
Abstract
Aims/hypothesis
Augmented formation of reactive oxygen species (ROS) induced by hyperglycaemia has been suggested to contribute to the development of diabetic nephropathy. This study was designed to evaluate the influence of streptozotocin (STZ)-induced diabetes mellitus, as well as the effects of preventing excessive ROS formation by α-tocopherol treatment, on regional renal blood flow, oxygen tension and oxygen consumption in anaesthetized Wistar Furth rats.
Methods
Non-diabetic and STZ-diabetic rats were investigated after 4 weeks with or without dietary treatment with the radical scavenger DL-α-tocopherol (vitamin E, 5%). A laser-Doppler technique was used to measure regional renal blood flow, whilst oxygen tension and consumption were measured using Clark-type microelectrodes.
Results
Renal oxygen tension, but not renal blood flow, was lower throughout the renal parenchyma of diabetic rats when compared to non-diabetic control rats. The decrease in oxygen tension was most pronounced in the renal medulla. Renal cellular oxygen consumption was markedly increased in diabetic rats, predominantly in the medullary region. Diabetes increased lipid peroxidation and protein carbonylation in the renal medulla. Treatment with α-tocopherol throughout the course of diabetes prevented diabetes-induced disturbances in oxidative stress, oxygen tension and consumption. The diabetic animals had a renal hypertrophy and a glomerular hyperfiltration, which were unaffected by α-tocopherol treatment.
Conclusions/interpretation
We conclude that oxidative stress occurs in kidneys of diabetic rats predominantly in the medullary region and relates to augmented oxygen consumption and impaired oxygen tension in the tissue.
Similar content being viewed by others

There are numerous hypotheses on the mechanisms involved in the onset and progression of diabetes-induced renal disturbances [1]. The Diabetes Control and Complication Trial (DCCT) showed that the degree of hyperglycaemia is an important predictor of subsequent diabetic renal complications [2]. However, a unifying mechanism has recently been put forward [3]. According to their hypothesis, normalizing the increased mitochondrial concentration of reactive oxygen species (ROS) prevents all major pathways for diabetes-induced damage. The putative role of ROS in the development of diabetic complications has been investigated for several decades [4, 5, 6, 7]. Levels of ROS are increased by metabolic changes in diabetes, including auto-oxidation and increased advanced glycation [4, 8]. Unsaturated fatty acids and proteins are major targets for ROS [9, 10]. ROS cause vascular endothelium abnormalities, react with nitric oxide (NO) in a diffusion-limited rate to generate peroxynitrite, thus depleting bioavailable NO and increasing the reactivity to vasoconstrictors [9]. Recent investigations suggest that excessive formation of ROS may cause vasoconstriction in the renal microvasculature [11].
Renal medullary blood flow is normally low in order to preserve osmotic gradients and enhance urinary concentration [12, 13]. At the same time, medullary oxygen consumption is high, since the medullary thick ascending limb is responsible for the generation of an osmotic gradient by active reabsorption of sodium. This results in a distinct gradient in oxygen tension from the renal cortex to the medulla [14, 15]. Previous studies have shown that renal proximal tubular cells from diabetic animals have an increased oxygen consumption [16, 17], whereas little is known concerning oxygen consumption in renal medullary cells.
Our study was designed to evaluate the influence of streptozotocin (STZ)-induced diabetes mellitus, with and without dietary treatment of the antioxidant α-tocopherol, on regional renal blood flow, oxygen tension and oxygen consumption in Wistar Furth rats. The degrees of lipid peroxidation and protein carbonylation were used as markers of oxidative stress within the renal tissue.
Materials and methods
Animals
Male Wistar-Furth rats, weighing 280 to 300 g, were purchased from B&K Universal (Sollentuna, Sweden) and had free access to water and rat chow (R3, Ewos, Södertälje, Sweden) throughout the study. All experiments were approved by the local animal ethics committee for Uppsala University.
The animals were divided into four experimental groups: Age-matched non-diabetic and diabetic animals were fed either R3 pellets or 5% (w/w) DL-α-tocopherol (Merck Eurolab, Stockholm, Sweden) supplemented to the powdered pellets throughout a 4-week experimental period. Animals from each of the four groups were then subjected to measurements of renal blood flow and oxygen tension or in vitro oxygen consumption. Renal tissue from the animals was frozen in liquid nitrogen and stored at −70°C for later analysis of α-tocopherol, lipid peroxidation, measured as thiobarbituric acids reactive substances (TBARS), and carbonylated proteins.
Diabetes induction
Diabetes mellitus was induced by an intravenous injection of streptozotocin (STZ, 45 mg/kg; Sigma-Aldrich, St. Louis, Mo., USA). Blood glucose concentrations were determined from blood samples obtained from the cut tip of the tail in all animals (MediSense, Bedford, Mass., USA). Animals were considered diabetic if blood glucose concentrations rose to greater than or equal to 15 mmol/l within 48 h after STZ-injection. The blood glucose concentrations and body weights were monitored weekly throughout the course of the study.
Regional renal oxygen tension and blood flow
The animals were anaesthetized with an intraperitoneal injection of thiobutabarbital (Inactin, Research Biochemicals International, Natick, Mass., USA; 120 mg/kg BW non-diabetic, 80 mg/kg BW diabetic animals), placed on an operating table maintained at 37°C and tracheostomized. Polyethylene catheters were placed in both femoral arteries and in the right femoral vein. One arterial catheter was used to monitor blood pressure (Statham P23dB, Statham Laboratories, Los Angeles, Calif., USA), whereas the other was used for blood sampling. Saline was infused into the vein to compensate for body fluid loss and the urinary bladder was catheterized to allow urinary drainage. The left kidney was exposed by a left subcostal flank incision, immobilized in a plastic cup and embedded in cotton wool soaked in saline. The surface of the kidney was covered with paraffin oil and the left ureter was catheterized for urine collection.
The animals were allowed a 60-min equilibration period followed by a period of 60 min to determine glomerular filtration rate (GFR) by measuring inulin clearance. For this purpose, 3H-inulin (185 kBq/ml; American Radiolabeled Company, St. Louis, Mo., USA) dissolved in saline was initially given as a bolus dose of 185 kBq and then infused (5 ml/kg/h) intravenously. Urine and arterial blood samples were taken for subsequent analyses. Renal oxygen tension was thereafter measured using modified Clark-type microelectrodes (4–6 µm o.d., Unisense, Aarhus, Denmark) [18]. The electrodes were two-point calibrated in water saturated with either Na2S2O5 or air at 37°C. Microelectrodes were then inserted into the renal tissue using a micromanipulator. A linear correlation was obtained between oxygen tension and the electric current. Oxygen tension measurements were carried out at one millimetre increments from the renal cortex to the inner part of the medulla (total depth of 5 millimetres from the renal surface). This procedure was repeated more than or equal to five times in each left kidney. The averages for each respective depth from one animal were then considered as one experiment in the statistical analysis. After the oxygen tension measurements, regional renal blood flow was measured with laser-Doppler flowmetry (probe 0.45 mm, PF 4001-2, Perimed, Stockholm, Sweden), by advancing the probe into the renal tissue in the same manner as the oxygen tension electrodes. After the experiment, animals were killed by an intravenous injection of KCl whereafter the kidney was dissected to verify the sites of the measurements. If any site was found not to be correctly located, the recording was discarded. The distances from the renal surface to the border between cortex and medulla and from the renal surface to the urinary calyces were determined using a stereomicroscope with a line ruler.
Isolation of renal cortical and outer medullary cells
The buffer solution had, if not stated otherwise, the following composition in mmol/l: 113.0 NaCl, 4.0 KCl, 27.2 NaHCO3, 1.0 KH2PO4, 1.2 MgCl2, 1.0 CaCl2, 10.0 HEPES, 0.5 Ca lactate, 2.0 glutamine. The buffer osmolality was adjusted to 298±2 mOsm as estimated with a freezing-point osmometer (Model 3MO; Advanced Instruments, Mass., USA), and the pH was adjusted to 7.40. Streptomycin (VWR International, Stockholm, Sweden) was added to a final concentration of 50 U/ml. For non-diabetic rats, the buffer contained 5.8 mmol/l glucose and for diabetic animals the buffer contained 23.2 mmol/l glucose (similar to the blood glucose concentrations in these latter animals). The renal cells were isolated as previously described [17, 19]. In brief, the rats were anaesthetized with thiobutabarbital, the kidneys were excised and the renal capsule was removed. The kidneys were placed on ice and the cortex and outer medulla were dissected under a stereomicroscope. Kidneys from two rats of the same group were pooled to increase the working material. The renal tissue was minced through a metallic mesh strainer and immediately placed in an ice-cooled buffer solution (cf. above) containing 0.05% (wt/vol) collagenase (Sigma-Aldrich). Thereafter, the minced tissue was incubated at 37°C, while the buffer was equilibrated with 95% O2/5% CO2. At least once every 5 min, the buffer-tissue solution was manually agitated. After incubation, the cell suspension was cooled on ice and filtrated through graded filters with pore sizes of 180, 75, 53 and 38 µm, respectively. After filtration, the cells were pelleted by a slow centrifugation (100 g, 4 min) and re-suspended in a collagenase-free buffer. The rinsing procedure was repeated three times to ensure that no collagenase remained in the final cell suspension. The suspension was kept on ice until oxygen consumption was measured.
Measurements of in vitro oxygen consumption
Oxygen consumption was measured as previously described [19]. In brief, a custom-made thermostatically controlled (37°C) gas-tight plexiglas chamber with a total volume of 1100 ml was used. The chamber was continuously stirred with an air driven magnetic stirrer. A modified Unisense 500 oxygen-sensing electrode (Unisense), calibrated with air-equilibrated buffer solution as 228 µmol/l O2 and Na2S2O5-saturated buffer as zero, was used to measure oxygen consumption. After the calibration, 100 µl of cell suspension was injected into the chamber and the rate of oxygen disappearance was recorded. At the end of each experiment, a 100 µl sample was taken to determine the protein concentration using DC Protein Assay (Bio-Rad Laboratories, Hercules, Calif., USA). The oxygen consumption was calculated as the disappearance rate of oxygen adjusted for protein concentration.
Cortical cells from non-diabetic rats were injected into the chamber and exposed to the following conditions: 5.8 mmol/l glucose (low glucose), 23.2 mmol/l glucose (high glucose) and 5.8 mmol/l glucose plus 1 mmol/l ouabain (low glucose + ouabain). Cortical cells from diabetic rats were exposed to 23.2 mmol/l glucose, 5.8 mmol/l glucose and 23.2 mmol/l glucose plus 1 mmol/l ouabain. Medullary cells from non-diabetic animals were only exposed to 5.8 mmol/l glucose. We did not carry out additional experiments on medullary cells as the amount of cells that could be isolated from the renal outer medulla was small. Medullary cells from diabetic animals were for the same reason only exposed to 23.2 mmol/l glucose.
Measurements of α-tocopherol, TBARS and protein carbonyls
Tissue samples were thawed and homogenized in ice-cold distilled water (0.2 g/ml). Tissue concentrations of α-tocopherol was measured as previously described [20, 21]. In brief, 500 µl of tissue homogenate was mixed with 500 µl methanol (VWR International), 2 ml of hexane was added and the samples were manually agitated for 3 min and thereafter centrifuged at 3000 rpm for 10 min. The hexane phase was collected for further analysis. The samples were separated with HPLC using a Spherisorb amino-column (4.6×250 mm, Phase Separation, Deeside, UK). The system was isocratic with isooctane/tert-butyl-methyl-ether/methanol (75/25/5) as mobile phase at a flow of 1 ml/min. The effluent was analyzed fluorometrically (Shimadzu RF-10A, Shimadzu, Kyoto, Japan) with an excitation wavelength of 295 nm and an emission wavelength of 327 nm. The sample values were compared to standard values from α-tocopherol (VWR International).
Thiobarbituric acids reactive substances (TBARS) were determined fluorometrically after boiling the samples with thiobarbituric acid. Of the tissue homogenate 200 µl was heated to 97°C for 60 min together with 250 µl 42 mmol/l thiobarbituric acid (Merck, Darmstadt, Germany). Standard samples were prepared from malondialdehyde-bis-(diethylacetate) (Merck-Schuchardt, Schuchardt, Germany). The samples were subsequently cooled on ice and precipitated with a mixture of methanol and 1 mol/l NaOH (91:9) and centrifuged at 3000 rpm for 5 min. Fluorescence was then measured in the supernatant; excitation wavelength was 532 nm and emission wavelength 553 nm, using Perkin-Elmer LS 5B luminescence spectrophotometer (Perkin-Elmer/Cetus, Norwalk, Conn., USA).
Concentrations of protein carbonyls in the renal medulla were determined using a commercially available ELISA kit (Zentech PC Test, Zenith Technology, Dunedin, New Zealand). In brief, a 5 µl homogenized sample was incubated for 45 min with a 2,4-dinitrophenylhydrazine solution yielding 2,4-dinitrophenylhydrazone groups on the protein carbonyls. The carbonylated proteins were then bound to a 96-well plate and antibodies directed against the hydrazone groups were used in the ELISA. Serum albumin was used as standard with five different concentrations of hypochlorous acid-oxidized protein. Oxidation of O-phenylenediamine by HPR was used as chromatogenic reaction, thereafter the absorbance was measured at 490 nm in a spectrophotometer.
Measurements of blood and urine parameters
The radioactivity of 3H-inulin in plasma (10 µl) and urine (1 µl) was measured by liquid scintillation. The GFR was then calculated as the clearance of 3H-inulin. The urine volumes were measured gravimetrically, the osmolality by use of a freezing point technique (Model 3MO, Advanced Instruments, Mass., USA) and the urinary sodium and potassium concentrations by use of flame photometry (IL543, Instrumentation Lab, Milan, Italy). At the end of each experiment, an arterial blood sample was collected for analysis of haematocrit (hcr) and blood gases. Mean arterial blood pressure of less than 90 mmHg, pH less than 7.30, pO2 less than 9.0 kPa, pCO2 greater than 6.8 kPa, or hcr less than 40 were set as exclusion criteria from the study.
Statistical analysis
All values are given as means ± SEM. Multiple comparisons between data were done using ANOVA followed by Fisher's protected least significant difference (PLSD) test (Statview, Abacus Concepts, Berkeley, Calif., USA). A p value of less than 0.05 was considered statistically significant.
Results
During the four-week experimental period, normoglycaemic animals continued to gain weight, whereas animals given STZ decreased in body weight by about 20% (Table 1). Blood glucose concentrations increased to greater than 15 mmol/l within 48 h after STZ-injection and remained at about 25 mmol/l throughout the course of the study (Table 1). Treatment with α-tocopherol had no effect in non-diabetic animals, but slightly increased body weights and decreased blood glucose concentrations in diabetic animals. Mean arterial blood pressure, haematocrit, arterial blood pH, blood pO2 and blood pCO2 was similar in all groups (Table 1).
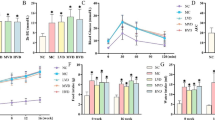
There was a pronounced blood flow gradient between the cortical and medullary regions in both non-diabetic and diabetic animals (Fig. 1). No difference in local renal blood flow was observed between diabetic and control animals. α-tocopherol treatment did not influence regional renal blood flow in either non-diabetic or diabetic animals. Oxygen tension in the renal tissue was found to be highest in the cortex (depth of 1mm) and in the outer stripe of the outer medulla (depth of 3 mm; Fig. 2). There was an evident decrease in tissue oxygen tension in the diabetic animals throughout the renal parenchyma, most pronounced in the medullary region. There was no correlation between the degree of hyperglycaemia and the decrease in renal tissue oxygen tension in the diabetic animals (data not shown). Treatment of diabetic animals with α-tocopherol fully prevented the decrease in tissue oxygen tension in the renal medulla, but only partially in the renal cortex.

Blood flow at different depths from the tissue surface in kidneys from non-diabetic control (filled circles), non-diabetic animals treated with vitamin E (empty circles), diabetic animals (filled triangles) and diabetic animals treated with vitamin E (empty triangles). The anatomical borders between the renal cortex and medulla (CMB) and between the outer and inner medulla (OM/IM) are delineated with broken lines

Tissue oxygen tension at different depths from the tissue surface in kidneys from non-diabetic control (filled circles), non-diabetic animals fed vitamin E (empty circles), diabetic animals (filled triangles) and diabetic animals fed vitamin E (empty triangles). The anatomical borders between the renal cortex and medulla (CMB) and between the outer and inner medulla (OM/IM) are delineated with broken lines. * denotes p<0.05 when compared to non-diabetic control animals, † p<0.05 for α-tocopherol treated diabetic animals compared to α-tocopherol treated non-diabetic animals and ‡ p<0.05 for α-tocopherol treated diabetic animals compared to untreated diabetic animals. The values represent means ± SEM for 7–10 animals
Renal oxygen consumption was not altered by acutely changing the glucose concentration in the medium (data not shown). Therefore, data from experiments carried out at different glucose concentrations on cells from the same group were pooled (Table 2). Renal cortical and preferentially medullary cells from diabetic rats had increased oxygen consumption when compared to corresponding cells from non-diabetic rats. Treatment of diabetic animals with α-tocopherol prevented the increase in oxygen consumption for both cortical and medullary cells. The Na+/K+-ATPase activity, measured as ouabain sensitive oxygen consumption, was 34% of the total oxygen consumption for cortical cells from non-diabetic animals and 40% for cortical cells from diabetic animals. After ouabain treatment, the oxygen consumption of cortical cells from diabetic and non-diabetic cells did not differ. Both non-diabetic and diabetic animals treated with α-tocopherol had a similar degree of ouabain sensitive oxygen consumption in their renal cortical cells as corresponding cells from untreated non-diabetic animals.
In all groups, higher α-tocopherol concentrations were found in the renal medulla than in the renal cortex (Table 3). No difference in either liver α-tocopherol or renal α-tocopherol concentrations was observed between un-supplemented non-diabetic and diabetic animals. In the non-diabetic and diabetic animals treated with α-tocopherol, increased α-tocopherol concentrations were recorded in the renal cortex, the renal medulla and the liver. In the medullary region, but not in the cortical region, TBARS concentrations were increased in diabetic animals compared with non-diabetic animals (Table 3). α-tocopherol treatment decreased TBARS in the renal cortex in both non-diabetic and diabetic animals, whereas TBARS in the renal medulla were only decreased by α-tocopherol treatment in diabetic animals. Diabetic rats given α-tocopherol did not differ in medullary TBARS levels compared to non-diabetic animals. Diabetic rats had increased concentrations of protein carbonyls in their renal medulla compared to control animals, an elevation which could be prevented by α-tocopherol treatment (Table 3). In non-diabetic animals, α-tocopherol treatment did not influence renal medullary protein carbonyl concentrations.
Renal weight and GFR were increased in both untreated and α-tocopherol treated diabetic animals when compared to non-diabetic animals (Table 4). α-tocopherol treatment slightly increased renal weights in non-diabetic animals, but diabetic animals treated with α-tocopherol had a more profound renal weight increase. Urine flow rate was about 10 times higher in diabetic animals compared to non-diabetic animals (Table 4). α-tocopherol treatment slightly reduced the urine flow rate in diabetic animals, but not in non-diabetic animals. The urinary excretion of osmotically active particles was markedly increased in all diabetic animals (Table 4). The urinary excretion of sodium tended to be increased in all diabetic animals, but only the α-tocopherol treated animals had an increase in sodium excretion compared with control animals (Table 4). The urinary excretion of potassium was increased in all diabetic animals compared with control rats (Table 4).
Discussion
Renal oxygen tension was substantially decreased throughout the renal parenchyma, but was more pronounced in the renal medulla of the diabetic animals. This diabetes-induced decrease could not be attributed to blood flow changes, but instead a ROS-dependent increase in cellular oxygen consumption. The scavenging of ROS by α-tocopherol treatment prevented both changes in renal oxygen tension and consumption. α-tocopherol treatment also slightly decreased blood glucose concentrations and increased body weights in diabetic animals. However, such small changes in blood glucose concentrations cannot explain the pronounced improvement in tissue oxygen tension in α-tocopherol treated diabetic animals, especially since no correlation between blood glucose concentrations and tissue oxygen tension was seen in the diabetic animals.
The significance of the diabetes-induced decrease in renal oxygen tension for renal function is unknown, but prevalence of tissue hypoxia has been postulated to be associated with both development of diabetic neuropathy and retinopathy [22, 23]. Surgically induced renal ischaemia has also been shown to accelerate the progression of nephropathy in STZ-diabetic rats [24]. In the kidney, the renal medullary region is particularly vulnerable to decreased tissue oxygen tension because of its low blood perfusion and high basal oxygen consumption [25, 26]. Already during normal circumstances, the renal medulla works at the threshold of hypoxia [25, 26] and has an oxygen extraction rate of about 90% [27]. Measurements of renal oxygen tension during diabetic conditions have previously been lacking. A hypothesis for the progression of renal disease with loss of glomerular function based on the concept of chronic oxygen deprivation to the tubulointerstitial compartment has nevertheless been proposed [28, 29]. Not only hyperglycaemia, but also chronic hypoxia, could contribute to induce cellular proliferation and extracellular matrix synthesis through increases in the expression of protein kinase C, growth factors such as vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF) and transforming growth factor β1 (TGFβ1), and osteopontin in several cell systems, including renal cells (for a review see [30, 31, 32]. Previous studies have used pimonidazole as a marker for renal hypoxia [26, 33]. However, due to unspecific glucose-dependent binding of this marker [34] it was unsuitable for use in this study.
Renal cortical cells from diabetic rats had increased oxygen consumption consistent with previously published results [16, 17]. In our study, we also measured oxygen consumption of cells from the outer medulla and noted that these had an even more pronounced increase in oxygen consumption. The exact mechanism for the diabetes-induced increase in renal oxygen consumption is not fully understood. The major oxygen consuming mechanism in the kidney is tubular reabsorption of filtered electrolytes, primarily sodium [17]. Indeed, the GFR in untreated diabetic rats was increased by about 30% in the present study. However, diabetic animals treated with α-tocopherol did not differ in either renal cellular oxygen consumption or oxygen tension when compared to non-diabetic animals, although they had a similar hyperfiltration as untreated diabetic animals. This clearly suggests other mechanisms mediating the diabetes-induced increase in renal cellular respiration and concomitant decrease in renal oxygen tension.
Although there are conflicting data, previous studies have shown that hyperglycaemia could neutralize the effects of NO by a direct scavenging effect, through the formation of advanced glycation end-products that consume NO, or through the induction of ROS, which avidly degrade NO and limit its bioavailability [35, 36, 37]. NO has been shown to have an inhibitory effect on the oxygen metabolism in both renal proximal tubules and vascular endothelial cells [19, 38]. Moreover, depletion of bioavailable NO has been shown to increase mitochondrial oxygen consumption and superoxide generation [39]. A possible effect in hyperglycaemic animals is a decreased influence of NO on oxygen metabolism, thereby resulting in increased oxygen consumption. This hypothesis is supported by the fact that scavenging of ROS, by α-tocopherol treatment, was shown to prevent the increase in cellular oxygen consumption in this study. However, regional blood flow was unaffected in the diabetic animals, which would not have been expected in view of the pronounced vasodilatory effects of NO. A perhaps even more plausible mechanism, resulting in augmented oxygen consumption of cells from diabetic animals, is the upregulation of Na+/K+-ATPase activity [16, 17]. When the flux through the Na+/glucose co-transporter is enhanced, Na+/K+-ATPase activity will increase as a result of increased intracellular levels of Na+ [17]. Indeed, the increase in renal cortical oxygen consumption could be prevented by ouabain-treatment. Chronic hyperglycaemia could also augment the consumption of oxygen by enhanced expression of uncoupling protein-2 and increased levels of free fatty acid which uncouple oxygen consumption from ATP production [16]. Furthermore, induction of gluconeogenesis, which is commonly seen in manifest diabetes [40, 41], might cause cellular oxygen consumption to increase [42].
α-tocopherol is well known for its ability to scavenge radicals and is essential for the defence against oxidation damage [43]. Conflicting results have been reported on changes in α-tocopherol concentrations in whole kidneys of hyperglycaemic animals [44, 45]. The highest concentration of α-tocopherol recorded in the kidney in our study was found in the renal medulla of both non-diabetic and diabetic animals. Interestingly, exogenous treatment with α-tocopherol markedly increased α-tocopherol concentrations in the renal medulla of all treated animals, but was more pronounced in the diabetic animals (increased five-fold). Previous studies have shown that lipid peroxidation, measured as TBARS, and carbonylated proteins are good markers for radical damage to lipids and proteins, respectively [9, 46]. An increased concentration of protein carbonyls is closely associated with poor diabetic control and correlates positively to HbA1c [47]. Both the concentrations of TBARS and protein carbonyls were higher in the renal medulla of diabetic animals, indicating increased oxidative stress. Medullary TBARS were increased five-fold, while there was no increase in cortical concentrations of TBARS. This suggests that the major target for ROS-induced renal damage during diabetes is the medullary region. Treatment of diabetic animals with α-tocopherol markedly decreased both concentrations of TBARS and protein carbonyls in the renal tissue.
The renal weight and GFR were increased in both untreated and α-tocopherol treated diabetic animals, suggesting that a ROS-independent mechanism mediates these changes in this experimental model. Previous studies in STZ-diabetic rats regarding the influence of α-tocopherol on GFR and renal weight have yielded conflicting results. In some studies, the increase in GFR and renal weight could be prevented by α-tocopherol, whereas in others no such effects were seen [48, 49, 50]. The differences could be attributed to different experimental settings where the severity of diabetes and the amount of α-tocopherol varied. α-tocopherol treatment also slightly increased renal weight in non-diabetic animals. The exact mechanisms for this increase remain to be determined. It should be noted that the distance from the renal surface to the corticomedullary border did not differ, neither did the distance from the renal surface to the papilla, between diabetic and non-diabetic animals. The renal hypertrophy seen in diabetic animals in the present study is instead attributed to a larger papillary area and tissue growth in the renal apices.
The urinary excretion of sodium, potassium and osmotically active particles were increased in diabetic animals and could not be prevented by the treatment with α-tocopherol. The most probable mechanism accounting for the increased excretion in the diabetic rats is the increased GFR and subsequently increased urinary production, which could not be normalized by α-tocopherol treatment.
In conclusion, oxidative stress occurred in renal tissue of diabetic rats predominantly in the medullary region and was related to augmented oxygen consumption and impaired oxygen tension in the tissue. These diabetic-induced alterations could be prevented by treatment with the ROS-scavenger α-tocopherol. The consequences of a chronic decrease in oxygen tension for the renal function are worthy of further studies.
Abbreviations
- ROS:
-
Reactive oxygen species
- TBARS:
-
thiobarbituric acids reactive substances
- NO:
-
nitric oxide
- STZ:
-
streptozotocin
References
King GL, Brownlee M (1996) The cellular and molecular mechanisms of diabetic complications. Endocrinol Metab Clin North Am 25:255–270
Group TDCaCTR (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
Nishikawa T, Edelstein D, Du XL et al. (2000) Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 404:787–790
Baynes JW (1991) Role of oxidative stress in development of complications in diabetes. Diabetes 40:405–412
Baynes JW, Thorpe SR (1999) Role of oxidative stress in diabetic complications: a new perspective on an old paradigm. Diabetes 48:1–9
Oberley LW (1988) Free radicals and diabetes. Free Radic Biol Med 5:113–124
Ceriello A, Morocutti A, Mercuri F et al. (2000) Defective intracellular antioxidant enzyme production in type 1 diabetic patients with nephropathy. Diabetes 49:2170–2177
Wolff SP (1993) Diabetes mellitus and free radicals. Free radicals, transition metals and oxidative stress in the aetiology of diabetes mellitus and complications. Br Med Bull 49:642–652
Cameron NE, Cotter MA (1999) Effects of antioxidants on nerve and vascular dysfunction in experimental diabetes. Diabetes Res Clin Pract 45:137–146
Miyata T, Inagi R, Asahi K et al. (1998) Generation of protein carbonyls by glycoxidation and lipoxidation reactions with autoxidation products of ascorbic acid and polyunsaturated fatty acids. FEBS Lett 437:24–28
Schnackenberg CG (2002) Physiological and pathophysiological roles of oxygen radicals in the renal microvasculature. Am J Physiol Regul Integr Comp Physiol 282:R335–R342
Hansell P (1992) Evaluation of methods for estimating renal medullary blood flow. Ren Physiol Biochem 15:217–230
Sadowski J, Kompanowska-Jezierska E, Dobrowolski L, Walkowska A, Badzynska B (1997) Simultaneous recording of tissue ion content and blood flow in rat renal medulla: evidence on interdependence. Am J Physiol 273:F658–F662
Leichtweiss HP, Lubbers DW, Weiss C, Baumgartl H, Reschke W (1969) The oxygen supply of the rat kidney: measurements of intrarenal pO2. Pflugers Arch 309:328–349
Brezis M, Heyman SN, Epstein F (1994) Determinants of intrarenal oxygenation II. Hemodynamic effects. Am J Physiol 267:F1063–F1068
Baines A, Ho P (2002) Glucose stimulates O2 consumption, NOS, and Na/H exchange in diabetic rat proximal tubules. Am J Physiol Renal Physiol 283:F286–F293
Korner A, Eklof AC, Celsi G, Aperia A (1994) Increased renal metabolism in diabetes. Mechanism and functional implications. Diabetes 43:629–633
Liss P, Nygren A, Revsbech NP, Ulfendahl H (1997) Intrarenal oxygen tension measured by a modified Clark electrode at normal and low blood pressure and after injection of x-ray contrast media. Pflugers Arch 434:705–711
Koivisto A, Pittner J, Froelich M, Persson AE (1999) Oxygen-dependent inhibition of respiration in isolated renal tubules by nitric oxide. Kidney Int 55:2368–2375
Siman CM, Eriksson UJ (1997) Vitamin E decreases the occurrence of malformations in the offspring of diabetic rats. Diabetes 46:1054–1061
Cederberg J, Siman CM, Eriksson UJ (2001) Combined treatment with vitamin E and vitamin C decreases oxidative stress and improves fetal outcome in experimental diabetic pregnancy. Pediatr Res 49:755–762
Cameron NE, Cotter MA (1997) Metabolic and vascular factors in the pathogenesis of diabetic neuropathy. Diabetes 46 [Suppl 2]:S31–S37
Linsenmeier RA, Braun RD, McRipley MA et al. (1998) Retinal hypoxia in long-term diabetic cats. Invest Ophthalmol Vis Sci 39:1647–1657
Melin J, Hellberg O, Akyurek LM, Kallskog O, Larsson E, Fellstrom BC (1997) Ischemia causes rapidly progressive nephropathy in the diabetic rat. Kidney Int 52:985–991
Brezis M, Rosen S (1995) Hypoxia of the renal medulla—its implication for disease. New Engl J Med 332:647–655
Zhong Z, Arteel GE, Connor HD et al. (1998) Cyclosporin A increases hypoxia and free radical production in rat kidneys: prevention by dietary glycine. Am J Physiol 275:F595–F604
Brezis M, Rosen S, Silva P, Epstein FH (1984) Renal ischemia: a new perspective. Kidney Int 26:375–383
Fine LG, Orphanides C, Norman JT (1998) Progressive renal disease: the chronic hypoxia hypothesis. Kidney Int [Suppl 65]:S74–S78
Fine LG, Bandyopadhay D, Norman JT (2000) Is there a common mechanism for the progression of different types of renal diseases other than proteinuria? Towards the unifying theme of chronic hypoxia. Kidney Int [Suppl 75]:S22–S26
Sahai A, Mei C, Schrier RW, Tannen RL (1999) Mechanisms of chronic hypoxia-induced renal cell growth. Kidney Int 56:1277–1281
Orphanides C, Fine LG, Norman JT (1997) Hypoxia stimulates proximal tubular cell matrix production via a TGF-beta1-independent mechanism. Kidney Int 52:637–647
Sodhi CP, Phadke SA, Batlle D, Sahai A (2001) Hypoxia and high glucose cause exaggerated mesangial cell growth and collagen synthesis: role of osteopontin. Am J Physiol Renal Physiol 280:F667–F674
MacManus MP, Maxwell AP, Abram WP, Bridges JM (1989) The effect of hypobaric hypoxia on misonidazole binding in normal and tumour-bearing mice. Br J Cancer 59:349–352
Ling LL, Sutherland RM (1986) Modulation of the hypoxic toxicity and binding of misonidazole by glucose. Br J Cancer 54:911–917
Goligorsky MS, Chen J, Brodsky S (2001) Workshop: endothelial cell dysfunction leading to diabetic nephropathy : focus on nitric oxide. Hypertension 37:744–748
Vaziri ND, Ni Z, Oveisi F, Liang K, Pandian R (2002) Enhanced nitric oxide inactivation and protein nitration by reactive oxygen species in renal insufficiency. Hypertension 39:135–141
Droge W (2002) Free radicals in the physiological control of cell function. Physiol Rev 82:47–95
Clementi E, Brown GC, Foxwell N, Moncada S (1999) On the mechanism by which vascular endothelial cells regulate their oxygen consumption. Proc Natl Acad Sci USA 96:1559–1562
Brodsky SV, Gao S, Li H, Goligorsky MS (2002) Hyperglycemic switch from mitochondrial nitric oxide to superoxide production in endothelial cells. Am J Physiol Heart Circ Physiol 283:H2130–H2139
Smith OK, Long CN (1971) Renal gluconeogenesis in eviscerated diabetic rats. Proc Natl Acad Sci USA 68:1618–1622
Peroni O, Large V, Diraison F, Beylot M (1997) Glucose production and gluconeogenesis in postabsorptive and starved normal and streptozotocin-diabetic rats. Metabolism 46:1358–1363
Horstman P (1951) The oxygen consumption in diabetes mellitus. Acta Med Scand 139:326–330
Kinalski M, Sledziewski A, Telejko B et al. (2000) Lipid peroxidation and scavenging enzyme activity in streptozotocin-induced diabetes. Acta Diabetol 37:179–183
Sun F, Iwaguchi K, Shudo R, Nagaki Y, Tanaka K, Ikeda K, Tokumaru S, Kojo S (1999) Change in tissue concentrations of lipid hydroperoxides, vitamin C and vitamin E in rats with streptozotocin-induced diabetes. Clin Sci (Lond) 96:185–190
Elangovan V, Shohami E, Gati I, Kohen R (2000) Increased hepatic lipid soluble antioxidant capacity as compared to other organs of streptozotocin-induced diabetic rats: a cyclic voltammetry study. Free Radic Res 32:125–134
Cederberg J, Basu S, Eriksson UJ (2001) Increased rate of lipid peroxidation and protein carbonylation in experimental diabetic pregnancy. Diabetologia 44:766–774
Odetti P, Garibaldi S, Noberasco G, Aragno I, Valentini S, Traverso N, Marinari UM (1999) Levels of carbonyl groups in plasma proteins of type 2 diabetes mellitus subjects. Acta Diabetol 36:179–183
Koya D, Lee IK, Ishii H, Kanoh H, King GL (1997) Prevention of glomerular dysfunction in diabetic rats by treatment with d-alpha-tocopherol. J Am Soc Nephrol 8:426–435
Craven PA, DeRubertis FR, Kagan VE, Melhem M, Studer RK (1997) Effects of supplementation with vitamin C or E on albuminuria, glomerular TGF-beta, and glomerular size in diabetes. J Am Soc Nephrol 8:1405–1414
Kim SS, Gallaher DD, Csallany AS (2000) Vitamin E and probucol reduce urinary lipophilic aldehydes and renal enlargement in streptozotocin-induced diabetic rats. Lipids 35:1225–1237
Acknowledgements
The excellent technical assistance of A. Fasching and A. Nordin is gratefully acknowledged. This study was supported by The Swedish Diabetes Association, Svenska Barndiabetesfonden, the Knut and Alice Wallenberg Foundation, the Anér Foundation, the Selander Foundation, the Family Ernfors Foundation, Konsul Ture Carlsson Foundation, the Swedish Medical Society, the Marcus and Amalia Wallenberg Foundation, The Apotekare Hedbergs Research Foundation, The Harald and Greta Jeansson Foundation and the Swedish Medical Research Council (9940, 10840, 12X-7457 and 12X-109).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Palm, F., Cederberg, J., Hansell, P. et al. Reactive oxygen species cause diabetes-induced decrease in renal oxygen tension. Diabetologia 46, 1153–1160 (2003). https://doi.org/10.1007/s00125-003-1155-z
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-003-1155-z