 (COVID-19) is gradually being comprehended. A high number of thrombotic episodes are reported, along with the mortality benefits of heparin. COVID-19 can be viewed as a prothrombotic disease. We overviewed the available evidence to explore this possibility. We identified various histopathology reports and clinical case series reporting thromboses in COVID-19. Also, multiple coagulation markers support this. COVID-19 can be regarded as a risk factor for thrombosis. Applying the principles of Virchow’s triad, we described abnormalities in the vascular endothelium, altered blood flow, and platelet function abnormalities that lead to venous and arterial thromboses in COVID-19. Endothelial dysfunction, activation of the renin-angiotensin-aldosterone system (RAAS) with the release of procoagulant plasminogen activator inhibitor (PAI-1), and hyperimmune response with activated platelets seem to be significant contributors to thrombogenesis in COVID-19. Stratifying risk of COVID-19 thromboses should be based on age, presence of comorbidities, D-dimer, CT scoring, and various blood cell ratios. Isolated heparin therapy may not be sufficient to combat thrombosis in this disease. There is an urgent need to explore newer avenues like activated protein C, PAI-1 antagonists, and tissue plasminogen activators (tPA). These should be augmented with therapies targeting RAAS, antiplatelet drugs, repurposed antiinflammatory, and antirheumatic drugs.
Key Points • Venous and arterial thromboses in COVID-19 can be viewed through the prism of Virchow’s triad. • Endothelial dysfunction, platelet activation, hyperviscosity, and blood flow abnormalities due to hypoxia, immune reactions, and hypercoagulability lead to thrombogenesis in COVID-19. • There is an urgent need to stratify COVID-19 patients at risk for thrombosis using age, comorbidities, D-dimer, and CT scoring. • Patients with COVID-19 at high risk for thrombosis should be put on high dose heparin therapy. |
Similar content being viewed by others
Introduction
Coronavirus disease 2019 (COVID-19) has swept through the world in the last 6 months with 6,194,533 confirmed cases, including 376,320 deaths, being reported to the World Health Organization (WHO) by the 3rd of June, 2020 [1]. COVID-19 is caused by the SARS-CoV-2 virus, a member of the Coronaviridae family that include the SARS-CoV and the MERS-CoV viruses that were responsible for outbreaks of severe respiratory illnesses. Initially recognized as an acute respiratory distress syndrome (ARDS) [2], it was soon realized that heart, brain, and kidney involvement was also common in COVID-19 [3,4,5]. A hyperimmune response referred to as cytokine release syndrome is associated with high mortality [6]. Interestingly, a larger than expected number of thrombotic events were being reported in COVID-19 patients. Advanced age and comorbidities are predictors of increased mortality in COVID-19 [7], which may be associated with susceptibility to thrombosis in these individuals.
Though several viruses are linked to haemorrhagic fevers [8], there are few viruses known to cause thrombosis. Cytomegalovirus has been implicated in thrombosis while varicella-zoster has been linked to both thrombosis and haemorrhage [9]. There is a minimal risk of thromboembolism linked to respiratory viral illnesses in the community [10].
Initial autopsies from COVID-19 patients showed microthrombi in the lung vasculature [11]. The report of heparin having a mortality benefit in a subgroup of patients drew attention to the thrombosis prevalent in COVID-19 [12]. COVID-19 patients are reported to suffer from hypoxemia, particularly in conditions associated with high glycosylated haemoglobin [13]. In the early stages of the disease, lung compliance is not reduced during mechanical ventilation [14]. The lung compliance of 16 patients had been studied and found atypical for ARDS [15]. Thus the hypoxia at the initial stage may be more due to other factors than ARDS. The loss of oxygen transfer capacity of haemoglobin and impaired gas exchange in alveoli with microthrombi can explain this. The microthrombi may cause altered lung perfusion and hypoxic vasoconstriction, worsening the hypoxemia [16]. Prone positioning may help by improving ventilation-perfusion ratio by changing vascular flow distribution in the pulmonary vessels, and not by recruitment as in the case of ARDS [17].
Elevated troponins, indicating cardiac damage, are common findings during the pandemic [18]. These raised troponins are also a predictor of mortality [19]. The cardiomyopathy seems to be an immune or hormone-driven pathology rather than due to direct invasion of the SARS-CoV-2 virion in the myocardium [20].
Venous thromboembolism is frequent in COVID-19. Deep vein thrombosis was detected by Doppler ultrasound in 15% of COVID-19 patients with pneumonia and elevated D-dimer [21]. Some patients present with resistance to conventional heparin therapy [22]. Patients with diabetes who develop COVID-19 seem to be at a higher risk of thrombosis [23]. Other evidence for the role of thrombosis is the presence of chilblain-like lesions in the periphery of COVID-19 patients that might be explained by vasculopathy [24]. Also, an atypical Kawasaki-like syndrome reported in COVID-19 may again be evidence of medium vessel vasculopathy [25].
In this article, we aim to overview thrombosis as an integral part of COVID-19 and characterize systemic features of thrombosis through the prism of Virchow’s triad. Additionally, we explore therapeutics most likely to be effective in this prothrombotic disease.
Search strategy
We performed comprehensive searches through the Scopus and MEDLINE/PubMed databases which were restricted to English sources. No time limitation was set since items on COVID-19 accumulate on a daily basis. Although all article types were processed, preference was given to items with higher evidence base. The following main keywords were employed: “COVID-19”, “SARS-CoV-2”, “thrombosis”, “rheumatic disease”, “immunity”, and “antithrombotic agents”. Overall, we adhered to the recommendations on writing narrative biomedical reviews [26].
COVID-19 as a risk factor of thromboembolism
Autopsies have been performed in a very limited number of COVID-19 cases [27]. Various studies reporting histopathology from autopsies or other sources in COVID-19 are analysed in Table 1 [28,29,30,31,32,33,34,35,36,37,38]. Most of these studies demonstrate venous thromboembolism and microthrombi in arterioles and venules.
There are numerous reports of patients with COVID-19 presenting with both arterial (stroke, myocardial infarction) and venous thrombosis (deep vein thrombosis, pulmonary thromboembolism, venous sinus thrombosis). Many of these patients had traditional risk factors for thrombosis. Perhaps the most important risk factors in the context of COVID-19 are obesity and poorly controlled diabetes mellitus that may aggravate physiological processes such as pregnancy and result in venous and arterial thromboses [39, 40].
Interestingly, pregnancy in women infected with the coronavirus may also increase the risk of placental thrombosis. A case series of 20 pregnant women with COVID-19 reported foetal vascular malperfusion or foetal vascular thrombosis in 10 mainly because of intravascular fibrin deposition, though clinical significance of this placental phenomenon remained uncertain [41].
We are summarising studies and case series (with at least three patients) demonstrating clinical thrombotic episodes in COVID-19 patients as Table 2 [42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57]. As apparent from Table 2, numerous thromboembolic episodes occurred despite prophylactic, or even therapeutic anticoagulation. The rate of pulmonary thromboembolism detected in the intensive care setting is above 20% while in nonCOVID-19 cases, it is usually less than 2% [58]. Besides conventional computerised tomography (CT), lung ultrasound was also able to detect peripheral pulmonary thrombosis confirmed by contrast-enhanced ultrasound [59]. Other lung ultrasounds have reported subpleural “consolidations” that might be microinfarcts of 3–5 mm size [60].
Virchow’s triad in COVID-19
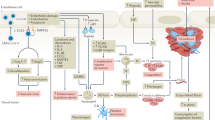
Virchow’s triad (Fig. 1) comprises of vascular damage, altered blood flow, and hypercoagulability of blood. These factors are active in varying degrees in venous thrombosis [61, 62], atrial fibrillation [63], myocardial infarction [64], and stroke [65]. The significance of this triad is that it unifies the inflammatory and the coagulation pathways in the genesis of clotting [63,64,65]. One classic example of Virchow’s triad explaining thrombosis in vascular disease is the case of Behcet disease where abnormalities in the vessel wall and in the blood flow, as well as of hypercoagulability have been described [66]. Each of these components is explored in the context of COVID-19.

Virchow’s triad in the thrombogenesis in COVID-19. Virchow’s triad consists of abnormal vessel wall (endotheliitis, endothelial dysfunction with loss of glycocalyx, endothelial disruption), abnormal flow (due to hyper-viscosity, immune activation, high fibrinogen, impaired microcirculation due to hypoxia and turbulent flow due to microthrombi), and hypercoagulable state (inhibition of plasminogen system due to unopposed canonical renin-angiotensin pathway, platelet dysfunction, complement activation (not shown), and hyperimmune response)
The primary function of the endothelium is the maintenance of nonturbulent blood flow with homeostatic mechanisms to prevent thrombosis and inflammation [67, 68]. The structure of endothelium is different in different tissue as required for specialised function as determined by local need [69]. The endothelium can undergo considerable proliferative changes as well as plastic changes [70]. Most diseases, including viral infections, affect the vascular endothelium and lead to endothelial dysfunction [71].
Vessel wall abnormalities in COVID-19
The endothelium has a glycocalyx layer and secretes tPA (tissue plasminogen activator) that prevents binding of platelets or initiation of the coagulation cascade [67, 72]. Previous, in the SARS outbreak, SARS-CoV virion was detectable in endothelial cells [73]. The ACE2 receptor for SARS-CoV-2 is present in endothelial cells [74]. With this background in mind, initial electron microscopy studies were done, and these studies have demonstrated SARS-CoV-2 like virion in endothelial cells [75, 76]. The seminal study exhibiting intussusceptive angiogenesis in COVID-19 has also demonstrated endothelial invasion by SARS-CoV-2, with subsequent severe endothelial injury and associated disruption of cellular membranes [38]. The endothelial dysfunction leads to the loss of the fibrinolytic function of these cells, predisposing to thrombus formation [77]. Endothelial disruption leads to massive release on von-Willebrand factor (vWF) from Weibel-Palade bodies that have been reported in COVID-19 [78]. All these endothelial factors can initiate thrombosis.
The propagation of thrombosis can be aided by the inflammation induced by endothelial dysfunction. Endothelial cells release interleukin-6 (IL-6) in response to the virus invasion that amplifies the host immune response, even to the state of cytokine storm syndrome [79]. Though immune complex vasculitis has been postulated as a pathological mechanism for COVID-19, the evidence is limited at present [80]. Severe COVID-19 leads to a cytokine storm and coagulopathy is a known consequence of acute sepsis [81]. This underlying coagulation cascade activation predisposes to sepsis-induced coagulopathy (SIC) and disseminated intravascular coagulation (DIC) [82].
Abnormal blood flow
COVID-19 has been linked to hyperviscosity [83]. In a study of 15 patients, all had blood viscosity exceeding 95th percentile of normal, and the plasma viscosity correlated with the sequential organ failure score (SOFA). Hyperviscosity not only directly predisposes to thrombosis but also induces endothelial injury and dysfunction [84]. Fibrinogen is a major determinant of blood viscosity, and high levels have been reported in COVID-19 [85,86,87]. Fibrinogen-to-albumin ratio (FAR) has been shown to be a predictor of disease progression in multivariate Cox analysis [85].
Another critical area for turbulent blood flow is the microcirculation. The presence of microthrombi and reactionary angiogenesis can lead to impaired microcirculation in COVID-19 [88]. The presence of arterial and venous occlusion has already been discussed (Tables 1 and 2), and the presence of such thrombus acts as a perpetrator of abnormal blood flow and further thrombosis.
Though there is no known association between COVID-19 and aneurysm formation, there may be accelerated thrombus formation in preexisting aneurysms. At least two cases of aortic aneurysm rupture during active COVID-19 infections have been reported [89, 90]. A similar report exists for thrombosis in a popliteal aneurysm [91]. This can be due to the hypercoagulable state of COVID-19 superimposed on the turbulent flow in the aneurysm.
Hypercoagulable state
The loss of the protective endothelium with its glycocalyx layer, low levels of tPA, and the inhibition of the clot-lysing system lead to a prothrombotic state. This can be augmented by platelet dysfunction, complement activation, and systemic immune reactions.
Inhibition of the plasminogen system
When SARS-CoV-2 enters a susceptible cell via the ACE2 (angiotensin converting enzyme-2) receptor, there is an internalisation of ACE2 and degradation in the lysosome [92]. ACE2 is a negative regulator of the canonical renin-angiotensin-aldosterone system (RAAS). The loss of the protective ACE2/angiotensin1–7/Mas receptor axis leads to unopposed action of angiotensin II via the angiotensin type 1 receptor (AT1R) that is further augmented by angiotensin IV [93].
Blockade of the angiotensin1–7/Mas by receptor knock-out [94] or pharmacological blockade [95] in murine experiments has accentuated thrombus formation. Directly blocking ACE2 increases thrombus size in animal models while amplifying its function can have antithrombotic effects [96]. ACE2 contributes by activation of tissue plasminogen activator (tPA). Angiotensin II and AT1R activation lead to the formation and release of plasminogen activator inhibitor-1 (PAI-1) from endothelial and smooth muscle cells [97, 98]. Thus the loss of ACE2 alters the PAI-1/tPA balance to a prothrombotic state. There is evidence from a cohort of 44 patients with COVID-19 that the plasminogen pathway is dysfunctional with the complete lack of lysis of clot at 30 min seen in 57% [99].
Platelet dysfunction
The platelet-to-lymphocyte ratio (PLR) is an established marker of inflammation and can predict immune suppression and thrombosis in neoplastic diseases [100]. PLR is raised in COVID-19 and predicts severe disease therein [101, 102]. Both thrombocytosis and thrombocytopenia can occur in COVID-19. A multivariate analysis from China had shown that platelet counts of more than 135x109/L predicted nonsevere disease [85]. Interestingly, bleeding manifestations are rare even in the presence of DIC with thrombocytopenia [103]. Absence of bleeding is more indicative of thrombotic microangiopathy [104].
Platelet activation can take place via the angiotensin II/AT1R pathways. Platelets have AT1R that cause increased adherence in response to angiotensin II [105]. AT1R activation causes the release of PAI-1 from platelet alpha-granules, smooth muscle cells, hepatocytes, and adipocytes [106]. Moreover, the loss of the protective angiotensin1–7/Mas pathway can increase platelet aggregation. Platelets have Mas receptor that modulates thrombosis via the release of nitric oxide (NO) [107].
Another pathway for platelet activation in COVID-19 is via the altered ACE1/ACE2 function. Bradykinin is cleaved by ACE1 into metabolites such as vasoactive bradykinin (1–8) and des-Arg9-bradykinin that are further degraded by ACE2. In the absence of ACE2, des-Arg9-bradykinin accumulation can activate both neutrophils and platelets into an inflammatory phenotype [108]. The activated platelets have a greater tendency to attach to the vascular endothelium [109]. COVID-19 patients have both increased clot strength as well as platelet contribution to clots [87]. Thus platelet activation has a specific role in COVID-19-induced thrombosis.
Complement activation
Complement activation has been demonstrated in COVID-19 skin biopsies, and autopsy and has been shown to be associated with microthrombosis [32]. Complement activation is an integral part of thrombogenesis at its various stages [110, 111]. It may be difficult to delineate cause-effect relationships. However, targeting complement in COVID-19 has been proposed [112, 113]. Four patients with severe COVID-19 have been successfully treated by eculizumab, a monoclonal antibody that targets complement protein C5 [114].
Another possibility is immune complex vasculitis. If the immune complex deposition reported in COVID-19 is due to immune complex vasculitis, it will be more likely to respond to a complement inhibitor eculizumab. The Kawasaki-like syndrome reported in children with COVID-19 may be due to this immune phenomenon [25]. COVID-19 can mimic a variety of vasculitides [115].
Hyperimmune response and thrombosis
DIC is often presented in the later stages of severe sepsis and is characterized by thrombosis in the microcirculation [116]. Presence of only laboratory abnormalities in various coagulation tests without evidence of thrombosis or bleeding manifestation has been termed SIC. This nomenclature attempts at detection and correction of these abnormalities before DIC sets in irreversibly. It has been recognized that SIC and DIC are an invariable component of the hyperimmune response. Thus, they are included in the diagnosis of the macrophage activation–like syndrome (MALS) [117]. Severe COVID-19 has a similar hyperimmune syndrome referred to by various names [6]. It is rational to suggest that the hyperimmune activation itself has a role in perpetuating the widespread thrombosis in this disease. Both activated platelets and leucocytes can activate complement and augment the coagulation cascades.
Antiphospholipid antibodies
A study of antiphospholipid antibodies in three patients with severe COVID-19 spawned interest if SARS-CoV-2 was precipitating antiphospholipid syndrome and related thrombogenesis [118]. Antiphospholipid antibodies are not uncommon in situations of severe sepsis and often do not have any pathological role [119]. Furthermore, a prospective cohort study of 24 patients with COVID-19 who had venous thromboembolism revealed weakly positive anticardiolipin IgM and antiβ2-glycoprotein I IgM in only 2 patients [120]. Another retrospective cohort study of 56 COVID-19 patients pointed to an independent association of anticardiolipin IgG with low oxygen saturation and other severe disease manifestations [121]. Only 1 patient with anticardiolipin IgG had a stroke in that study. There are no studies specifically examining shifts in titres of antiphospholipid antibodies in patients with lupus and other prothrombotic rheumatic diseases overlapping with COVID-19. Available retrospective analyses may suggest that antiphospholipid antibodies alone rarely lead to thromboses even in the course severe inflammatory and immune disturbances due to the virus. Overall, the role of antiphospholipid antibodies and antiphospholipid syndrome in the thrombogenesis due to COVID-19 remains uncertain [122].
Targeting thrombosis in severe COVID-19
It is apparent that thrombosis plays a major role in the pathogenesis of severe COVID-19. Thrombogenesis in such a critical condition should be targeted at multiple levels since only anticoagulation seems insufficient.
Heparin
The initial evidence for mortality benefit with heparin was found in patients with a SIC score ≥ 4 or D-dimer > 6-fold of the upper limit of normal [12]. A larger retrospective study with the data of 17 Spanish hospitals has also confirmed the mortality benefit of heparin after adjusting for age and gender [123]. Beyond the benefit of anticoagulation, heparin also has antiarrhythmic properties [124] and can even oppose classical RAAS activation [125].
The International Society of Thrombosis and Hemostasis (ISTH) have suggested that patients with raised D-dimers (defined as three- to fourfold above the upper range of normal), should be admitted even in the absence of other features because this signifies increased thrombin generation. They have also recommended low-molecular-weight heparin (LMWH) for all admitted patients, including noncritically ill patients [126]. The problem, however, lies in that several studies have shown that thrombosis occurs in patients with severe COVID-19 despite LMWH therapy at therapeutic doses (Table 2).
Overall, clinicians utilizing anticoagulation therapy COVID-19 should also carefully weigh risks of thrombosis and bleeding, particularly in patients with low platelet counts [127].
Tissue plasminogen activator
Thrombolysis can be life-saving in myocardial infarction, ischemic stroke, and pulmonary thromboembolism. One prospective study of 24 patients in intensive care (without COVID-19 or cardiac disease) revealed that tissue factor and PAI-1 rise with the development of ARDS [128].
There are two case series encompassing a total of eight cases of ARDS due to COVID-19 who benefitted from tPA administration [129, 130]. After tPA infusion, oxygen requirements improves, allowing to avoid intubation [130]. An in silico model has also demonstrated mortality benefit with the use of tPA [131]. There is a proposal to use nebulised tPA for ADRS due to COVID-19 [132].
PAI-1 antagonists
Thrombosis can be initiated due to high levels of PAI-1 after the loss of ACE2/angiotensin1–7/Mas pathway. As such, PAI-1 antagonists may offer some benefits. Angiotensin-converting enzyme inhibitors (ACE-I), insulin-sensitizing agents (including metformin and thiazolidinediones), and hormone-replacement therapy in women can have mild to moderate reduction of PAI-1 levels [133]. There are direct PAI-1 antagonists known, but none has been cleared for human use yet [134, 135].
Activated protein C (APC) was initially shown to have benefit in severe sepsis in the protein C Worldwide Evaluation in Severe Sepsis (PROWESS) trial [136]. Later trials did not support the results of this study, and APC went into disuse [137]. It may be worthwhile to try APC to maintain the plasminogen pathway in severe COVID-19.
Antiplatelet drugs
Antiplatelet drugs can potentiate the action of anticoagulation. In a proof-of-concept study, five COVID-19 patients with pulmonary infiltrates and D-dimer > 3 times the upper limit of normal were given oral clopidogrel and infusions of acetylsalicylic acid and tirofiban, on a background of fondaparinux. After 48 h of infusion, the patients had better PaO2/FiO2 ratio as compared with controls that persisted till 7 days [138].
RAAS inhibitors
There was some controversy regarding the use of ACE1-inhibitors (ACE-I) and angiotensin II-receptor blockers (ARBs) in patients with COVID-19 since both are known to increase ACE2 level that is the receptor for SARS-CoV-2 [139]. However, since the primary pathology is due to the loss of ACE2, it stands to reason that ACE-I and ARBs can be helpful [140].
At least two metaanalyses have shown that the use of ACE-I/ARBs is not associated with more severe disease but lead to decreased mortality [141, 142]. However, both these reviews had included a study that has been retracted [143]. A third metaanalysis that has not included this retracted study has also reported mortality benefit of ACE-I/ARBs [144].
There is evidence from cardiology that RAAS inhibitors can reduce thrombosis [145]. Even in animal models, the use of ACE-I and ARBs have been shown to abrogate thrombosis [146, 147]. Both losartan and ramipril have synergistic antiplatelet action when given with dual antiplatelet drugs given postmyocardial infarction [148]. Thus, ACE-I/ARBs can potentiate other antithrombotic therapies in COVID-19.
Targeting complement
Eculizumab can be successfully used in COVID-19 [114]. Complement activation has been demonstrated in COVID-19 patients, but it is difficult to discern whether this is a cause or effect of the pathogenesis [149]. Murine models of various coronavirus infections do not support the therapeutic inhibition of complement factors C3 or C4 [150].
Immune therapy
Timely use of various modalities of immune therapies may decrease viral load and improve survival rates in COVID-19 [151]. While interventions such as intravenous immunoglobulin (IVIg) therapy may save lives of patients with Kawasaki-like syndrome, its use may also increase thrombosis risk [152]. This risk may be due to increased blood viscosity [153] or due to factor (F) XI in substantial quantities in the IVIg products [154].
A Cochrane living systematic review has identified 32 patients across eight studies reporting low evidence of the effectiveness of convalescence plasma therapy in the middle of May 2020 [155]. However, these data are preliminary, and outcomes of clinical trials are awaited [156]. Transfusion-related acute lung injury (TRALI) is a rare complication of IVIg therapy [157], and it has even been reported with convalescence plasma therapy, too [158].
Antirheumatic drugs for preventing thrombosis
The use of various disease-modifying antirheumatic drugs (DMARDs) has been suggested for COVID-19 [159,160,161]. There is controversy about the utility of hydroxychloroquine (HCQ) in COVID-19. The prolonged use of HCQ in systemic lupus erythematosus and rheumatoid arthritis proved effective for reducing cardiovascular risk [162]. HCQ has been shown to reduce levels of tissue factor and related thrombotic pathways in antiphospholipid syndrome [163].
Colchicine exerts some cardiovascular protective effects in neutrophilic disease entities and in other clinical conditions [164]. It is also viewed as a drug candidate for repurposing therapies and using in COVID-19 [165]. Interleukin-1 and interleukin-6 are potential targets in COVID-19 and both can be attenuated by colchicine [166]. Beyond reducing antiinflammatory cytokines, colchicine also has favourable effects on peptides associated with vascular health such as oxidized low-density lipoprotein receptor and phosphodiesterase 5A [167]. In this randomized controlled trial, colchicine had antithrombotic effects in the sera of obese patients [167].
Baricitinib is another repurposed antirheumatic drug [168]. Patients on baricitinib may be at risk for venous thromboembolism [169]. This risk may be amplified in COVID-19. Similarly, there is a theoretical risk of thrombosis with tocilizumab since it alters the lipid profile. However, metaanalysis has shown that it may have better cardiovascular safety as compared with other biological DMARDs [170].
Interestingly, a recent description of mild cases of COVID-19 in 3 different patients with Behçet disease with history of venous thromboses, ankylosing spondylitis and rheumatoid arthritis pointed to positive effects of previously prescribed antiTNF therapies which possibly protected from cytokine storm and related vascular events [171].
Glucocorticoids are associated with an increased risk of thrombosis. Although confounding effect cannot be eliminated, there is a pathobiological basis for this association [172]. The initial report of the COVID-19 Global Rheumatology Alliance has demonstrated an association of prednisolone doses of more than 10 mg with hospitalisation due to COVID-19 [173].
Conclusion
Available evidence points to a propensity for thrombosis in COVID-19. Bleeding is rare even in the setting of thrombocytopenia and DIC [103]. Thrombosis can occur despite prophylactic and therapeutic use of heparin. Manifestations such as ARDS and cardiac compromise are better explained by microthrombi and RAAS activation than by isolated immune activation. These provide compelling reasons to augment anticoagulation with other synergistic therapies such as immune therapies, DMARDs, antiplatelet drugs, RAAS antagonists, statins, and activators of the plasmin system (active thrombolysis therapy).
There is an urgent need to stratifying patients at high risk of thrombosis, particularly the elderly, those with comorbidities, high D-dimer, high chest CT (computed tomography) scores, and shifted blood cell ratios, high ferritin, PAI-1, and IL-6.
Targeting COVID-19 is inchoate without reducing the risk for thrombosis. Thrombotic risk reduction must take into account each facet of Virchow’s triad. The endothelial dysfunction leading to PAI-1/tPA imbalance can be corrected by APC and PAI-1 antagonists. Blood flow abnormalities and microthrombi can be prevented by high dose heparin and RAAS inhibitors including ACE-I/ARBs and statins. Platelet activation and hyperinflammation leading to hyperviscosity can be dealt with by antiinflammatory and antiplatelet drugs.
References
COVID-19 situation reports. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports. Accessed 3 Jun 2020
Cohen J, Normile D (2020) New SARS-like virus in China triggers alarm. Science 367:234–235. https://doi.org/10.1126/science.367.6475.234
Huang C, Wang Y, Li X et al (2020) Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet Lond Engl 395:497–506. https://doi.org/10.1016/S0140-6736(20)30183-5
Zhou F, Yu T, Du R et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet Lond 395:1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3
Garaci F, Di Giuliano F, Picchi E et al (2020) Venous cerebral thrombosis in COVID-19 patient. J Neurol Sci 414:116871. https://doi.org/10.1016/j.jns.2020.116871
Moore JB, June CH (2020) Cytokine release syndrome in severe COVID-19. Science 368:473–474. https://doi.org/10.1126/science.abb8925
Zhou F, Yu T, Du R et al (2020) Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet 395:1054–1062. https://doi.org/10.1016/S0140-6736(20)30566-3
Paessler S, Walker DH (2013) Pathogenesis of the viral hemorrhagic fevers. Annu Rev Pathol 8:411–440. https://doi.org/10.1146/annurev-pathol-020712-164041
Goeijenbier M, van Wissen M, van de Weg C, Jong E, Gerdes VEA, Meijers JCM, Brandjes DPM, van Gorp ECM (2012) Review: viral infections and mechanisms of thrombosis and bleeding. J Med Virol 84:1680–1696. https://doi.org/10.1002/jmv.23354
Smeeth L, Cook C, Thomas S et al (2006) Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet Lond 367:1075–1079. https://doi.org/10.1016/S0140-6736(06)68474-2
Lax SF, Skok K, Zechner P, Kessler HH, Kaufmann N, Koelblinger C, Vander K, Bargfrieder U, Trauner M (2020) Pulmonary arterial thrombosis in COVID-19 with fatal outcome: results from a prospective, single-center, Clinicopathologic Case Series. Ann Intern Med. https://doi.org/10.7326/M20-2566
Tang N, Bai H, Chen X et al (2020) Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost. https://doi.org/10.1111/jth.14817
Wang Z, Du Z, Zhu F (2020) Glycosylated hemoglobin is associated with systemic inflammation, hypercoagulability, and prognosis of COVID-19 patients. Diabetes Res Clin Pract 164:108214. https://doi.org/10.1016/j.diabres.2020.108214
Beloncle FM, Pavlovsky B, Desprez C, Fage N, Olivier PY, Asfar P, Richard JC, Mercat A (2020) Recruitability and effect of PEEP in SARS-Cov-2-associated acute respiratory distress syndrome. Ann Intensive Care 10:55. https://doi.org/10.1186/s13613-020-00675-7
Gattinoni L, Coppola S, Cressoni M, Busana M, Rossi S, Chiumello D (2020) COVID-19 does not Lead to a “typical” acute respiratory distress syndrome. Am J Respir Crit Care Med 201:1299–1300. https://doi.org/10.1164/rccm.202003-0817LE
Dunham-Snary KJ, Wu D, Sykes EA, Thakrar A, Parlow LRG, Mewburn JD, Parlow JL, Archer SL (2017) Hypoxic pulmonary vasoconstriction: from molecular mechanisms to medicine. Chest 151:181–192. https://doi.org/10.1016/j.chest.2016.09.001
Sklar MC, Patel BK, Beitler JR, Piraino T, Goligher E (2019) Optimal ventilator strategies in acute respiratory distress syndrome. Semin Respir Crit Care Med 40:81–93. https://doi.org/10.1055/s-0039-1683896
Fan H, Zhang L, Huang B, Zhu M, Zhou Y, Zhang H, Tao X, Cheng S, Yu W, Zhu L, Chen J (2020) Cardiac injuries in patients with coronavirus disease 2019: not to be ignored. Int J Infect Dis 96:294–297. https://doi.org/10.1016/j.ijid.2020.05.024
Vrsalovic M, Vrsalovic Presecki A (2020) Cardiac troponins predict mortality in patients with COVID-19: a meta-analysis of adjusted risk estimates. J Inf Secur. https://doi.org/10.1016/j.jinf.2020.05.022
Zhou R (2020) Does SARS-CoV-2 cause viral myocarditis in COVID-19 patients? Eur Heart J. https://doi.org/10.1093/eurheartj/ehaa392
Demelo-Rodríguez P, Cervilla-Muñoz E, Ordieres-Ortega L, Parra-Virto A, Toledano-Macías M, Toledo-Samaniego N, García-García A, García-Fernández-Bravo I, Ji Z, de-Miguel-Diez J, Álvarez-Sala-Walther LA, del-Toro-Cervera J, Galeano-Valle F (2020) Incidence of asymptomatic deep vein thrombosis in patients with COVID-19 pneumonia and elevated D-dimer levels. Thromb Res 192:23–26. https://doi.org/10.1016/j.thromres.2020.05.018
Stoneham SM, Milne KM, Nuttal E, Frew GH, Sturrock BR, Sivaloganathan H, Ladikou EE, Drage S, Phillips B, Chevassut TJT, Eziefula AC (2020) Thrombotic risk in COVID-19: a case series and case-control study. Clin Med Lond:clinmed.2020–clinmed.0228. https://doi.org/10.7861/clinmed.2020-0228
(2020) Analysis on 54 mortality cases of Coronavirus Disease 2019 in the Republic of Korea from January 19 to March 10, 2020. J Korean Med Sci 35. https://doi.org/10.3346/jkms.2020.35.e132
Galván Casas C, Català A, Carretero Hernández G et al (2020) Classification of the cutaneous manifestations of COVID-19: a rapid prospective nationwide consensus study in Spain with 375 cases. Br J Dermatol. https://doi.org/10.1111/bjd.19163
Licciardi F, Pruccoli G, Denina M, Parodi E, Taglietto M, Rosati S, Montin D (2020) SARS-CoV-2-induced Kawasaki-like Hyperinflammatory syndrome: a novel COVID phenotype in children. Pediatrics:e20201711. https://doi.org/10.1542/peds.2020-1711
Gasparyan AY, Ayvazyan L, Blackmore H, Kitas GD (2011) Writing a narrative biomedical review: considerations for authors, peer reviewers, and editors. Rheumatol Int 31:1409–1417. https://doi.org/10.1007/s00296-011-1999-3
Salerno M, Sessa F, Piscopo A et al (2020) No autopsies on COVID-19 deaths: a missed opportunity and the lockdown of science. J Clin Med 9. https://doi.org/10.3390/jcm9051472
Tian S, Hu W, Niu L, Liu H, Xu H, Xiao SY (2020) Pulmonary pathology of early-phase 2019 novel coronavirus (COVID-19) pneumonia in two patients with lung cancer. J Thorac Oncol 15:700–704. https://doi.org/10.1016/j.jtho.2020.02.010
Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, Liu S, Zhao P, Liu H, Zhu L, Tai Y, Bai C, Gao T, Song J, Xia P, Dong J, Zhao J, Wang FS (2020) Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med 8:420–422. https://doi.org/10.1016/S2213-2600(20)30076-X
Wichmann D, Sperhake J-P, Lütgehetmann M, Steurer S, Edler C, Heinemann A, Heinrich F, Mushumba H, Kniep I, Schröder AS, Burdelski C, de Heer G, Nierhaus A, Frings D, Pfefferle S, Becker H, Bredereke-Wiedling H, de Weerth A, Paschen HR, Sheikhzadeh-Eggers S, Stang A, Schmiedel S, Bokemeyer C, Addo MM, Aepfelbacher M, Püschel K, Kluge S (2020) Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med. https://doi.org/10.7326/M20-2003
Barton LM, Duval EJ, Stroberg E, Ghosh S, Mukhopadhyay S (2020) COVID-19 Autopsies, Oklahoma, USA. Am J Clin Pathol 153:725–733. https://doi.org/10.1093/ajcp/aqaa062
Magro C, Mulvey JJ, Berlin D, Nuovo G, Salvatore S, Harp J, Baxter-Stoltzfus A, Laurence J (2020) Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res 220:1–13. https://doi.org/10.1016/j.trsl.2020.04.007
Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, Xiao SY (2020) Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol 33:1007–1014. https://doi.org/10.1038/s41379-020-0536-x
Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, Yi F, Yang HC, Fogo AB, Nie X, Zhang C (2020) Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int 98:219–227. https://doi.org/10.1016/j.kint.2020.04.003
Nunes Duarte-Neto A, de Almeida Monteiro RA, da Silva LFF, Malheiros DMAC, de Oliveira EP, Theodoro Filho J, Pinho JRR, Soares Gomes-Gouvêa M, Salles APM, de Oliveira IRS, Mauad T, do Nascimento Saldiva PH, Dolhnikoff M (2020) Pulmonary and systemic involvement of COVID-19 assessed by ultrasound-guided minimally invasive autopsy. Histopathology. https://doi.org/10.1111/his.14160
Mulvey JJ, Magro CM, Ma LX, Nuovo GJ, Baergen RN (2020) Analysis of complement deposition and viral RNA in placentas of COVID-19 patients. Ann Diagn Pathol 46:151530. https://doi.org/10.1016/j.anndiagpath.2020.151530
Menter T, Haslbauer JD, Nienhold R, Savic S, Hopfer H, Deigendesch N, Frank S, Turek D, Willi N, Pargger H, Bassetti S, Leuppi JD, Cathomas G, Tolnay M, Mertz KD, Tzankov A (2020) Post-mortem examination of COVID19 patients reveals diffuse alveolar damage with severe capillary congestion and variegated findings of lungs and other organs suggesting vascular dysfunction. Histopathology. https://doi.org/10.1111/his.14134
Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, Vanstapel A, Werlein C, Stark H, Tzankov A, Li WW, Li VW, Mentzer SJ, Jonigk D (2020) Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. https://doi.org/10.1056/NEJMoa2015432
Martinelli I, Ferrazzi E, Ciavarella A, Erra R, Iurlaro E, Ossola M, Lombardi A, Blasi F, Mosca F, Peyvandi F (2020) Pulmonary embolism in a young pregnant woman with COVID-19. Thromb Res 191:36–37. https://doi.org/10.1016/j.thromres.2020.04.022
Ahmed I, Azhar A, Eltaweel N, Tan BK (2020) First Covid-19 maternal mortality in the UK associated with thrombotic complications. Br J Haematol 190:e37–e38. https://doi.org/10.1111/bjh.16849
Baergen RN, Heller DS (2020) Placental pathology in Covid-19 positive mothers: preliminary findings. Pediatr Dev Pathol 23:177–180. https://doi.org/10.1177/1093526620925569
Morassi M, Bagatto D, Cobelli M, D’Agostini S, Gigli GL, Bnà C, Vogrig A (2020) Stroke in patients with SARS-CoV-2 infection: case series. J Neurol. https://doi.org/10.1007/s00415-020-09885-2
Yaghi S, Ishida K, Torres J et al (2020) SARS2-CoV-2 and stroke in a New York healthcare system. Stroke:STROKEAHA120030335. https://doi.org/10.1161/STROKEAHA.120.030335
Tunç A, Ünlübaş Y, Alemdar M, Akyüz E (2020) Coexistence of COVID-19 and acute ischemic stroke report of four cases. J Clin Neurosci. https://doi.org/10.1016/j.jocn.2020.05.018
Beyrouti R, Adams ME, Benjamin L, Cohen H, Farmer SF, Goh YY, Humphries F, Jäger HR, Losseff NA, Perry RJ, Shah S, Simister RJ, Turner D, Chandratheva A, Werring DJ (2020) Characteristics of ischaemic stroke associated with COVID-19. J Neurol Neurosurg Psychiatry:jnnp-2020-323586. https://doi.org/10.1136/jnnp-2020-323586
Kihira S, Schefflein J, Chung M et al (2020) Incidental COVID-19 related lung apical findings on stroke CTA during the COVID-19 pandemic. J Neurointerv Surg. https://doi.org/10.1136/neurintsurg-2020-016188
Avula A, Nalleballe K, Narula N, Sapozhnikov S, Dandu V, Toom S, Glaser A, Elsayegh D (2020) COVID-19 presenting as stroke. Brain Behav Immun 87:115–119. https://doi.org/10.1016/j.bbi.2020.04.077
Lodigiani C, Iapichino G, Carenzo L, Cecconi M, Ferrazzi P, Sebastian T, Kucher N, Studt JD, Sacco C, Alexia B, Sandri MT, Barco S, Humanitas COVID-19 Task Force (2020) Venous and arterial thromboembolic complications in COVID-19 patients admitted to an academic hospital in Milan, Italy. Thromb Res 191:9–14. https://doi.org/10.1016/j.thromres.2020.04.024
Llitjos J-F, Leclerc M, Chochois C, Monsallier JM, Ramakers M, Auvray M, Merouani K (2020) High incidence of venous thromboembolic events in anticoagulated severe COVID-19 patients. J Thromb Haemost. https://doi.org/10.1111/jth.14869
Middeldorp S, Coppens M, van Haaps TF, Foppen M, Vlaar AP, Müller MCA, Bouman CCS, Beenen LFM, Kootte RS, Heijmans J, Smits LP, Bonta PI, van Es N (2020) Incidence of venous thromboembolism in hospitalized patients with COVID-19. J Thromb Haemost. https://doi.org/10.1111/jth.14888
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers D, Kant KM, Kaptein FHJ, van Paassen J, Stals MAM, Huisman MV, Endeman H (2020) Confirmation of the high cumulative incidence of thrombotic complications in critically ill ICU patients with COVID-19: an updated analysis. Thromb Res 191:148–150. https://doi.org/10.1016/j.thromres.2020.04.041
Poyiadji N, Cormier P, Patel PY et al (2020) Acute pulmonary embolism and COVID-19. Radiology 201955:201955. https://doi.org/10.1148/radiol.2020201955
Helms J, Tacquard C, Severac F et al (2020) High risk of thrombosis in patients with severe SARS-CoV-2 infection: a multicenter prospective cohort study. Intensive Care Med 46:1089–1098. https://doi.org/10.1007/s00134-020-06062-x
Grillet F, Behr J, Calame P et al (2020) Acute pulmonary embolism associated with COVID-19 pneumonia detected by pulmonary CT angiography. Radiology 201544. https://doi.org/10.1148/radiol.2020201544
Bompard F, Monnier H, Saab I et al (2020) Pulmonary embolism in patients with Covid-19 pneumonia. Eur Respir J. https://doi.org/10.1183/13993003.01365-2020
Griffin DO, Jensen A, Khan M, Chin J, Chin K, Saad J, Parnell R, Awwad C, Patel D (2020) Pulmonary embolism and increased levels of D-dimer in patients with Coronavirus disease. Emerg Infect Dis 26. https://doi.org/10.3201/eid2608.201477
Leonard-Lorant I, Delabranche X, Severac F, Helms J, Pauzet C, Collange O, Schneider F, Labani A, Bilbault P, Moliere S, Leyendecker P, Roy C, Ohana M (2020) Acute pulmonary embolism in COVID-19 patients on CT angiography and relationship to D-dimer levels. Radiology 201561:201561. https://doi.org/10.1148/radiol.2020201561
Lim W, Meade M, Lauzier F et al (2015) Failure of anticoagulant thromboprophylaxis: risk factors in medical-surgical critically ill patients*. Crit Care Med 43:401–410. https://doi.org/10.1097/CCM.0000000000000713
Tee A, Wong A, Yusuff T et al (2020) Contrast-enhanced ultrasound (CEUS) of the lung reveals multiple areas of microthrombi in a COVID-19 patient. Intensive Care Med. https://doi.org/10.1007/s00134-020-06085-4
Peng Q-Y, Wang X-T, Zhang L-N, Chinese Critical Care Ultrasound Study Group (CCUSG) (2020) Findings of lung ultrasonography of novel corona virus pneumonia during the 2019-2020 epidemic. Intensive Care Med 46:849–850. https://doi.org/10.1007/s00134-020-05996-6
Lurie JM, Png CYM, Subramaniam S, Chen S, Chapman E, Aboubakr A, Marin M, Faries P, Ting W (2019) Virchow’s triad in “silent” deep vein thrombosis. J Vasc Surg Venous Lymphat Disord 7:640–645. https://doi.org/10.1016/j.jvsv.2019.02.011
Alturki N, Alkahtani M, Daghistani M, Alyafi T, Khairy S, Ashi M, Aljuffri A (2019) Incidence and risk factors for deep vein thrombosis among pediatric burn patients. Burns 45:560–566. https://doi.org/10.1016/j.burns.2018.09.032
Watson T, Shantsila E, Lip GYH (2009) Mechanisms of thrombogenesis in atrial fibrillation: Virchow’s triad revisited. Lancet Lond 373:155–166. https://doi.org/10.1016/S0140-6736(09)60040-4
Raggi P, Genest J, Giles JT, Rayner KJ, Dwivedi G, Beanlands RS, Gupta M (2018) Role of inflammation in the pathogenesis of atherosclerosis and therapeutic interventions. Atherosclerosis 276:98–108. https://doi.org/10.1016/j.atherosclerosis.2018.07.014
Stoll G, Nieswandt B (2019) Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat Rev Neurol 15:473–481. https://doi.org/10.1038/s41582-019-0221-1
La Regina M, Gasparyan AY, Orlandini F, Prisco D (2010) Behçet’s disease as a model of venous thrombosis. Open Cardiovasc Med J 4:71–77. https://doi.org/10.2174/1874192401004020071
Pober JS, Sessa WC (2007) Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7:803–815. https://doi.org/10.1038/nri2171
Teuwen L-A, Geldhof V, Pasut A, Carmeliet P (2020) COVID-19: the vasculature unleashed. Nat Rev Immunol 20:389–391. https://doi.org/10.1038/s41577-020-0343-0
Gross PL, Aird WC (2000) The endothelium and thrombosis. Semin Thromb Hemost 26:463–478. https://doi.org/10.1055/s-2000-13202
Pearson JD (2015) Plasticity of adult endothelium: how frequent and to what extent? Cardiovasc Res 108:319–320. https://doi.org/10.1093/cvr/cvv240
Rajendran P, Rengarajan T, Thangavel J et al (2013) The vascular endothelium and human diseases. Int J Biol Sci 9:1057–1069. https://doi.org/10.7150/ijbs.7502
Urano T, Suzuki Y (2012) Accelerated fibrinolysis and its propagation on vascular endothelial cells by secreted and retained tPA. J Biomed Biotechnol 2012:208108. https://doi.org/10.1155/2012/208108
Ye J, Zhang B, Xu J et al (2007) Molecular pathology in the lungs of severe acute respiratory syndrome patients. Am J Pathol 170:538–545. https://doi.org/10.2353/ajpath.2007.060469
Hamming I, Timens W, Bulthuis MLC et al (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637. https://doi.org/10.1002/path.1570
Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, Tan W, China Novel Coronavirus Investigating and Research Team (2020) A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727–733. https://doi.org/10.1056/NEJMoa2001017
Varga Z, Flammer AJ, Steiger P et al (2020) Endothelial cell infection and endotheliitis in COVID-19. Lancet 395:1417–1418. https://doi.org/10.1016/S0140-6736(20)30937-5
Suzuki Y, Yasui H, Brzoska T, Mogami H, Urano T (2011) Surface-retained tPA is essential for effective fibrinolysis on vascular endothelial cells. Blood 118:3182–3185. https://doi.org/10.1182/blood-2011-05-353912
Escher R, Breakey N, Lämmle B (2020) Severe COVID-19 infection associated with endothelial activation. Thromb Res 190:62. https://doi.org/10.1016/j.thromres.2020.04.014
McGonagle D, Sharif K, O’Regan A, Bridgewood C (2020) The role of cytokines including Interleukin-6 in COVID-19 induced pneumonia and macrophage activation syndrome-like disease. Autoimmun Rev 102537. https://doi.org/10.1016/j.autrev.2020.102537
Roncati L, Ligabue G, Fabbiani L, Malagoli C, Gallo G, Lusenti B, Nasillo V, Manenti A, Maiorana A (2020) Type 3 hypersensitivity in COVID-19 vasculitis. Clin Immunol Orlando Fla 108487:108487. https://doi.org/10.1016/j.clim.2020.108487
Semeraro N, Ammollo CT, Semeraro F, Colucci M (2015) Coagulopathy of acute Sepsis. Semin Thromb Hemost 41:650–658. https://doi.org/10.1055/s-0035-1556730
McGonagle D, O’Donnell JS, Sharif K et al (2020) Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol 2:e437–e445. https://doi.org/10.1016/S2665-9913(20)30121-1
Maier CL, Truong AD, Auld SC et al (2020) COVID-19-associated hyperviscosity: a link between inflammation and thrombophilia? Lancet Lond. https://doi.org/10.1016/S0140-6736(20)31209-5
Forconi S, Pieragalli D, Guerrini M, Galigani C, Cappelli R (1987) Primary and secondary blood hyperviscosity syndromes, and syndromes associated with blood hyperviscosity. Drugs 33(Suppl 2):19–26. https://doi.org/10.2165/00003495-198700332-00006
Bi X, Su Z, Yan H et al (2020) Prediction of severe illness due to COVID-19 based on an analysis of initial fibrinogen to albumin ratio and platelet count. Platelets:1–6. https://doi.org/10.1080/09537104.2020.1760230
Han H, Yang L, Liu R, Liu F, Wu KL, Li J, Liu XH, Zhu CL (2020) Prominent changes in blood coagulation of patients with SARS-CoV-2 infection. Clin Chem Lab Med 58:1116–1120. https://doi.org/10.1515/cclm-2020-0188
Ranucci M, Ballotta A, Di Dedda U et al (2020) The procoagulant pattern of patients with COVID-19 acute respiratory distress syndrome. J Thromb Haemost. https://doi.org/10.1111/jth.14854
Bray MA, Sartain SA, Gollamudi J, Rumbaut RE (2020) MICROVASCULAR thrombosis: experimental and clinical implications. Transl Res J Lab Clin Med. https://doi.org/10.1016/j.trsl.2020.05.006
Rinaldi LF, Marazzi G, Marone EM (2020) Endovascular treatment of a ruptured pararenal abdominal aortic aneurysm in a COVID-19 patient: suggestions and case report. Ann Vasc Surg 66:18–23. https://doi.org/10.1016/j.avsg.2020.05.011
Shih M, Swearingen B, Rhee R (2020) Ruptured abdominal aortic aneurysm treated with endovascular repair in a patient with active COVID-19 infection during the pandemic. Ann Vasc Surg 66:14–17. https://doi.org/10.1016/j.avsg.2020.05.001
Powezka K, Khan T, Narlawar R, Antoniou GA (2020) Ruptured popliteal artery aneurysm complicated with acute respiratory distress syndrome secondary to SARS-CoV-2 infection. Ann Vasc Surg 66:24–27. https://doi.org/10.1016/j.avsg.2020.05.012
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 181:271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052
Verdecchia P, Cavallini C, Spanevello A, Angeli F (2020) The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur J Intern Med 76:14–20. https://doi.org/10.1016/j.ejim.2020.04.037
Fraga-Silva RA, Da Silva DG, Montecucco F et al (2012) The angiotensin-converting enzyme 2/angiotensin-(1-7)/mas receptor axis: a potential target for treating thrombotic diseases. Thromb Haemost 108:1089–1096. https://doi.org/10.1160/TH12-06-0396
Fang C, Stavrou E, Schmaier AA et al (2013) Angiotensin 1-7 and mas decrease thrombosis in Bdkrb2−/− mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood 121:3023–3032. https://doi.org/10.1182/blood-2012-09-459156
Fraga-Silva RA, Sorg BS, Wankhede M, deDeugd C, Jun JY, Baker MB, Li Y, Castellano RK, Katovich MJ, Raizada MK, Ferreira AJ (2010) ACE2 activation promotes antithrombotic activity. Mol Med Camb Mass 16:210–215. https://doi.org/10.2119/molmed.2009.00160
Nishimura H, Tsuji H, Masuda H, Nakagawa K, Nakahara Y, Kitamura H, Kasahara T, Sugano T, Yoshizumi M, Sawada S, Nakagawa M (1997) Angiotensin II increases plasminogen activator inhibitor-1 and tissue factor mRNA expression without changing that of tissue type plasminogen activator or tissue factor pathway inhibitor in cultured rat aortic endothelial cells. Thromb Haemost 77:1189–1195
van Leeuwen RT, Kol A, Andreotti F, Kluft C, Maseri A, Sperti G (1994) Angiotensin II increases plasminogen activator inhibitor type 1 and tissue-type plasminogen activator messenger RNA in cultured rat aortic smooth muscle cells. Circulation 90:362–368. https://doi.org/10.1161/01.cir.90.1.362
Wright FL, Vogler TO, Moore EE et al (2020) Fibrinolysis shutdown correlates to thromboembolic events in severe COVID-19 infection. J Am Coll Surg. https://doi.org/10.1016/j.jamcollsurg.2020.05.007
Gasparyan AY, Ayvazyan L, Mukanova U, Yessirkepov M, Kitas GD (2019) The platelet-to-lymphocyte ratio as an inflammatory marker in rheumatic diseases. Ann Lab Med 39:345–357. https://doi.org/10.3343/alm.2019.39.4.345
Qu R, Ling Y, Zhang Y-H-Z et al (2020) Platelet-to-lymphocyte ratio is associated with prognosis in patients with coronavirus disease-19. J Med Virol. https://doi.org/10.1002/jmv.25767
Yang A-P, Liu J-P, Tao W-Q, Li H-M (2020) The diagnostic and predictive role of NLR, d-NLR and PLR in COVID-19 patients. Int Immunopharmacol 84:106504. https://doi.org/10.1016/j.intimp.2020.106504
Connors JM, Levy JH (2020) COVID-19 and its implications for thrombosis and anticoagulation. Blood. 135:2033–2040. https://doi.org/10.1182/blood.2020006000
Becker RC (2020) COVID-19 update: Covid-19-associated coagulopathy. J Thromb Thrombolysis:1–14. https://doi.org/10.1007/s11239-020-02134-3
Nagai M, Terao S, Vital SA, Rodrigues SF, Yilmaz G, Granger D (2011) Role of blood cell-associated angiotensin II type 1 receptors in the cerebral microvascular response to ischemic stroke during angiotensin-induced hypertension. Exp Transl Stroke Med 3:15. https://doi.org/10.1186/2040-7378-3-15
Yamamoto K, Takeshita K, Kojima T et al (2005) Aging and plasminogen activator inhibitor-1 (PAI-1) regulation: implication in the pathogenesis of thrombotic disorders in the elderly. Cardiovasc Res 66:276–285. https://doi.org/10.1016/j.cardiores.2004.11.013
Fraga-Silva RA, Pinheiro SVB, Gonçalves ACC, Alenina N, Bader M, Souza Santos RA (2008) The antithrombotic effect of angiotensin-(1-7) involves mas-mediated NO release from platelets. Mol Med 14:28–35. https://doi.org/10.2119/2007-00073.Fraga-Silva
Sodhi CP, Wohlford-Lenane C, Yamaguchi Y et al (2018) Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am J Phys Lung Cell Mol Phys 314:L17–L31. https://doi.org/10.1152/ajplung.00498.2016
Waldner MJ, Baethmann A, Uhl E, Lehmberg J (2012) Bradykinin-induced leukocyte- and platelet-endothelium interactions in the cerebral microcirculation. Brain Res 1448:163–169. https://doi.org/10.1016/j.brainres.2012.02.010
Afshar-Kharghan V (2017) Complement and clot. Blood 129:2214–2215. https://doi.org/10.1182/blood-2017-03-771501
Foley JH, Walton BL, Aleman MM, O'Byrne AM, Lei V, Harrasser M, Foley KA, Wolberg AS, Conway EM (2016) Complement activation in arterial and venous thrombosis is mediated by plasmin. EBioMedicine 5:175–182. https://doi.org/10.1016/j.ebiom.2016.02.011
Risitano AM, Mastellos DC, Huber-Lang M, Yancopoulou D, Garlanda C, Ciceri F, Lambris JD (2020) Complement as a target in COVID-19? Nat Rev Immunol 20:343–344. https://doi.org/10.1038/s41577-020-0320-7
Campbell CM, Kahwash R (2020) Will complement inhibition be the new target in treating COVID-19 related systemic thrombosis? Circulation. 141:1739–1741. https://doi.org/10.1161/CIRCULATIONAHA.120.047419
Diurno F, Numis FG, Porta G et al (2020) Eculizumab treatment in patients with COVID-19: preliminary results from real life ASL Napoli 2 Nord experience. Eur Rev Med Pharmacol Sci 24:4040–4047. https://doi.org/10.26355/eurrev_202004_20875
Zhang W, Zhao Y, Zhang F, Wang Q, Li T, Liu Z, Wang J, Qin Y, Zhang X, Yan X, Zeng X, Zhang S (2020) The use of anti-inflammatory drugs in the treatment of people with severe coronavirus disease 2019 (COVID-19): the perspectives of clinical immunologists from China. Clin Immunol 214:108393. https://doi.org/10.1016/j.clim.2020.108393
Iba T, Levi M, Levy JH (2020) Sepsis-induced coagulopathy and disseminated intravascular coagulation. Semin Thromb Hemost 46:89–95. https://doi.org/10.1055/s-0039-1694995
Karakike E, Giamarellos-Bourboulis EJ (2019) Macrophage activation-like syndrome: a distinct entity leading to early death in sepsis. Front Immunol 10:55. https://doi.org/10.3389/fimmu.2019.00055
Zhang Y, Xiao M, Zhang S, Xia P, Cao W, Jiang W, Chen H, Ding X, Zhao H, Zhang H, Wang C, Zhao J, Sun X, Tian R, Wu W, Wu D, Ma J, Chen Y, Zhang D, Xie J, Yan X, Zhou X, Liu Z, Wang J, du B, Qin Y, Gao P, Qin X, Xu Y, Zhang W, Li T, Zhang F, Zhao Y, Li Y, Zhang S (2020) Coagulopathy and Antiphospholipid antibodies in patients with Covid-19. N Engl J Med 382:e38. https://doi.org/10.1056/NEJMc2007575
Asherson RA, Cervera R (2003) Antiphospholipid antibodies and infections. Ann Rheum Dis 62:388–393. https://doi.org/10.1136/ard.62.5.388
Galeano-Valle F, Oblitas CM, Ferreiro-Mazón MM, Alonso-Muñoz J, del Toro-Cervera J, di Natale M, Demelo-Rodríguez P (2020) Antiphospholipid antibodies are not elevated in patients with severe COVID-19 pneumonia and venous thromboembolism. Thromb Res 192:113–115. https://doi.org/10.1016/j.thromres.2020.05.017
Bertin D, Brodovitch A, Beziane A, Hug S, Bouamri A, Mege JL, Bardin N (2020) Anti-cardiolipin IgG autoantibodies are an independent risk factor of COVID-19 severity. Arthritis Rheum. https://doi.org/10.1002/art.41409
Mehta S, Bhandari S, Mehta S (2020) Cautious interpretation of antiphospholipid antibodies in COVID-19. Clin Chim Acta Int J Clin Chem 509:166. https://doi.org/10.1016/j.cca.2020.06.024
Ayerbe L, Risco C, Ayis S (2020) The association between treatment with heparin and survival in patients with Covid-19. J Thromb Thrombolysis. https://doi.org/10.1007/s11239-020-02162-z
Menezes-Rodrigues FS, Padrão Tavares JG, Pires de Oliveira M, Guzella de Carvalho R, Ruggero Errante P, Omar Taha M, Fagundes DJ, Caricati-Neto A (2020) Anticoagulant and antiarrhythmic effects of heparin in the treatment of COVID-19 patients. J Thromb Haemost. https://doi.org/10.1111/jth.14902
Ahmed S, Anirvan P (2020) Reply to rheumatologists’ perspective on coronavirus disease 19: is heparin the dark horse for COVID-19? Clin Rheumatol 39:2099–2100. https://doi.org/10.1007/s10067-020-05145-w
Thachil J, Tang N, Gando S, Falanga A, Cattaneo M, Levi M, Clark C, Iba T (2020) ISTH interim guidance on recognition and management of coagulopathy in COVID-19. J Thromb Haemost 18:1023–1026. https://doi.org/10.1111/jth.14810
Gavioli EM, Sikorska G, Man A et al (2020) Current perspectives of anticoagulation in patients with COVID-19. J Cardiovasc Pharmacol. https://doi.org/10.1097/FJC.0000000000000861
Ozolina A, Sarkele M, Sabelnikovs O, Skesters A, Jaunalksne I, Serova J, Ievins T, Bjertnaes LJ, Vanags I (2016) Activation of coagulation and fibrinolysis in acute respiratory distress syndrome: a prospective pilot study. Front Med 3:64. https://doi.org/10.3389/fmed.2016.00064
Wang J, Hajizadeh N, Moore EE, McIntyre RC, Moore PK, Veress LA, Yaffe MB, Moore HB, Barrett CD (2020) Tissue plasminogen activator (tPA) treatment for COVID-19 associated acute respiratory distress syndrome (ARDS): a case series. J Thromb Haemost. https://doi.org/10.1111/jth.14828
Christie DB, Nemec HM, Scott AM et al (2020) Early outcomes with utilization of tissue plasminogen activator in COVID-19 associated respiratory distress: a series of five cases. J Trauma Acute Care Surg. https://doi.org/10.1097/TA.0000000000002787
Choudhury R, Barrett CD, Moore HB et al (2020) Salvage use of tissue plasminogen activator (tPA) in the setting of acute respiratory distress syndrome (ARDS) due to COVID-19 in the USA: a Markov decision analysis. World J Emerg Surg 15. https://doi.org/10.1186/s13017-020-00305-4
Whyte CS, Morrow GB, Mitchell JL et al (2020) Fibrinolytic abnormalities in acute respiratory distress syndrome (ARDS) and versatility of thrombolytic drugs to treat COVID-19. J Thromb Haemost. https://doi.org/10.1111/jth.14872
Vaughan DE (2011) PAI-1 antagonists: the promise and the peril. Trans Am Clin Climatol Assoc 122:312–325
Gong L, Proulle V, Fang C, Hong Z, Lin Z, Liu M, Xue G, Yuan C, Lin L, Furie B, Flaumenhaft R, Andreasen P, Furie B, Huang M (2016) A specific plasminogen activator inhibitor-1 antagonist derived from inactivated urokinase. J Cell Mol Med 20:1851–1860. https://doi.org/10.1111/jcmm.12875
Yu B, Zhang G, Jin L et al (2017) Inhibition of PAI-1 activity by Toddalolactone as a mechanism for promoting blood circulation and removing stasis by Chinese herb Zanthoxylum nitidum var. tomentosum. Front Pharmacol 8:489. https://doi.org/10.3389/fphar.2017.00489
Bernard GR, Vincent JL, Laterre PF, LaRosa S, Dhainaut JF, Lopez-Rodriguez A, Steingrub JS, Garber GE, Helterbrand JD, Ely EW, Fisher CJ Jr, Recombinant human protein C Worldwide Evaluation in Severe Sepsis (PROWESS) study group (2001) Efficacy and safety of recombinant human activated protein C for severe sepsis. N Engl J Med 344:699–709. https://doi.org/10.1056/NEJM200103083441001
Ikezoe T (2015) Thrombomodulin/activated protein C system in septic disseminated intravascular coagulation. J Intensive Care 3:1. https://doi.org/10.1186/s40560-014-0050-7
Viecca M, Radovanovic D, Forleo GB, Santus P (2020) Enhanced platelet inhibition treatment improves hypoxemia in patients with severe Covid-19 and hypercoagulability. A case control, proof of concept study. Pharmacol Res. https://doi.org/10.1016/j.phrs.2020.104950
Thomas G (2020) Renin-angiotensin system inhibitors in COVID-19. Cleve Clin J Med. https://doi.org/10.3949/ccjm.87a.ccc009
Vaduganathan M, Vardeny O, Michel T, McMurray JJV, Pfeffer MA, Solomon SD (2020) Renin–angiotensin–aldosterone system inhibitors in patients with Covid-19. N Engl J Med 382:1653–1659. https://doi.org/10.1056/NEJMsr2005760
Zhang X, Yu J, Pan L-Y, Jiang H-Y (2020) ACEI/ARB use and risk of infection or severity or mortality of COVID-19: a systematic review and meta-analysis. Pharmacol Res 158:104927. https://doi.org/10.1016/j.phrs.2020.104927
Pirola CJ, Sookoian S (2020) Estimation of RAAS-inhibitor effect on the COVID-19 outcome: a meta-analysis. J Inf Secur. https://doi.org/10.1016/j.jinf.2020.05.052
Mehra MR, Desai SS, Kuy S, Henry TD, Patel AN (2020) Retraction: cardiovascular disease, drug therapy, and mortality in Covid-19. N Engl J Med. DOI: 10.1056/NEJMoa2007621. N Engl J Med 382:2582. https://doi.org/10.1056/NEJMc2021225
Guo X, Zhu Y, Hong Y (2020, 1979) Decreased mortality of COVID-19 with renin-angiotensin-aldosterone system inhibitors therapy in patients with hypertension:a meta-analysis. Hypertension. https://doi.org/10.1161/HYPERTENSIONAHA.120.15572
Suo Y, Zhang Z, Fu H, Zhang Y, Yuan M, Wang Y, Goudis CA, Tse G, Liu T, Li G (2018) Inhibition of renin-angiotensin axis reduces the risk of thrombus formation in the left atrial appendage in patients with hypertension complicated by atrial fibrillation. J Renin-Angiotensin Aldosterone Syst 19:19. https://doi.org/10.1177/1470320318782623
Schmaier AH (2016) A novel antithrombotic mechanism mediated by the receptors of the Kallikrein/Kinin and renin-angiotensin systems. Front Med 3:61. https://doi.org/10.3389/fmed.2016.00061
Düsing R (2016) Pharmacological interventions into the renin-angiotensin system with ACE inhibitors and angiotensin II receptor antagonists: effects beyond blood pressure lowering. Ther Adv Cardiovasc Dis 10:151–161. https://doi.org/10.1177/1753944716644130
Marinšek M, Sinkovič A (2016) Ramipril and losartan exert a similar long-term effect upon markers of heart failure, endogenous fibrinolysis, and platelet aggregation in survivors of ST-elevation myocardial infarction: a single centre randomized trial. Biomed Res Int 2016:9040457–9040457. https://doi.org/10.1155/2016/9040457
Cugno M, Meroni PL, Gualtierotti R, Griffini S, Grovetti E, Torri A, Panigada M, Aliberti S, Blasi F, Tedesco F, Peyvandi F (2020) Complement activation in patients with COVID-19: a novel therapeutic target. J Allergy Clin Immunol. https://doi.org/10.1016/j.jaci.2020.05.006
Maglakelidze N, Manto KM, Craig TJ (2020) A review: does complement or the contact system have a role in protection or pathogenesis of COVID-19? Pulm Ther. https://doi.org/10.1007/s41030-020-00118-5
Gasparyan AY, Misra DP, Yessirkepov M, Zimba O (2020) Perspectives of immune therapy in coronavirus disease 2019. J Korean Med Sci 35:e176. https://doi.org/10.3346/jkms.2020.35.e176
Branch DR (2019) Serologic problems associated with administration of intravenous immune globulin (IVIg). Immunohematology 35:13–15
Bentley P, Rosso M, Sadnicka A, Israeli-Korn S, Laffan M, Sharma P (2012) Intravenous immunoglobulin increases plasma viscosity without parallel rise in blood pressure. J Clin Pharm Ther 37:286–290. https://doi.org/10.1111/j.1365-2710.2011.01287.x
Drelich DA, Bray PF, Herman JH (2012) Immunoglobulin therapy and thrombosis: coincidence or causation? Transfusion (Paris) 52:2075–2077. https://doi.org/10.1111/j.1537-2995.2012.03868.x
Valk SJ, Piechotta V, Chai KL et al (2020) Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a rapid review. Cochrane Database Syst Rev 5:CD013600. https://doi.org/10.1002/14651858.CD013600
Bloch EM, Shoham S, Casadevall A et al (2020) Deployment of convalescent plasma for the prevention and treatment of COVID-19. J Clin Invest 130:2757–2765. https://doi.org/10.1172/JCI138745
Baudel JL, Vigneron C, Pras-Landre V, Joffre J, Marjot F, Ait-Oufella H, Bigé N, Maury E, Guidet B, Fain O, Mekinian A (2020) Transfusion-related acute lung injury (TRALI) after intravenous immunoglobulins: French multicentre study and literature review. Clin Rheumatol 39:541–546. https://doi.org/10.1007/s10067-019-04832-7
Chun S, Chung CR, Ha YE, Han TH, Ki CS, Kang ES, Park JK, Peck KR, Cho D (2016) Possible transfusion-related acute lung injury following convalescent plasma transfusion in a patient with Middle East respiratory syndrome. Ann Lab Med 36:393–395. https://doi.org/10.3343/alm.2016.36.4.393
Alijotas-Reig J, Esteve-Valverde E, Belizna C, Selva-O'Callaghan A, Pardos-Gea J, Quintana A, Mekinian A, Anunciacion-Llunell A, Miró-Mur F (2020) Immunomodulatory therapy for the management of severe COVID-19. Beyond the anti-viral therapy: A comprehensive review. Autoimmun Rev 102569:102569. https://doi.org/10.1016/j.autrev.2020.102569
Misra DP, Agarwal V, Gasparyan AY, Zimba O (2020) Rheumatologists’ perspective on coronavirus disease 19 (COVID-19) and potential therapeutic targets. Clin Rheumatol 39:2055–2062. https://doi.org/10.1007/s10067-020-05073-9
Perricone C, Triggianese P, Bartoloni E, Cafaro G, Bonifacio AF, Bursi R, Perricone R, Gerli R (2020) The anti-viral facet of anti-rheumatic drugs: lessons from COVID-19. J Autoimmun 111:102468. https://doi.org/10.1016/j.jaut.2020.102468
Gasparyan AY, Ayvazyan L, Cocco G, Kitas GD (2012) Adverse cardiovascular effects of antirheumatic drugs: implications for clinical practice and research. Curr Pharm Des 18:1543–1555. https://doi.org/10.2174/138161212799504759
Schreiber K, Breen K, Parmar K, Rand JH, Wu XX, Hunt BJ (2018) The effect of hydroxychloroquine on haemostasis, complement, inflammation and angiogenesis in patients with antiphospholipid antibodies. Rheumatol Oxf Engl 57:120–124. https://doi.org/10.1093/rheumatology/kex378
Gasparyan AY, Ayvazyan L, Yessirkepov M, Kitas GD (2015) Colchicine as an anti-inflammatory and cardioprotective agent. Expert Opin Drug Metab Toxicol 11:1781–1794. https://doi.org/10.1517/17425255.2015.1076391
Deftereos S, Giannopoulos G, Vrachatis DA et al (2020) Colchicine as a potent anti-inflammatory treatment in COVID-19: can we teach an old dog new tricks? Eur Heart J Cardiovasc Pharmacother. https://doi.org/10.1093/ehjcvp/pvaa033
Piantoni S, Patroni A, Toniati P, Furloni R, Franceschini F, Andreoli L, Scarsi M (2020) Why not to use colchicine in COVID-19? An old anti-inflammatory drug for a novel auto-inflammatory disease. Rheumatol Oxford 59:1769–1770. https://doi.org/10.1093/rheumatology/keaa217
Demidowich AP, Levine JA, Apps R et al (2005) Colchicine’s effects on metabolic and inflammatory molecules in adults with obesity and metabolic syndrome: results from a pilot randomized controlled trial. Int J Obes 2020:1–7. https://doi.org/10.1038/s41366-020-0598-3
Richardson P, Griffin I, Tucker C, Smith D, Oechsle O, Phelan A, Rawling M, Savory E, Stebbing J (2020) Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet 395:e30–e31. https://doi.org/10.1016/S0140-6736(20)30304-4
Sepriano A, Kerschbaumer A, Smolen JS, van der Heijde D, Dougados M, van Vollenhoven R, McInnes IB, Bijlsma JW, Burmester GR, de Wit M, Falzon L, Landewé R (2020) Safety of synthetic and biological DMARDs: a systematic literature review informing the 2019 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis 79:760–770. https://doi.org/10.1136/annrheumdis-2019-216653
Castagné B, Viprey M, Martin J, Schott AM, Cucherat M, Soubrier M (2019) Cardiovascular safety of tocilizumab: a systematic review and network meta-analysis. PLoS One 14:e0220178. https://doi.org/10.1371/journal.pone.0220178
Brito CA, Paiva JG, Pimentel FN, Guimarães RS, Moreira MR (2020) COVID-19 in patients with rheumatological diseases treated with anti-TNF. Ann Rheum Dis:annrheumdis-2020-218171. https://doi.org/10.1136/annrheumdis-2020-218171
Johannesdottir SA, Horváth-Puhó E, Dekkers OM, Cannegieter SC, Jørgensen JOL, Ehrenstein V, Vandenbroucke JP, Pedersen L, Sørensen HT (2013) Use of glucocorticoids and risk of venous thromboembolism: a nationwide population-based case-control study. JAMA Intern Med 173:743–752. https://doi.org/10.1001/jamainternmed.2013.122
Gianfrancesco M, Hyrich KL, Al-Adely S et al (2020) Characteristics associated with hospitalisation for COVID-19 in people with rheumatic disease: data from the COVID-19 Global Rheumatology Alliance physician-reported registry. Ann Rheum Dis 79:859–866. https://doi.org/10.1136/annrheumdis-2020-217871
Author information
Authors and Affiliations
Contributions
The conception and design of the study–SA, AYG; acquisition of data, analysis, and interpretation of data–SA, OZ, AYG. Drafting the article–SA, AYG; revising it critically for important intellectual content–OZ, AYG. Final approval of the version to be submitted–SA, OZ, AYG. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved–SA, AYG, OZ.
Corresponding author
Ethics declarations
Disclosures
None.
Disclaimer
All views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of any institution or association.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Ahmed, S., Zimba, O. & Gasparyan, A.Y. Thrombosis in Coronavirus disease 2019 (COVID-19) through the prism of Virchow’s triad. Clin Rheumatol 39, 2529–2543 (2020). https://doi.org/10.1007/s10067-020-05275-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10067-020-05275-1