
Abstract
mRNA vaccines have become a versatile technology for the prevention of infectious diseases and the treatment of cancers. In the vaccination process, mRNA formulation and delivery strategies facilitate effective expression and presentation of antigens, and immune stimulation. mRNA vaccines have been delivered in various formats: encapsulation by delivery carriers, such as lipid nanoparticles, polymers, peptides, free mRNA in solution, and ex vivo through dendritic cells. Appropriate delivery materials and formulation methods often boost the vaccine efficacy which is also influenced by the selection of a proper administration route. Co-delivery of multiple mRNAs enables synergistic effects and further enhances immunity in some cases. In this chapter, we overview the recent progress and existing challenges in the formulation and delivery technologies of mRNA vaccines with perspectives for future development.
You have full access to this open access chapter, Download chapter PDF
Similar content being viewed by others

1 Introduction
Since the first use of in vitro transcribed messenger RNA (mRNA) to express an exogenous protein in mice in 1990 (Wolff et al. 1990), mRNA has evolved into a versatile platform spanning many therapeutic and prophylactic fields (Hajj and Whitehead 2017; Xiong et al. 2018; Li et al. 2019; Patel et al. 2019b; Pardi et al. 2020; Weng et al. 2020). In particular, numerous mRNA vaccines are being developed to tackle infectious diseases and various types of cancer, with many advancing to different stages of clinical trials (Pardi et al. 2018).
Several features of in vitro transcribed mRNA contribute to its vaccine potential. First, the development process of an mRNA vaccine can be much faster than conventional protein vaccines (DeFrancesco 2017). In response to the pandemic of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) in 2020, an mRNA vaccine was administrated to the first volunteer in a phase 1 clinical trial within ten weeks after the sequence of the viral genome was revealed (Lurie et al. 2020). Second, in vitro transcription reaction is easy to conduct, has a high yield, and can be scaled up (Pardi et al. 2018). Advanced industrial setup can manufacture mRNA up to kilogram scales (Versteeg et al. 2019). Third, mRNA vaccine enables the synthesis of antigen proteins in situ, eliminating the need for protein purification and long-term stabilization which are challenging for some antigens. Fourth, transportation and storage of mRNA may be easier than protein-based vaccines, since RNA, if protected properly against ribonucleases (RNases), is less prone to degradation compared to proteins (Stitz et al. 2017; Zhang et al. 2019). Because of these advantages, mRNA vaccines have great potential to be manufactured and deployed in a timely manner in response to rapid infectious disease outbreaks.
Despite mRNA’s appealing features and advances in the field, in vivo delivery of mRNA remains challenging. The first challenge is the instability of mRNA mostly due to enzymatic degradation by RNases. RNases are present ubiquitously throughout the body to degrade exogenous RNAs (Gupta et al. 2012). And mRNA, consisting of hundreds to thousands of nucleotides, has to reach the cytosol in full length for active translation. Hence, protection against RNases is critical for most in vivo delivery strategies. Secondly, efficient intracellular delivery of mRNA is another challenge owing to the negative charge and large size of mRNA molecules. The negative charge prevents most mRNA from translocating across the negatively charged cell membrane. The large size makes efficient encapsulation and delivery more challenging than other payloads, such as small molecules, siRNAs, and antisense oligonucleotides (ASOs). Various delivery strategies have been investigated to address these obstacles with different delivery materials, formulation methods, and routes of administrations.
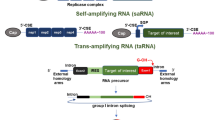
The mRNAs used as vaccines can be categorized into conventional mRNAs and self-amplifying mRNAs. Conventional mRNAs are similar to endogenous mRNAs in mammalian cells, consisting of a 5’ cap, 5’ UTR, coding region, 3’ UTR, and a polyadenylated tail (Pardi et al. 2018; Kowalski et al. 2019). The typical size is 1–5 k nucleotides. When delivered to the cytosol, this type of mRNA is translated until its degradation without additional replication. On the other hand, self-amplifying mRNAs are derived from the genomes of single-stranded RNA viruses, such as alphaviruses (Brito et al. 2015). Besides encoding proteins of interest, self-amplifying mRNAs encode replication machinery consisting of several viral non-structural proteins (nsPs) to replicate themselves. Therefore, their typical size is approximately 8–12 k nucleotides, larger than the conventional mRNA vaccine. When delivered to the cytosol, self-amplifying mRNAs replicate themselves while expressing the designated proteins in a relatively large amount (Iavarone et al. 2017). More importantly, self-amplifying mRNAs are unique for vaccine applications because of their self-adjuvant nature (Maruggi et al. 2019). Many factors involved in their self-replication process, such as the double-stranded RNA (dsRNA) intermediate of replication (von Herrath and Bot 2003) and the nsPs in the replication machinery (Maruggi et al. 2013), could stimulate interferon-mediated immune responses (Pepini et al. 2017).
Three major types of proteins are encoded by mRNA vaccines: antigens (Grunwitz and Kranz 2017; Zhang et al. 2019), neutralizing antibodies (Stadler et al. 2017; Tiwari et al. 2018), and proteins with immunostimulatory activity (Bonehill et al. 2008; Manara et al. 2019). Antigens or neutralizing antibodies induce specific immune responses, while proteins with immunostimulatory activity, such as CD70 (Van Lint et al. 2012) and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Manara et al. 2019) boost innate and/or adaptive immunity.
Advances in recent years made mRNA a promising vaccine platform. For example, chemical modifications of RNA using nucleotide analogs, such as pseudouridine, dramatically increased protein production in vivo by diminishing the translation inhibition triggered by the unmodified nucleotides (Kariko et al. 2008; Warren et al. 2010). High-performance liquid chromatography (HPLC) purification further increased the purity and translation capability of mRNA by removing the byproducts from in vitro transcription, such as dsRNA, which could induce inhibition of mRNA translation (Karikó et al. 2011; Weissman et al. 2013). Lipid and lipid-derived nanoparticles (LNPs) were previously used to deliver small molecule drugs and siRNAs (Brito et al. 2015; Ickenstein and Garidel 2019). The adaptation of LNPs for mRNA delivery greatly enhanced the delivery efficiency of mRNA both in vitro and in vivo (Dimitriadis 1978; Malone et al. 1989; Martinon et al. 1993). The use of new formulation technologies, such as continuous-flow microfluidic devices, enabled reproducible production of nanoparticles at various scales with controllable sizes (Jahn et al. 2008; Valencia et al. 2012).
In this chapter, we summarize the routes of administrations for mRNA vaccines, discuss mRNA delivery carriers and their corresponding formulation methods, and overview the challenges and future development of mRNA vaccines. A comprehensive overview of recent advances in mRNA vaccine delivery may facilitate the future development of novel delivery strategies and effective mRNA vaccines.
2 Administration Routes for mRNA Vaccines
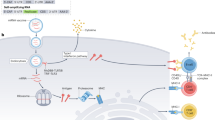
The administration route for mRNA vaccines plays an important role in determining vaccination efficacy (Eggert et al. 1999). Figure 1 depicts the most commonly used injection routes, including intradermal (ID), subcutaneous (SC), intramuscular (IM), intranodal (IN), and intravenous (IV) administration (Verbeke et al. 2019b). Other routes, such as intranasal injection (Lorenzi et al. 2010; Li et al. 2017a), intravaginal injection (Lindsay et al. 2020), and intratumoral injection (Scheel et al. 2006; Van Lint et al. 2016), were also tested. Since the immune cells and lymphoid organs are the common vaccination targets, the anatomical and physiological properties of the vaccination sites (skin, muscle, lymphoid organ, and systemic circulation) may affect the safety and efficacy of a vaccine (Johansen and Kündig 2015). Such information is useful for the selection of administration route when the type (conventional or self-amplifying) and the delivery format (carrier-mediated, naked, or cell-based) of the mRNA vaccine are chosen.

Common routes for the delivery of mRNA vaccines
Intradermal (ID) injection delivers mRNA vaccines directly into the dermis region, which is dense connective tissue (Fig. 1a). Antigen-presenting cells (APCs) in the dermis tissue, such as dermal dendritic cells (DCs) and macrophages (Kashem et al. 2017), can internalize and process the mRNA vaccine. The vascular and lymphatic vessels in this layer of skin also help transport mRNA vaccines and APCs to the draining lymph nodes to activate T and B cells (Kashem et al. 2017; Melo et al. 2019). Because of these properties, ID injection was tested in clinical studies for delivering mRNA vaccine encoding rabies virus glycoprotein (Alberer et al. 2017). Their results showed ID vaccination by a needle-free device could induce better antibody response than IM injection. Although ID injection has preferential access to immune cells and lymphoid organs, and has shown vaccination efficacy, this method has been limited by its small injection volume and high risk of local adverse effect including swelling, pain, erythema, and pruritus (Engmann et al. 1998; Diehl et al. 2001; Rini et al. 2016; Sienkiewicz and Palmunen 2017). To increase injection volume and mRNA dose, patients received multiple ID injections at different sites per visit in a clinical trial (Sebastian et al. 2019).
Subcutaneous (SC) injection administers mRNA vaccines to the subcutis region under the epidermis and dermis (Fig. 1b). This layer of skin is mainly composed of a loose network of adipose tissues and few immune cells compared to the dermis (Ibrahim 2010). Comparing to ID injection, the loose adipose tissue at the SC injection site permits a larger injection volume (Sienkiewicz and Palmunen 2017), causing less pain and lower pressure (Johansen and Kündig 2015). In addition, the larger injection volume may compensate for the less efficient draining activity in this layer of skin (Johansen and Kündig 2015). It is noteworthy that the absorption rate in the SC area is slow, which may cause unintended degradation of the mRNA vaccine (Gradel et al. 2018).
Intramuscular (IM) injection delivers the vaccine into muscles, a deeper tissue under the dermal and subcutaneous layer (Fig. 1c). Muscles contain a large network of blood vessels that can help recruit and recirculate different types of immune cells, such as the infiltrating APCs, to the injection site (Liang et al. 2017). A recent study indicated that after IM injection of LNPs-encapsulated mRNA, the radiolabeled mRNA was detected at the site of injection and draining lymph node for at least 28 h (Lindsay et al. 2019). Detailed flow cytometry analysis showed that APCs in muscle as well as APCs and B cells in draining lymph nodes contained the radiolabeled mRNA (Lindsay et al. 2019). The volume of IM injection is larger than that of ID injection in humans (Sienkiewicz and Palmunen 2017). Additionally, IM injection may cause milder local side effects compared with ID and SC routes. Thus, IM injection is the most widely used administration route for adjuvanted vaccines (Moyer et al. 2016).
Intranodal (IN) injection directly introduces mRNA vaccines to the peripheral lymphoid organs where APCs and primed T or B cells interact (Fig 1e). IN injection is considered to be an efficient way of vaccination, since the APCs in lymphoid organs can readily engulf the injected mRNA vaccine (Kreiter et al. 2010; Bialkowski et al. 2016; Joe et al. 2019). Even though studies reported increased vaccination efficacy by IN delivery route compared with other injection methods for DNA, peptide and protein vaccines (Senti and Kündig 2015), side-by-side comparison between IN delivery and other routes for mRNA vaccine remains limited (Kreiter et al. 2010). In addition, IN injection was seldom used mostly because of the relatively complicated procedure (Johansen and Kundig 2014; Senti and Kündig 2015). For example, IN injection needs ultrasound guidance in human (Senti and Kündig 2015).
Mucosal delivery of mRNA vaccines was studied because of the accessible APCs in lymphoid organs at the mucosal sites and their protective roles against various pathogens. Among the mucosal administration routes, intranasal and intravaginal administrations were utilized to deliver mRNA vaccines (Lorenzi et al. 2010; Li et al. 2017a; Lindsay et al. 2020). Intranasal injection delivers mRNA vaccines to the nasal mucosa and nasal associated lymphoid tissue (NALT), both of which contain rich APCs and related immune cells (Lobaina Mato 2019). As a result, the intranasal delivery of antigen-encoding mRNA was reported to induce humoral and cell-mediated immunity (Lorenzi et al. 2010; Zhuang et al. 2020). Intranasal delivery can also apply mRNA to the lung through the trachea. Similar to the nasal mucosa and NALT, the immature and activated APCs available in the lung can engulf and process the mRNA vaccine (Stehle et al. 2018). Additionally, an mRNA vaccine was delivered to the lung epithelial cells via intranasal injection and expressed neutralizing antibodies against virus infection (Tiwari et al. 2018). Intravaginal injection is another approach to deliver mRNA vaccines to the site of infection to express neutralizing antibodies. In one report, the intravaginal delivery of the mRNA vaccine encoding an anti-HIV antibody induced high levels of antibody expression in the reproductive tract of sheep and rhesus macaques (Lindsay et al. 2020). Immunofluorescence staining showed the expressed antigen was mainly found in cervical epithelial cells and stromal cells (Lindsay et al. 2020).
Intravenous (IV) injection delivers mRNA vaccines into the systemic circulation (Fig. 1d). The volume of IV injection is the largest among the delivery routes mentioned above (Diehl et al. 2001). The total amount of protein produced via IV administration is often the highest compared to other routes (Pardi et al. 2015). Thus, the IV route was chosen for delivering mRNA encoding a neutralizing antibody when a functional concentration of the antibody was required in circulation (Kose et al. 2019). Generally, IV injected LNP delivers mRNA to the liver, and more specifically, to hepatocytes, Kupffer cells, and liver endothelial cells depending on different types of delivery carriers (Pardi et al. 2015; Conway et al. 2019). Moreover, IV injections may also allow the direct access of mRNA vaccines to immune cells and lymphoid organs in the circulatory system, which may enhance the vaccination efficacy (Kranz et al. 2016). For example, previous studies found IV injection of mRNA vaccine targeted spleen DCs and induced immune response against tumors in mice (Kranz et al. 2016). Despite the advantages of IV injection mentioned above, the plasma proteins, enzymes, and mechanical forces in the bloodstream may hinder the vaccine delivery (Reichmuth et al. 2016). Furthermore, the administration of mRNA and delivery carriers to the circulation may introduce systemic side effects, such as spleen injury and lymphocyte depletion (Reichmuth et al. 2016; Sedic et al. 2017).
In summary, the biological features of different administration routes may impact the safety and efficacy of vaccination. Table 1 summarizes the features of several major delivery routes of mRNA vaccines. For a given combination of mRNA-type and delivery carrier, a careful comparison of several administration routes will help determine the most appropriate injection method and promote the development of an effective mRNA vaccine.
3 Delivery Strategies for mRNA Vaccines
Researchers have investigated many methods to deliver mRNA vaccines. For example, delivery carriers, such as lipid-derived and polymer-derived materials, dramatically increased cellular uptake of RNAs, thus receiving tremendous attention in recent years (Reichmuth et al. 2016; Kowalski et al. 2019; Riley et al. 2019). mRNA vaccines were also delivered as free mRNA (Fleeton et al. 2001; Edwards et al. 2017). Additionally, dendritic cells were loaded with mRNA vaccines ex vivo and transferred to the hosts (Benteyn et al. 2015). In this section, we focus on the technologies for formulating and delivering mRNA vaccines in carrier-mediated, naked, and DC-based forms.
3.1 Delivery Carriers of mRNA Vaccines
3.1.1 Lipid-based Delivery
Lipids, lipid-like compounds, and lipid derivatives have been widely used to formulate lipid and lipid-derived nanoparticles (LNPs) for in vivo delivery of mRNA vaccines (Midoux and Pichon 2015; Reichmuth et al. 2016; Corthésy and Bioley 2018; Pardi et al. 2018; Li et al. 2019). LNPs are generally defined as nano-sized particulate systems that are composed of synthetic or physiological lipid materials (Ganesan and Narayanasamy 2017). Table 2 lists representative in vivo delivery of mRNA vaccines by LNPs. LNPs are developed for mRNA vaccine delivery for the following two main reasons. Firstly, LNPs can encapsulate RNA molecules, protecting RNA from enzymatic degradation (Midoux and Pichon 2015). The reported mRNA encapsulation efficiency by LNP was usually high, indicating the mRNA molecules were mostly encapsulated (Richner et al. 2017a, b). Secondly, LNPs can effectively deliver mRNA molecules into the cell cytosol through a series of endocytosis mechanisms (Sahay et al. 2010). For example, it was reported that the surface adsorption of apolipoprotein E (apoE) on LNP might facilitate its intracellular delivery via low-density lipoprotein receptor-mediated clathrin-dependent endocytosis (Basak et al. 2012). This endocytosis process transported the mRNA-loaded LNPs into cell membrane-bound vesicles, including endosomes and lysosomes (Sahay et al. 2010; Patel et al. 2019c). Eventually, the LNPs helped translocate mRNA cargos into the cytosol for protein expression (Midoux and Pichon 2015).
The LNPs usually contain one or more of the functional lipid components that are crucial for the intracellular RNA delivery described above (Midoux and Pichon 2015; Kowalski et al. 2019; Verbeke et al. 2019b). The cationic or ionizable lipid materials, such as 1,2-di-O-octadecenyl-3-trimethylammonium propane (DOTMA), N,N-Dimethyl-2,3-bis[(9Z,12Z)-octadeca-9,12-dienyloxy]propan-1-amine (DLinDMA), and N1,N3,N5-tris(3-(didodecylamino)propyl)benzene-1,3,5-tricarboxamide (TT3) usually contain one or multiple amino groups (Semple et al. 2010; Jayaraman et al. 2012; Billingsley et al. 2020; Zeng et al. 2020). These lipid materials can be positively charged at a certain pH to encapsulate the negatively charged RNA molecules via electrostatic interactions and help interact with the cell membrane on target cells. Previous studies indicated the final step of RNA release from LNPs into the cytosol might involve the membrane disruption of endosomes (Cullis and Hope 2017). In this process, the ionizable cationic lipids were suggested to interact with anionic lipid on endosome membrane and form disruptive non-bilayer structures, which finally released the encapsulated RNA into the cytosol (Cullis and Hope 2017). Furthermore, the structure–activity relationship of the lipids head and tail for RNA delivery and endosomal escape was studied (Sato et al. 2019). The results indicated that the hydrophilic head group in lipid materials might determine the acid dissociation constant (pKa) and influence the delivery efficiency (Sato et al. 2019). Besides, modification of fatty acids structures in hydrophobic tails may also affect the delivery efficiency (Sabnis et al. 2018; Sato et al. 2019). Even though the membrane disruptive features of lipid materials improve the delivery efficiency, these synthetic materials may cause side effects in vivo (Pun and Hoffman 2013; Sedic et al. 2017). The helper lipids, such as 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE), 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), and cholesterol, stabilize LNPs structures and facilitate endosome escape (Cheng and Lee 2016). The PEG-lipid conjugates could stabilize the nanoparticles during preparation and provide a hydrophilic outer layer that prolongs the circulation time after in vivo administration (Ambegia et al. 2005; Heyes et al. 2005; Cheng and Lee 2016; Kowalski et al. 2019). In addition to these functions, the engineered ionizable lipid materials containing cyclic amino head groups, isocyanide linker, and two unsaturated alkyl tails were reported to provide adjuvant activities independent of the encapsulated mRNA (Miao et al. 2019). These cyclic amino head groups directly bound the STING (stimulator of interferon genes) protein and triggered the downstream signaling pathway, leading to an elevated innate response. After SC injection of an antigen-coding mRNA encapsulated by such LNP into mice, the researchers observed the upregulation of antigen-specific T cells and inhibition of tumor growth (Miao et al. 2019).
The formulation methods of lipid-based mRNA vaccines mainly include thin-film hydration (Akbarzadeh et al. 2013; Kranz et al. 2016), direct mixing (Borrego et al. 2017), ethanol injection (Geall et al. 2012), and continuous-flow microfluidic device (Chen et al. 2012; Kose et al. 2019). Among these methods, the continuous-flow microfluidic device emerges as a prevalent method to prepare RNA encapsulated nanoparticle, especially LNP, for in vivo use (Liu et al. 2018; Kowalski et al. 2019). These chip-based microfluidic devices mix two laminar flows, the RNA-containing aqueous phase and the carriers-containing solvent phase, through a confined microchannel equipped with chaotic mixers at a controlled speed, leading to rapid diffusion, change of polarity and self-assembly of mRNA-LNP at the interface (Belliveau et al. 2012; Yanez Arteta et al. 2018). The resulting lipid nanoparticles are relatively homogeneous formulation and usually show spherical and multilamellar morphology (Yanez Arteta et al. 2018). Compared to other preparation methods, the use of continuous-flow microfluidic devices increases reproducibility, improves molecular stability, reduces the chance of contamination, and is easily scaled up for preclinical and clinical studies (Damiati et al. 2018; Liu et al. 2018).
The delivery routes of lipid-based mRNA vaccines include IM, ID, SC, IN, and IV injection (Midoux and Pichon 2015). Delivery routes can affect the in vivo distribution pattern and expression kinetics of encapsulated mRNA vaccines (Pardi et al. 2015). Local injections, such as IM, ID, SC, and IN administrations, deliver LNPs mRNA vaccine to resident/infiltrating APCs and related immune cells, stimulating strong and prolonged local expression (Kreiter et al. 2010; Pardi et al. 2015; Hassett et al. 2019). Thus, local injections were utilized to deliver most LNP encapsulated antigen-encoding mRNA vaccines (see examples in Table 2). For example, TT3-LNPs were used to deliver mRNAs encoding the full-length spike protein or its receptor binding domain of SARS-CoV-2. After IM administration, the expression of both antigens was observed in muscle tissues (Zeng et al. 2020). The resident APCs in the skin, muscle, and lymph node can process the expressed antigens and capture mRNA nanoparticles (Moyer et al. 2016). The activated APCs can be recruited to the injection site to process the mRNA vaccine. Additionally, the lymph vessels may directly transport small-sized lipid nanoparticles to draining lymph nodes (Moyer et al. 2016). Systemic injection, like IV injection, often leads to liver accumulation of the LNP-delivered mRNA vaccine and can generate a relatively large amount of protein compared with local injection methods (Pardi et al. 2015). Therefore, IV injection is often used to deliver mRNAs encoding antibodies when the high functional concentration of neutralizing antibodies is required in the bloodstream (Kose et al. 2019). Furthermore, IV injection of LNPs-delivered mRNA may also target the spleen by changing the formulation ratio (Kranz et al. 2016).
Overall, LNPs-based mRNA vaccines have shown efficacy in preventing infectious diseases and treating cancers in preclinical and early-stage clinical studies (Bahl et al. 2017; John et al. 2018; Kose et al. 2019; Liang et al. 2017; Pardi et al. 2017a, b; Thran et al. 2017). In a recent phase 1 clinical trial, the safety, tolerability, and immunogenicity of LNPs-based Zika mRNA vaccine were evaluated after IM injection (NCT04064905). Another phase 2 clinical trial aimed at testing the efficacy of a personalized mRNA cancer vaccine through IM injection started in July 2019 (NCT03897881). With further improvement, LNPs may facilitate the development of more effective mRNA vaccines.
3.1.2 Polymer-based Delivery
Polymeric materials, including polyamines, dendrimers, and copolymers, are functional materials capable of delivering mRNA vaccines. Similar to functional lipid-based carriers, polymers can also protect RNA from RNase-mediated degradation and facilitate intracellular delivery (Kowalski et al. 2019). However, the formulation of polymer-based mRNA nanoparticles tends to have high polydispersity (Kowalski et al. 2019). To stabilize the formulation and improve the safety profile, structural modification of polymer materials, such as incorporating lipid chains, hyperbranched groups, and biodegradable subunits, has been explored (Dong et al. 2016; Kaczmarek et al. 2016; Patel et al. 2019a).
Cationic polymers, such as polyethylenimine (PEI), polyamidoamine (PAMAM) dendrimer, and polysaccharide, condensed and delivered negatively charged RNA molecules (McCullough et al. 2014; Chahal et al. 2016; Vogel et al. 2018; Blakney et al. 2020; Son et al. 2020). PEI was one widely used polymeric material for mRNA vaccine delivery. PEI formulations were often prepared by direct mixing PEI solution with RNA solution. For example, a PEI formulation delivered an mRNA encoding HIV gp120 and triggered specific antibodies against HIV infections after intranasal vaccination in mice (Li et al. 2017b). Later on, a PEI formulation of self-amplifying mRNA encoding the hemagglutinin antigens from several influenza virus strains stimulated high antibody titer after IM immunization in mice and protected mice against virus challenge (Vogel et al. 2018). More recently, a PEI-based formulation with mRNAs encoding HIV-1 Gag and Pol proteins induced specific CD8+ and CD4+ T-cell responses against HIV infections upon IM vaccination in mice (Moyo et al. 2019). Even though PEI formulation showed in vivo efficacy, the potential toxicity may impede its development (Kowalski et al. 2019). PAMAM dendrimer is another cationic polymer material. Antigen-encoding self-amplifying mRNAs formulated by PAMAM dendrimer protected mice from lethal challenge of Ebola, H1N1 influenza, Toxoplasma gondii, respectively, after IM administration (Chahal et al. 2016). IM vaccination of a similar dendrimer formulation with self-amplifying mRNAs encoding premembrane (prM) and envelope (E) proteins of Zika virus elicited specific IgG and CD8+ T-cell responses in mice (Chahal et al. 2017). Of note, the microfluidic mixing method was used by the above two studies to formulate the mRNA vaccines. Another report used chitosan, a polysaccharide material, to condense self-amplifying mRNAs encoding influenza virus hemagglutinin and nucleoprotein (McCullough et al. 2014). After SC injection, the expressed antigen was detected in DCs (McCullough et al. 2014). Moreover, a recent study reported that a cationic copolymer material co-delivering one mRNA encoding OVA and a CpG ssDNA adjuvant eliminated OVA-expressing lymphoma tumor from mice after either SC or IV administration (Haabeth et al. 2018).
Besides cationic polymer materials, anionic polymers, such as PLGA, were also used to deliver mRNA vaccines. Since an anionic polymer was not able to efficiently encapsulate the negatively charged mRNA molecules, cationic lipid materials were added to create lipid–polymer hybrid formulations (Yang et al. 2011; Islam et al. 2018; Kong et al. 2019). For example, a cationic lipid, N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryloxy-carbonyl aminoethyl) ammonium bromide (BHEM-Chol), was mixed with a block copolymer poly(ethylene glycol)-block-poly(lactic-co-glycolic acid) (PEG-b-PLGA) and PLGA to form a lipid–polymer hybrid emulsion for mRNA delivery (Fan et al. 2018). This formulation delivered OVA-mRNA and delayed OVA-expressing lymphoma growth in mice after IV injection (Fan et al. 2018).
In general, the mRNA vaccines delivered by polymer materials showed therapeutic effects in preclinical studies. New functional polymers, with improved biodegradability and delivery efficiency, are needed for clinical translation of the polymer-based mRNA vaccines.
3.1.3 Peptide-based Delivery
Various peptides are used as carriers to deliver mRNA vaccines. Peptides themselves are also a large class of vaccine agents, which have been reviewed in the literature (Li et al. 2014; Hos et al. 2018; Reche et al. 2018).
Peptides, when used as the primary carrier for RNA delivery, should be positively charged. Cationic peptides contain many lysine and arginine residues that provide positively charged amino groups, therefore enabling complexing with nucleic acids through electrostatic interactions (Grau et al. 2018; Qiu et al. 2019). The ratio of positively charged amino groups on the peptide to the negatively charged phosphate groups on the RNA affects nanocomplex formation. Increasing the ratio of charged amino to phosphate groups from 1:1 to 10:1 was reported to afford smaller particle size, larger zeta potential, and higher encapsulation efficiency (Udhayakumar et al. 2017).
Protamine is a cationic peptide used in many early studies for the delivery of mRNA vaccines. In solution, protamine and mRNA spontaneously form a complex, the size of which is dependent on NaCl concentration (Sköld et al. 2015). Protamine possesses two features beneficiary for mRNA vaccines. Firstly, protamine protects mRNA. In the presence of protamine, antigen-encoding mRNA was more resistant to RNase degradation, suggesting better stability in vitro (Hoerr et al. 2000). In another study, protamine maintained the vaccine efficacy in mice by protecting the mRNA encoding rabies virus glycoprotein during harsh storage conditions: long-term in high temperature or cycles of temperature variation (Stitz et al. 2017). Secondly, the protamine-mRNA complex has adjuvant activity. The protamine-mRNA complex is immunogenic through activation of TLR7, likely owing to its structural similarity with condensed viral RNA genome (Scheel et al. 2005; Fotin-Mleczek et al. 2011). When an irrelevant β-galactosidase mRNA was complexed with protamine and injected into glioblastoma tumor, the anti-tumor effect rivalled two uncomplexed nucleic acid adjuvants (CpG ssDNA and polyI:C dsRNA). The side effect of an enlarged spleen associated with CpG ssDNA was avoided (Scheel et al. 2006). However, mRNA was translated poorly when in complex with protamine (Scheel et al. 2004, 2005), limiting the expression of the encoded antigen and reducing its potential as an independent mRNA carrier. Therefore, further development used the protamine-mRNA complex as an adjuvant in combination with another naked mRNA to express an antigen in animal models and human trials (Fotin-Mleczek et al. 2011; Kallen et al. 2013). This method will be further discussed in Sect. 3.4, “Co-delivery of mRNA vaccines” below.
Cationic cell-penetrating peptides (CPPs) can complex with RNA. Although many CPPs were used in gene therapies [reviewed by (Kang et al. 2019)], only a few CPPs delivered mRNA vaccines. RALA peptide (sequence: N-WEARLARALARALARHLARALARALRACEA-C) is an amphipathic arginine-rich CPP with positively charged arginine residues on one side and neutral leucine residues on the other side (McCarthy et al. 2014). It condensed a modified OVA-mRNA into nanocomplex, transfected dendritic cells and induced OVA-specific cytotoxic T-cell activation upon intradermal injection into mice (Udhayakumar et al. 2017). The amphipathic feature enhanced the endosomal escape of mRNA as RALA peptide selectively disrupted the endosome membrane at low pH (McCarthy et al. 2014). Another amphipathic CPP, LAH4-L1 (sequence: N- KKALLAHALHLLALLALHLAHALKKA-C) facilitated the binding of antigen-encoding mRNA to negatively charged polylactic acid nanoparticle (Coolen et al. 2019). The resulting nanoparticle induced innate and specific immune responses in primary human DC upon in vitro delivery. The mechanism study suggested mRNA complex was taken up by phagocytosis and clathrin-dependent endocytosis followed by endosomal escape (Coolen et al. 2019). In another report, a truncated 9-aa cationic CPP (N-RKKRRQRRR-C) derived from HIV Tat protein was fused to the C terminus of a tumor epitope antigen Melan-A (sequence: N-ELAGIGILTV-C) (Haenssle et al. 2010). The fusion peptide formed complexes with polyI:C dsRNA adjuvant. Transfection of immature DCs in vitro with the complex led to DC maturation and IL-12 secretion. The matured DC activated co-cultured antigen-specific lymphocytes from human donors (Haenssle et al. 2010). Furthermore, protamine-CPP fusion protein combines cationic and cell-penetrating features. A short CPP called Xentry (sequence: N-LCLRPVG-C) was fused to truncated protamine (sequence: N-RSQSRSRYYRQRQRSRRRRRRS-C) and delivered a protein-coding mRNA into several human cell lines in vitro (Bell et al. 2018).
Anionic peptides were also utilized to deliver mRNA vaccines in vitro. Anionic peptides cannot complex RNA due to their negative charges. Therefore, they were conjugated to positively charged polymers which served as scaffolds for RNA encapsulation. For example, an OVA-mRNA was first encapsulated with a random copolymer p(HPMA-DMAE-co-PDTEMA-co-AzEMAm) (pHDPA) containing azide group (Lou et al. 2019). Next, an anionic peptide, named GALA (sequence: N-WEAALAEALAEALAEHLAEALAEALEALAA-OH-C), was conjugated to the azide groups on pHDPA by click chemistry through a BCN-PEG linker. The resulted particle showed similar delivery efficiency to macrophages and DCs as lipofectamine 2000 with lower cytotoxicity. Mechanism of action studies suggested the GALA peptide facilitated the cell uptake and release of mRNA into the cytosol through binding to sialic acid groups on the DC surface (Lou et al. 2019).
In summary, protamine was the only peptide carrier evaluated in clinical trials of mRNA vaccines. In these trials, the protamine-mRNA complex and a naked mRNA were injected simultaneously via ID or IM routes (Rausch et al. 2014; Alberer et al. 2017; Sabari et al. 2019). Although well-tolerated in patients, these mRNA vaccines did not induce sufficient immune responses against the designated vaccine targets in the trials (Rausch et al. 2014; Kubler et al. 2015; Alberer et al. 2017; Sebastian et al. 2019).
3.1.4 Virus-Like Replicon Particle
Viral particles can package and deliver antigen-encoding self-amplifying mRNA into cytoplasm like a virus in a method called virus-like self-amplifying mRNA particle, i.e., virus-like replicon particle (VRP) (Lundstrom 2016). Self-amplifying mRNA then self-replicates and efficiently expresses the designated antigens. The viral structural proteins necessary for particle formation are expressed from packaging (helper) cell lines in trans to package self-amplifying mRNAs (Harvey et al. 2004; Li et al. 2017b). The viral particle and self-amplifying mRNA pair can be selected from either the same or different virus species (Dorange et al. 2004). Some VRPs are replication-competent but attenuated (Fuchs et al. 2015; Marzi et al. 2015), while other VRPs only engage in one cycle of transduction because the genetic regions encoding envelope and capsid proteins necessary for the viral infection are absent from the self-amplifying mRNAs (Lundstrom 2016). The advantage of VRPs arises from the efficient cytoplasmic delivery of RNA payload by viral vectors (Usme-Ciro et al. 2013). This ability is attributed to the fact that viruses have evolved to internalize and release their genomes into cells via many different pathways with high efficiency (Vázquez-Calvo et al. 2012). Many ssRNA viruses including alphaviruses, flaviviruses, measles viruses, and rhabdoviruses were used as VRP vaccines (Lundstrom 2016). For example, a Venezuelan equine encephalitis virus (VEEV) self-amplifying mRNA-based VRP expressing two dengue virus antigens was used to immunize non-human primates intradermally and protected them in the viral challenge (White et al. 2013). A Kunjin virus-derived VRP expressing GM-CSF was injected intratumorally, leading to complete removal of the primary tumor in more than half of the mice with colon carcinoma and OVA-expressing melanoma. Metastases to the lung were also reduced (Hoang-Le et al. 2009). Many more viral infections, bacterial infections, and various cancers were targeted using engineered VRP vaccines which were reviewed by Lundstrom (Lundstrom 2016).
However, there are two challenges for VRP-based mRNA vaccines. The first challenge is to scale up the production which is limited by the process of generating VRPs from packaging cell lines (Morrison and Plotkin 2016). Large-scale production of VRPs may require a special manufacturing process (Rauch et al. 2018). Another challenge is the antibody production against the viral vectors, which was reported in several clinical trials (Bernstein et al. 2009; Morse et al. 2010; Wecker et al. 2012; Fuchs et al. 2015). Although such anti-vector antibodies likely hindered two human trials of VRP-based anti-HIV-1 vaccines (Wecker et al. 2012; Fuchs et al. 2015), these antibodies did not prevent the development of specific immunity against the designated antigens in another two vaccine trials against cytomegalovirus (Bernstein et al. 2009) and cancer (Morse et al. 2010). Therefore, future development of VRP-based mRNA vaccines should improve efficacy and manufacture scale while minimizing the anti-vector immunity.
3.1.5 Cationic Nanoemulsion
Cationic nanoemulsion (CNE) combines nanoemulsion with cationic lipids for RNA delivery. Nanoemulsion utilizes hydrophobic and hydrophilic surfactants to stabilize the oil core in the aqueous phase, thereby generating particles. Nanoemulsion can be induced by various methods, such as vigorous agitation, ultrasound, and microfluidics. (Gurpreet and Singh 2018). MF59 is an FDA-approved oil-in-water nanoemulsion adjuvant used with inactivated Flu vaccine for elders (Vesikari et al. 2012). The components of MF59 include a naturally occurring oil (Squalene), sorbitan trioleate (Span 85), polyoxyethylene sorbitan monooleate (Tween 80) and citrate buffer (Podda and Del Giudice 2003; Cioncada et al. 2017). MF59 nanoemulsion enhances the efficacy of vaccines through MyD88-mediated release of cytokines/chemokines and recruitment of immune cells, without triggering TLRs (Seubert et al. 2011; O’Hagan et al. 2012; Calabro et al. 2013). Incorporation of cationic lipids, e.g., DOTAP, in the squalene-based formulation creates positively charged CNE particles that can absorb negatively charged nucleic acids to the outer shell (Ott et al. 2002; Brito et al. 2015). Such surface interaction still protected mRNA from RNase degradation (Brito et al. 2014). Squalene-based CNEs and MF59 are similar in structure and formulation and displayed equivalent recruitments of immune cells (Brito et al. 2014). CNEs delivered self-amplifying mRNA vaccines against several viral and bacterial infections (Brito et al. 2014; Davis et al. 2014; Brazzoli et al. 2016; Maruggi et al. 2017; Samsa et al. 2019). For example, MF59-based CNE delivered three chimeric self-amplifying mRNA vaccines derived from VEEV and Sindbis virus (SINV) (Brito et al. 2014). The three self-amplifying mRNAs expressed antigens against the respiratory syncytial virus (RSV), human cytomegalovirus (hCMV), and human immunodeficiency virus (HIV), respectively. After IM injection, the vaccines induced high antigen-specific IgG titer and efficient leukocyte infiltration in mice, rabbits, and rhesus macaques (Brito et al. 2014). In one recent study developing a vaccine against VEEV, CNE delivered an engineered replication-defective VEEV-based self-amplifying mRNA without the capsid gene (Samsa et al. 2019). Upon IM injection into mice, the self-amplifying mRNA-CNE stimulated neutralizing IgG production and protected mice from lethal VEEV challenge (Samsa et al. 2019). Overall, CNE has shown its potential for the delivery of mRNA vaccines in preclinical studies. Its vaccine efficacy in human awaits further evaluation by clinical trials.
3.2 Naked mRNA Vaccines
The mRNA vaccines can be delivered without any additional carrier, namely in a naked format. This method dissolves mRNA into a buffer and then injects the mRNA solution directly. The feasibility of naked RNA delivery in vivo was reported in an early effort in which a naked mRNA was delivered to mice by intramuscular injection (Wolff et al. 1990). Although naked mRNA cannot diffuse across the membrane spontaneously, the mechanism(s) underlying its intracellular delivery remain debatable. Several studies proposed that naked mRNA was internalized via macropinocytosis (Diken et al. 2011; Selmi et al. 2016). Such a macropinocytosis pathway is highly active in macrophages (Redka et al. 2018) and immature dendritic cells (Kreiter et al. 2010; Diken et al. 2011; Lim and Gleeson 2011), both of which play critical roles in developing immune responses. Others speculated the cellular uptake of naked mRNA via mechanical forces. One possible force is the hydrostatic pressure formed after fast injection of a relatively large volume into small mammals. This pressure may disrupt the cell membrane and permit cytosolic delivery of nucleic acids (Stewart et al. 2018). Further study is needed to reveal the detailed mechanism(s) responsible for the delivery of naked mRNAs.
The naked mRNA vaccine has two prominent features. One feature is the ease to store and prepare. In the presence of a storage reagent, such as 10% trehalose, freeze-dried naked RNA remains stable in the refrigerator temperature (4 ℃) for up to 10 months (Jones et al. 2007). Before administration, the naked mRNA vaccine only needs to be dissolved into a buffer. No additional formulation is needed. The other feature of the naked mRNA vaccine, especially those made of unmodified nucleotides, is its intrinsic immunogenicity, which serves as a double-edged sword. On one side, the immunogenicity might benefit vaccination by providing some adjuvant activity. The exogenous RNAs could be detected by RNA sensors, such as TLRs, RIG-I, PKR, IFIT1, leading to activation of NF‑κB and type I interferon signaling pathways, and release of cytokines (Schlee and Hartmann 2016). A naked mRNA vaccine was reported to trigger some RNA sensors and induce innate responses (Edwards et al. 2017). Unmodified RNA was considered a strong stimulator of TLR3/7/8 (Kariko et al. 2005) and PKR (Anderson et al. 2010). On the other side, the activation of certain RNA sensors may inhibit mRNA translation in cell cytosol (Pardi et al. 2018). For instance, activated PKR inhibits cap-dependent translation by phosphorylating eukaryotic translation initiation factor 2A (eIF2A) (Anderson et al. 2010). Therefore, detailed characterizations are necessary for each specific naked mRNA vaccine (Pardi et al. 2018).
When developing naked mRNA vaccines, the buffer is an essential component to be chosen carefully. Ringer’s solution (Ringer 1882) and Ringer’s lactate (Hartmann and Senn 1932; Lee 1981) are two commonly used buffers for dissolving and diluting naked mRNA vaccines before injection. Both buffers contain calcium which was suggested to trigger the uptake of mRNA into human cells via a calcium-dependent route (Probst et al. 2007). Ringer’s solution was used in a clinical trial against melanoma (Sahin et al. 2017). In that trial, naked personalized mRNA vaccines were diluted in Ringer’s solution, injected into patients’ lymph nodes, and induced antigen-specific T-cell response (Sahin et al. 2017). Ringer’s lactate was used to dissolve the mRNA encoding influenza A hemagglutinin antigen (Edwards et al. 2017). This mRNA solution expressed the antigen and stimulated innate response by triggering cellular RNA sensors in mouse models and primary human peripheral blood mononuclear cells (PBMC) (Edwards et al. 2017).
Naked mRNA vaccines are more susceptible to the delivery obstacles, namely, RNase degradation and intracellular delivery (Singer and Linderman 1990; Canton 2018). However, the obstacles might be partially alleviated by local administration of naked mRNA vaccines via intramuscular (Ying et al. 1999; Fleeton et al. 2001), intradermal (Edwards et al. 2017), intranodal (Kreiter et al. 2010; Bialkowski et al. 2016; Joe et al. 2019), intratracheal (Tiwari et al. 2018) or intranasal (Lorenzi et al. 2010) routes to minimize the contact of mRNA with RNases in the bloodstream. Direct exposure of immune cells with a higher dose of naked mRNA enhanced expression (Diken et al. 2011; Lorenz et al. 2011; Selmi et al. 2016).
In recent clinical trials, naked mRNA vaccines were administered via ultrasound-guided intranodal injection (Sahin et al. 2017; Leal et al. 2018). Repeated IN injection of naked mRNAs was well-tolerated and induced a various degree of specific immune responses against tumor or HIV-1 (Sahin et al. 2017; Leal et al. 2018).
3.3 Dendritic Cells-Based mRNA Vaccines
Therapeutic vaccination needs to effectively elicit the body’s adaptive immunity. During the initial development of adaptive immune response, antigen-presenting cells (APCs) internalize, process and present antigens to functional lymphocytes. As the most efficient APCs, dendritic cells (DCs) can present antigens processed from various sources, for example, the captured microorganisms, virus-infected cells, and tumor cells (Wculek et al. 2019). Several special characteristics make DCs suitable vaccination targets, including their T-cell-oriented migration in the lymph nodes and high expression of major histocompatibility complex (MHC) molecules, co-stimulators, and cytokines (Garg et al. 2017). In addition, DCs can be loaded with various forms of antigens and stimulatory signals and are highly amenable to such modifications (Pardi et al. 2018). An early study revealed that the inoculation of antigen-pulsed DCs primed T-cell-dependent immune response (Inaba et al. 1990). A few years later, a DC-based mRNA vaccine was reported (Boczkowski et al. 1996). In that report, DCs were pulsed with the mRNAs expressing chicken ovalbumin (OVA). The tumor-bearing mice were then vaccinated with such mRNA-pulsed DCs and were protected against the subsequent challenge of OVA-expressing tumor cells (Boczkowski et al. 1996). From then on, preclinical and clinical studies began testing DC-based mRNA vaccines against infectious diseases and cancers.
Autologous DCs from primary human PBMC are the main sources for preparing mRNA-treated DCs for in vivo applications (Benteyn et al. 2015). For further stimulation and maturation, the DCs were transfected with mRNAs encoding specific antigens and maturation signals (Benteyn et al. 2015). Next, the mRNA-transfected DCs were validated with their phenotypes and functions, then re-introduced back to the patients to function as antigen-specific APCs (Benteyn et al. 2015).
To deliver mRNAs into DCs, several strategies, such as electroporation and lipid-derived carriers, were employed (Boczkowski et al. 1996; Van Tendeloo et al. 2001; De Temmerman et al. 2011). Electroporation is the most frequently used method for generating DC-based mRNA vaccines due to its high mRNA delivery efficiency (Van Tendeloo et al. 2001). Several DC-based mRNA vaccines prepared by electroporation were evaluated in clinical trials (Wilgenhof et al. 2013; Mitchell et al. 2015; Batich et al. 2017). Electroporation disrupted the cell membrane by an electric shock to enable intracellular nucleic acid delivery (Stewart et al. 2016). Important electrical characteristics, such as voltage, capacitance, and resistance, were adjusted to improve the delivery efficiency (Van Tendeloo et al. 2001; Derdelinckx et al. 2016). Other parameters, including electroporation solution, pulse time, cell number, density, and RNA quantity should also be optimized. Under the optimized condition, the mRNA-loaded DCs should maintain their biological properties, including cell phenotypes, maturation status, cytokine secreting ability, and antigen presentation function (Tuyaerts et al. 2002; Tateshita et al. 2019). Besides electroporation, lipid-derived carriers were also tested to deliver mRNA into DCs for the preparation of DC-based mRNA vaccines (De Temmerman et al. 2011; Tateshita et al. 2019). In one study, ionizable lipid-based LNPs were used to deliver tumor antigen OVA-mRNA into DCs. The resulting ex vivo DC-based mRNA vaccine showed prophylactic anticancer efficacy and inhibited the growth of OVA-expressing cancer cells in mice (Tateshita et al. 2019).
The routes for administration of mRNA-loaded DCs mainly include ID, SC, IV, and IN injections (Benteyn et al. 2015). These routes were chosen because they delivered mRNA-loaded DCs to where native DCs function in the body. Different routes of administration may exhibit different DCs distribution patterns in vivo. The distribution of mRNA-loaded DCs was compared in metastatic cancer patients after three ways of delivery: IV, ID, and SC injections (Morse et al. 1999). After IV injection, the DCs loaded with an Indium-111-labeled mRNA encoding a carcinoembryonic antigen localized to the lungs within one hour, followed by redistributing to other organs, including liver, spleen, and bone marrow. However, no DCs were found in local lymph nodes. After ID injection, a small number of DCs were detected in proximal lymph nodes in some patients. After SC injection, no radioactivity was observed in the draining lymph nodes. These results suggested IV and ID were superior to SC for administering mRNA-loaded DCs (Morse et al. 1999). Therefore, a combined IV and ID administration method was chosen in a clinical trial (Van Nuffel et al. 2012). The autologous DCs from one melanoma stage IV-M1c patient were electroporated with a mixture of three mRNAs encoding melanoma-associated antigens and three mRNAs encoding immunostimulatory proteins. After the combined IV and ID administration, the patient obtained a durable clinical response, including stable disease and partial response based on the response evaluation criteria in solid tumors (RECIST) (Eisenhauer et al. 2009). The tumor antigen-specific CD8+ and CD4+ T-cell response were also detected in peripheral blood and skin biopsies (Van Nuffel et al. 2012).
In summary, the DC-based mRNA vaccines have shown efficacy in many preclinical and clinical studies. In one recent clinical trial (NCT00639639), the long-term progression-free survival (PFS) and overall survival (OS) were significantly increased in glioblastoma patients who were injected intradermally with autologous DCs pulsed with an antigen-encoding mRNA(Batich et al. 2017).
Taken together, the formulation and delivery of mRNA vaccines have been extensively studied. The delivery formats and delivery materials described above have advanced to various stages of preclinical and clinical studies. However, each delivery technology has its advantages and challenges which are summarized in Table 3. Their readiness for human use is also listed.
3.4 Co-delivery of mRNA Vaccines
Several mRNA molecules can be co-delivered to trigger synergic effects in vaccination. Co-delivery of mRNA vaccines enables either assembly of protein complexes, generation of multivalent mRNA vaccines, or better immune response against one specific target. The co-delivered mRNAs can be a combination of conventional mRNAs and/or self-amplifying mRNAs. There are many co-delivery options. Several mRNAs can be delivered naked or formulated, complexed together or individually, and injected through different routes at different times. In this section, we summarize the recent results for the co-delivery of mRNA vaccines, including delivery formats, dose ratios, formulation methods, and injection routes of the components. Table 4 lists representative examples for different applications of co-delivered mRNA vaccines.
3.4.1 Co-delivery of mRNAs to Assemble Protein Complexes
Antibodies, such as immunoglobulin G (IgG), and some antigens are assembled from more than one single-chain protein subunits. Co-delivery of mRNAs is an option to express these multi-subunit proteins to provide passive immunity or stimulate adaptive immune responses. All subunits need to be translated into one cell and assembled into a complex in the endoplasmic reticulum (ER), followed by translocation to their destinations (Ellgaard et al. 2016). Several methods were used to co-deliver multiple nucleic acids into the same cell. A cationic copolymer co-formulated an mCherry mRNA with FITC-CpG ssDNA and delivered them to the same cells in vitro, according to the flow cytometry results (Haabeth et al. 2018). Electroporation delivered into K562 cells two mRNAs each of which encoding half of an engineered IgG against a tumor-associated antigen, the tight-junction proteins claudin 6 (Stadler et al. 2017). The secreted whole IgG complex was detected in the supernatant by immunoblotting and induced better cytotoxicity against tumor cells in vitro than a single-chain bi-specific antibody expressed from one mRNA (Stadler et al. 2017). In another study, two mRNAs encoding heavy and light chains of one IgG, palivizumab, were administrated as an intratracheal aerosol to mouse lungs against the respiratory syncytial virus (RSV) (Tiwari et al. 2018). The two mRNAs showed similar in vivo distribution on tissue and single-cell levels. All three delivery formats tested as aerosol, naked mRNA or two PEI-derived formulations (Viromer RED and in vivo-jetPEI), reduced RSV infection after viral challenge (Tiwari et al. 2018). To develop an anti-hCMV vaccine, six conventional mRNAs encoding five subunits of the hCMV pentameric complex (PC) and one glycoprotein, respectively, were co-delivered by LNP in equal mass (John et al. 2018). Such a delivery enabled PC expression in vitro and induced specific anti-PC antibody production in mice and monkeys after IM injection. Notably, the five subunits of hCMV PC need to be expressed and assembled into a complex by the same cell to be immunogenic (Macagno et al. 2010; Gerna et al. 2017). Manufacturing, transporting, and storing a purified PC protein vaccine while maintaining its stability is challenging (Nelson et al. 2018). Co-delivery of mRNAs circumvents such a challenge by enabling the PC production in the cells (John et al. 2018), potentially lowering the logistical requirement and reducing the vaccine cost. This study also indicated that LNP had the potential to deliver multiple conventional mRNAs into one cell both in vitro and in vivo (John et al. 2018).
3.4.2 Co-delivery of mRNAs Encoding Multiple Antigens
Two or more independent antigen-coding mRNAs can be co-delivered to enhance and broaden immune responses. To enhance immunity against one target, six VEEV self-amplifying mRNAs each encoding one antigen from the same parasite, Toxoplasma gondii, were co-formulated in an equal molar ratio by a PEI-based monodispersed ionizable dendrimer nanoparticle. IM injection of the co-formulated self-amplifying mRNA vaccine protected mice from the lethal challenge (Chahal et al. 2016). In another example, Sahin and colleagues simultaneously delivered two mRNAs encoding a total of ten neo-epitopes from melanoma (Sahin et al. 2017). A variety of responses were observed in all patients, ranging from epitope-induced T-cell response, reduced metastasis to progression-free survival (Sahin et al. 2017). To broaden immunity with a multivalent mRNA vaccine, three self-amplifying mRNAs encoding hemagglutinin (HA) from three different influenza virus strains were formulated by a medium-length PEI in equal mass, co-delivered to mice intramuscularly, and protected mice against viral challenge (Vogel et al. 2018).
When co-delivering several antigen-encoding mRNAs, one challenge is to elicit potent specific immune responses to every antigen. The immunostimulatory activity of each antigen may be different. For example, two influenza virus antigens, nucleoprotein, and matrix protein 1 (M1) were expressed from two self-amplifying mRNAs (Magini et al. 2016). The two self-amplifying mRNAs were mixed in an equal amount, formulated together by LNP and delivered into mice intramuscularly. The mouse group immunized with two co-delivered self-amplifying mRNAs showed similar immune response and protection as the group received the nucleoprotein self-amplifying mRNA alone. This was explained by the low immunostimulatory effect of the M1 antigen since the group injected with the M1 self-amplifying mRNA alone showed weaker immunogenicity and protection (Magini et al. 2016). A similar observation was reported in another study in 2018 (Vogel et al. 2018). One of the three hemagglutinins encoded by the self-amplifying mRNAs failed to reduce influenza viral RNA copies and elicit adequate protection in both monovalent and trivalent formats.
Even if each mRNA-encoded antigen triggers sufficient immune response when used alone, the co-delivery of several antigens may lead to competition in epitope presentation and diminished response. For example, one group generated seven antigen-encoding mRNAs in order to develop one anti-hCMV mRNA vaccine (John et al. 2018). One mRNA encoding a mutant pp65 antigen induced a strong specific cytotoxic T-cell response when used alone. However, after co-formulating this mRNA with six additional antigen-encoding mRNAs in equal mass by LNP and simultaneous intramuscular injection, the anti-pp65 response was barely above the negative control. Their further studies suggested the inhibition to pp65-specific response was likely due to the dominant response to the epitopes of the pentameric complex encoded by other co-delivered mRNAs. This epitope competition was alleviated by sequential injection of pp65-encoding mRNA on day 1 and all seven antigen-coding mRNAs on day 21 (John et al. 2018).
3.4.3 Co-delivery of mRNAs Encoding Antigens and Immunostimulatory Proteins
While antigen-encoding mRNAs trigger the adaptive immune response, co-delivery of mRNAs encoding immunostimulatory proteins boost innate response to enhance vaccine efficacy. For example, a recent vaccine study against influenza A virus employed two self-amplifying mRNAs: one encoding the influenza A virus nucleoprotein antigen and the other encoding murine immunostimulatory GM-CSF (Manara et al. 2019). The two self-amplifying mRNAs were formulated by CNE independently and mixed before simultaneous intramuscular injection to mice. Such injection enhanced immune cell recruitment to the muscle injection site, expanded the antigen-specific CD8+ T-cell counts and resulted in better survival upon the influenza challenge than other groups receiving a single antigen-encoding self-amplifying mRNA (Manara et al. 2019).
In one approach named TriMix, three protein-coding conventional mRNAs were used as immune-stimulators to enhance the dendritic cell-mediated immune response against cancer (Van Lint et al. 2012). The three proteins encoded by the mRNAs were CD40 ligand, constitutive active TLR4 and CD70. Co-electroporation of the three mRNAs in vitro outperformed any single-mRNA or two-mRNA electroporation for increasing the numbers of helper and cytotoxic T-cells (Bonehill et al. 2008). The three mRNAs were used in an equal mass ratio in mouse tumor models (Van Lint et al. 2012; Bialkowski et al. 2016; Van Lint et al. 2016; Guardo et al. 2017). However, the total amount of the three mRNAs varied depending on specific applications and delivery routes. One mRNA encoding an antigen was commonly mixed with the three mRNAs and administered simultaneously to initiate specific immunity. The dose of the antigen-encoding mRNA was equal or several-fold larger than each of the three mRNAs encoding immunostimulatory proteins (Van Lint et al. 2012; Dewitte et al. 2014; Bialkowski et al. 2016; Van Lint et al. 2016; Guardo et al. 2017). The TriMix and antigen-coding mRNA mixture were co-delivered in vitro and ex vivo by electroporation (Bonehill et al. 2008; Van Lint et al. 2012; Guardo et al. 2017) or sonoporation (Dewitte et al. 2014), and in vivo in 0.8 Ringer’s lactate through intradermal (Van Lint et al. 2012), intranodal (Van Lint et al. 2012; Bialkowski et al. 2016; Guardo et al. 2017) or intratumoral (Jeught et al. 2014; Van Lint et al. 2016) routes. The intranodal co-administration of the TriMix and antigen-encoding mRNAs was reported to be superior to the intradermal route for boosting antigen-induced specific tumor lysis (Van Lint et al. 2012).
Another method of co-delivering mRNAs vaccines was called RNActive (Fotin-Mleczek et al. 2011). This method utilizes one antigen-encoding mRNA: 50% was naked in Ringer’s lactate to express an antigen and 50% was complexed with protamine as an adjuvant (Kallen et al. 2013). This vaccine was formulated in two steps (Fotin-Mleczek et al. 2011). First, protamine in Ringer’s lactate was added to the mRNA in a 1:2 mass ratio to form a stable protamine-mRNA complex. Second, the naked antigen-coding mRNA was mixed with the complexed mRNA in a 1:1 mass ratio. The final mass ratio of free mRNA, complexed mRNA, and protamine was 2:2:1 in Ringer’s lactate. Based on this co-delivery method, vaccines against various types of viral infection (Petsch et al. 2012; Schnee et al. 2016; Alberer et al. 2017), and cancers (Weide et al. 2009; Fotin-Mleczek et al. 2014; Sebastian et al. 2014; Kubler et al. 2015; Hong et al. 2016) were developed. Further development of this method used four-to-six mRNAs encoding different tumor antigens against non-small cell lung cancer or prostate cancer (Kubler et al. 2015; Hong et al. 2016; Papachristofilou et al. 2019; Sebastian et al. 2019). In these studies, every antigen-encoding mRNA was a mixture of its free and protamine-complexed formats. The mRNAs were formulated and injected separately through the intradermal route into patients. The cancer vaccines were well-tolerated and induced immune responses. Interestingly, the delivery approach appeared to influence the vaccine efficacy of this co-delivery method. In the clinical trial evaluating RNActive mRNA vaccine against rabies virus, the needle-free injection induced neutralizing antibody titers in some participates, while needle-syringe injection was not effective. And using the needle-free injection, the ID route performed better than the IM route (Alberer et al. 2017). Yet, multiple clinical trials for the RNActive mRNA vaccines had shown moderate efficacy, such as a weaker antibody titer than available vaccines in the clinic against rabies virus (Alberer et al. 2017; Fooks et al. 2019) and low anti-tumor activity against several types of cancer (Rausch et al. 2014; Kubler et al. 2015; Sebastian et al. 2019). Subsequently, their next generation of rabies mRNA vaccine delivered by LNP is being tested in a clinical trial (NCT03713086). Additional clinical trials against cancers combined the RNActive mRNA vaccine with other therapies, such as radiation therapy (Sebastian et al. 2014; Papachristofilou et al. 2019) or checkpoint inhibitors (NCT03164772).
Overall, the co-delivery of multiple mRNAs is a promising vaccination strategy. However, optimization is essential to determine the appropriate antigens to be expressed, delivery material, formulation method, mass ratio of components, and administration route. It is also necessary to examine whether the antigens expressed from the co-delivered mRNAs interfere with each other. If such interference is detected, modification of vaccination procedure, such as injection time, is likely needed to improve immune response.
4 Current Challenges and Future Perspectives
While many carriers are effective in delivering mRNA vaccines in preclinical studies and clinical trials, there are still challenges to be addressed. The first challenge is delivery efficiency. During the delivery process, a large portion of RNA-loaded carriers is trapped in endosome/lysosome or recycled out of cells by exocytosis (Sahay et al. 2013; Sayers et al. 2019), reducing the effective amount of RNA reaching the cytosol. Future developments that enhance endosomal escape and reduce exocytosis of nanoparticles would likely improve delivery efficiency. The second challenge is targeting specific cell types in vivo. Current delivery technologies often deliver mRNA vaccines indistinguishably into many different cell types at the injection site, many of which contribute little to immune stimulation (Veiga et al. 2018). Active in vivo targeting to specific cell types of interest, e.g., dendritic cells, macrophages, B cells, and T cells, have the potential to enhance immunization efficacy (Fenton et al. 2017). The third challenge is the safety of the delivery vehicles. Delivery materials, such as cationic lipids and polymers, may induce high delivery efficiency through enhanced membrane fusion, disruption of the endosome, or other mechanisms that might be associated with cell stresses, leading to potential cytotoxicity (Lv et al. 2006; Xue et al. 2014). Although some approaches have been explored to reduce cytotoxicity, such as using biodegradable materials (Zhang et al. 2017) and masking cationic charges (Taratula et al. 2011), delivery systems with a broad therapeutic index are still in urgent demand. The fourth challenge is the applicability to human. Effective immunization observed in preclinical animal studies may or may not be applied to human. The mRNA dose necessary to induce sufficient immune response in mice and other animals might not be directly correlated to humans. The differences in the immune systems between human and other animal species may lead to distinct immune responses (Shay et al. 2013; Zschaler et al. 2014). Therefore, clinical trials are of paramount importance for assessing the efficacy of mRNA vaccines in humans. Chapter 7 of this book reviews the clinical development of mRNA vaccines in details.
Meanwhile, the molecular mechanisms of the delivery process demand further investigation (Sahay et al. 2013; Iavarone et al. 2017; Sayers et al. 2019). Regardless of the delivery formats, carrier materials, and administration routes, our knowledge is limited regarding the factors and pathways responsible for cellular uptake, cytosolic release, endosomal escape, lysosomal degradation, and exocytotic recycling of mRNA vaccines. A more profound understanding of these biological processes will facilitate the development of delivery materials and administration strategies, leading to more effective immunization by mRNA vaccines.
5 Conclusion
mRNA has demonstrated its potential as a vaccine platform. In clinical trials, mRNA vaccines encoding antigen proteins from rabies virus, influenza virus, and cancers induced humoral and cellular responses in healthy volunteers and patients (Alberer et al. 2017; Sahin et al. 2017; Feldman et al. 2019). However, improvements are still needed to optimize the safety profile and to increase the vaccination efficacy. When delivering mRNA vaccines, a comparison of several administration routes will help determine the most appropriate injection method and promote efficacy. The progress in the development of various delivery carriers has enabled numerous preclinical studies and clinical trials. LNPs represent one of the most advanced platforms among various carriers for mRNA vaccine delivery in vivo. DC-based mRNA vaccines have been tested in many clinical trials and have shown acceptable safety profiles (Garg et al. 2017), while therapeutic efficacy needs to be further increased (Perez and De Palma 2019). Besides the improvement in delivery carriers, co-delivery of mRNAs in vaccination can enhance efficacy and/or enable expression of antigen complexes. As the delivery methods and the vaccine formulations further advance, mRNA vaccines will become an important class of medicine to effectively tackle diverse health issues, such as infectious diseases and cancers.
References
Akbarzadeh A, Rezaei-Sadabady R, Davaran S et al (2013) Liposome: classification, preparation, and applications. Nanoscale Res Lett 8:102
Alberer M, Gnad-Vogt U, Hong HS et al (2017) Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 390:1511–1520
Ambegia E, Ansell S, Cullis P et al (2005) Stabilized plasmid–lipid particles containing PEG-diacylglycerols exhibit extended circulation lifetimes and tumor selective gene expression. Biochim et Biophys Acta (BBA)—Biomembr 1669:155–163
Anderson BR, Muramatsu H, Nallagatla SR et al (2010) Incorporation of pseudouridine into mRNA enhances translation by diminishing PKR activation. Nucleic Acids Res 38:5884–5892
Awasthi S, Hook LM, Pardi N et al. (2019) Nucleoside-modified mRNA encoding HSV-2 glycoproteins C, D, and E prevents clinical and subclinical genital herpes. Sci Immunol 4:eaaw7083
Bahl K, Senn JJ, Yuzhakov O et al (2017) Preclinical and clinical demonstration of immunogenicity by mRNA vaccines against H10N8 and H7N9 influenza viruses. Mol Ther 25:1316–1327
Basak JM, Verghese PB, Yoon H et al (2012) Low-density lipoprotein receptor represents an apolipoprotein E-independent pathway of Aβ uptake and degradation by astrocytes. J Biol Chem 287:13959–13971
Batich KA, Reap EA, Archer GE et al (2017) Long-term survival in glioblastoma with cytomegalovirus pp65-targeted vaccination. Clin Cancer Res 23:1898–1909
Bell GD, Yang Y, Leung E et al (2018) mRNA transfection by a Xentry-protamine cell-penetrating peptide is enhanced by TLR antagonist E6446. PLoS ONE 13:e0201464
Belliveau NM, Huft J, Lin PJC et al (2012) Microfluidic synthesis of highly potent limit-size lipid nanoparticles for in vivo delivery of siRNA. Mol Ther—Nucleic Acids 1:e37
Benteyn D, Heirman C, Bonehill A et al (2015) mRNA-based dendritic cell vaccines. Expert Rev Vaccines 14:161–176
Bernstein DI, Reap EA, Katen K et al (2009) Randomized, double-blind, Phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine 28:484–493
Bialkowski L, van Weijnen A, Van der Jeught K et al (2016) Intralymphatic mRNA vaccine induces CD8 T-cell responses that inhibit the growth of mucosally located tumours. Sci Rep 6:22509
Billingsley MM, Singh N, Ravikumar P et al (2020) Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano Lett 20:1578–1589
Blakney AK, Abdouni Y, Yilmaz G et al (2020) Mannosylated poly(ethylene imine) copolymers enhance saRNA uptake and expression in human skin explants. Biomacromolecules
Boczkowski D, Nair SK, Snyder D et al (1996) Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J Exp Med 184:465–472
Bonehill A, Tuyaerts S, Van Nuffel AMT et al (2008) Enhancing the T-cell stimulatory capacity of human dendritic cells by co-electroporation with CD40L, CD70 and constitutively active TLR4 encoding mRNA. Mol Ther 16:1170–1180
Borrego B, Blanco E, Rodríguez Pulido M et al (2017) Combined administration of synthetic RNA and a conventional vaccine improves immune responses and protection against foot-and-mouth disease virus in swine. Antiviral Res 142:30–36
Brazzoli M, Magini D, Bonci A et al (2016) Induction of broad-based immunity and protective efficacy by self-amplifying mRNA vaccines encoding influenza virus hemagglutinin. J Virol 90:332–344
Brito LA, Chan M, Shaw CA et al (2014) A cationic nanoemulsion for the delivery of next-generation RNA vaccines. Mol Ther 22:2118–2129
Brito LA, Kommareddy S, Maione D et al (2015) Self-amplifying mRNA vaccines. In: Huang L, Liu D, Wagner E (eds) Advances in genetics. Academic Press, pp 179–233
Calabro S, Tritto E, Pezzotti A et al (2013) The adjuvant effect of MF59 is due to the oil-in-water emulsion formulation, none of the individual components induce a comparable adjuvant effect. Vaccine 31:3363–3369
Canton J (2018) Macropinocytosis: new insights into its underappreciated role in innate immune cell surveillance. Front Immunol 9:2286
Chahal JS, Fang T, Woodham AW et al (2017) An RNA nanoparticle vaccine against Zika virus elicits antibody and CD8+ T cell responses in a mouse model. Sci Rep 7:252
Chahal JS, Khan OF, Cooper CL et al (2016) Dendrimer-RNA nanoparticles generate protective immunity against lethal Ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc Natl Acad Sci 113:E4133
Chen D, Love KT, Chen Y et al (2012) Rapid discovery of potent siRNA-containing lipid nanoparticles enabled by controlled microfluidic formulation. J Am Chem Soc 134:6948–6951
Cheng X, Lee RJ (2016) The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery. Adv Drug Deliv Rev 99:129–137
Cioncada R, Maddaluno M, Vo HTM et al (2017) Vaccine adjuvant MF59 promotes the intranodal differentiation of antigen-loaded and activated monocyte-derived dendritic cells. PLoS ONE 12:e0185843
Conway A, Mendel M, Kim K et al (2019) Non-viral delivery of zinc finger nuclease mRNA enables highly efficient <em> In Vivo </em> genome editing of multiple therapeutic gene targets. Mol Ther 27:866–877
Coolen AL, Lacroix C, Mercier-Gouy P et al (2019) Poly(lactic acid) nanoparticles and cell-penetrating peptide potentiate mRNA-based vaccine expression in dendritic cells triggering their activation. Biomaterials 195:23–37
Corthésy B, Bioley G (2018) Lipid-based particles: versatile delivery systems for mucosal vaccination against infection. Front Immunol 9
Cullis PR, Hope MJ (2017) Lipid nanoparticle systems for enabling gene therapies. Mol Ther 25:1467–1475
Damiati S, Kompella UB, Damiati SA et al (2018) Microfluidic devices for drug delivery systems and drug screening. Genes 9:103
Davis D, Verschoor EJ, Koopman G et al (2014) Potent immune responses in rhesus macaques induced by nonviral delivery of a self-amplifying RNA vaccine expressing HIV type 1 envelope with a cationic nanoemulsion. J Infect Dis 211:947–955
De Temmerman M-L, Dewitte H, Vandenbroucke RE et al (2011) mRNA-Lipoplex loaded microbubble contrast agents for ultrasound-assisted transfection of dendritic cells. Biomaterials 32:9128–9135
de Vries IJ, Lesterhuis WJ, Barentsz JO et al (2005) Magnetic resonance tracking of dendritic cells in melanoma patients for monitoring of cellular therapy. Nat Biotechnol 23:1407–1413
DeFrancesco L (2017) The ‘anti-hype’ vaccine. Nat Biotechnol 35:193–197
Derdelinckx J, Berneman ZN, Cools N (2016) GMP-grade mRNA electroporation of dendritic cells for clinical use In: Synthetic mRNA. Springer, pp 139–50
Dewitte H, Van Lint S, Heirman C et al (2014) The potential of antigen and TriMix sonoporation using mRNA-loaded microbubbles for ultrasound-triggered cancer immunotherapy. J Control Release: Official J Control Release Soc 194:28–36
Diehl KH, Hull R, Morton D et al (2001) A good practice guide to the administration of substances and removal of blood, including routes and volumes. J Appl Toxico Int J 21:15–23
Diken M, Kreiter S, Selmi A et al (2011) Selective uptake of naked vaccine RNA by dendritic cells is driven by macropinocytosis and abrogated upon DC maturation. Gene Ther 18:702–708
Dimitriadis GJ (1978) Translation of rabbit globin mRNA introduced by liposomes into mouse lymphocytes. Nature 274:923–924
Dong Y, Dorkin JR, Wang W et al (2016) Poly(glycoamidoamine) brushes formulated nanomaterials for systemic siRNA and mRNA delivery in vivo. Nano Lett 16:842–848
Dorange F, Piver E, Bru T et al (2004) Vesicular stomatitis virus glycoprotein: a transducing coat for SFV-based RNA vectors. J Gene Med 6:1014–1022
Doyle G, McCuteheon J (2015) Intravenous medications by direct IV route. In: Clinical procedures for safer patient care. BCcampus, Victoria, B.C., p 414
Edwards DK, Jasny E, Yoon H et al (2017) Adjuvant effects of a sequence-engineered mRNA vaccine: translational profiling demonstrates similar human and murine innate response. J Transl Med 15:1
Eggert AA, Schreurs MW, Boerman OC et al (1999) Biodistribution and vaccine efficiency of murine dendritic cells are dependent on the route of administration. Cancer Res 59:3340–3345
Eisenhauer EA, Therasse P, Bogaerts J et al (2009) New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45:228–247
Ellgaard L, McCaul N, Chatsisvili A et al (2016) Co- and post-translational protein folding in the ER. Traffic 17:615–638
Engmann L, Shaker A, White E et al (1998) Local side effects of subcutaneous and intramuscular urinary gonadotropins for ovarian stimulation in in vitro fertilization: a prospective, randomized study 11 supported by Organon, Cambridge, United Kingdom. Fertil Steril 69:836–840
Fan Y-N, Li M, Luo Y-L et al (2018) Cationic lipid-assisted nanoparticles for delivery of mRNA cancer vaccine. Biomater Sci 6:3009–3018
Feldman RA, Fuhr R, Smolenov I et al (2019) mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 37:3326–3334
Fenton OS, Kauffman KJ, Kaczmarek JC et al (2017) Synthesis and biological evaluation of ionizable lipid materials for the in vivo delivery of messenger RNA to B lymphocytes. Adv Mater 29:1606944
Fleeton MN, Chen M, Berglund P et al (2001) Self-replicative RNA vaccines elicit protection against influenza A virus, respiratory syncytial virus, and a tickborne encephalitis virus. J Infect Dis 183:1395–1398
Fooks AR, Banyard AC, Ertl HCJ (2019) New human rabies vaccines in the pipeline. Vaccine 37:A140–A145
Fotin-Mleczek M, Duchardt KM, Lorenz C et al (2011) Messenger RNA-based vaccines with dual activity induce balanced TLR-7 dependent adaptive immune responses and provide antitumor activity. J Immunother 34:1–15
Fotin-Mleczek M, Zanzinger K, Heidenreich R et al (2014) mRNA-based vaccines synergize with radiation therapy to eradicate established tumors. Radiat Oncol (London, England) 9:180
Fuchs JD, Frank I, Elizaga ML et al (20150 First-in-human evaluation of the safety and immunogenicity of a recombinant vesicular stomatitis virus human immunodeficiency virus-1 gag vaccine (HVTN 090). Open Forum Infect Dis 2:ofv082
Ganesan P, Narayanasamy D (2017) Lipid nanoparticles: different preparation techniques, characterization, hurdles, and strategies for the production of solid lipid nanoparticles and nanostructured lipid carriers for oral drug delivery. Sustain Chem Pharm 6:37–56
Garg AD, Coulie PG, Van den Eynde BJ et al (2017) Integrating next-generation dendritic cell vaccines into the current cancer immunotherapy landscape. Trends Immunol 38:577–593
Geall AJ, Verma A, Otten GR et al (2012) Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci 109:14604–14609
Gerna G, Revello MG, Baldanti F et al (2017) The pentameric complex of human cytomegalovirus: cell tropism, virus dissemination, immune response and vaccine development. J Gen Virol 98:2215–2234
Gradel AKJ, Porsgaard T, Lykkesfeldt J et al (2018) Factors affecting the absorption of subcutaneously administered insulin: effect on variability. J Diabetes Res 2018:1205121–21
Grau M, Walker PR, Derouazi M (2018) Mechanistic insights into the efficacy of cell penetrating peptide-based cancer vaccines. Cell Mol Life Sci CMLS 75:2887–2896
Grunwitz C, Kranz LM (2017) mRNA cancer vaccines-messages that prevail. Curr Top Microbiol Immunol 405:145–164
Guardo AC, Joe PT, Miralles L et al (2017) Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix). AIDS 31:321–332
Guevara ML, Jilesen Z, Stojdl D et al (2019) Codelivery of mRNA with α-galactosylceramide using a new lipopolyplex formulation induces a strong antitumor response upon intravenous administration. ACS Omega 4:13015–13026
Gupta SK, Haigh BJ, Griffin FJ et al (2013) The mammalian secreted RNases: mechanisms of action in host defence. Innate immunity 19:86–97
Gurpreet K, Singh S (2018) Review of nanoemulsion formulation and characterization techniques. Indian J Pharm Sci 80:781–789
Haabeth OAW, Blake TR, McKinlay CJ et al (2018) mRNA vaccination with charge-altering releasable transporters elicits human T cell responses and cures established tumors in mice. Proc Natl Acad Sci 115:E9153–E9161
Haenssle HA, Riedl P, Buhl T et al (2010) Intracellular delivery of major histocompatibility complex class I-binding epitopes: dendritic cells loaded and matured with cationic peptide/poly(I:C) complexes efficiently activate T cells. Exp Dermatol 19:19–28
Hajj KA, Whitehead KA (2017) Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat Rev Mater 2:17056
Hartmann AF, Senn MJ (1932) Studies in the metabolism of sodium r-lactate. I. response of normal human subjects to the intravenous injection of sodium r-lactate. J Clin Investig 11:327–335
Harvey TJ, Liu WJ, Wang XJ et al (2004) Tetracycline-inducible packaging cell line for production of flavivirus replicon particles. J Virol 78:531–538
Hassett KJ, Benenato KE, Jacquinet E et al (2019) Optimization of lipid nanoparticles for intramuscular administration of mRNA vaccines. Mol Ther Nucleic Acids 15:1–11
Heyes J, Palmer L, Bremner K et al (2005) Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J Control Release: Official J Control Release Soc 107:276–287
Hoang-Le D, Smeenk L, Anraku I et al (2009) A Kunjin replicon vector encoding granulocyte macrophage colony-stimulating factor for intra-tumoral gene therapy. Gene Ther 16:190–199
Hoerr I, Obst R, Rammensee H-G et al (2000) In vivo application of RNA leads to induction of specific cytotoxic T lymphocytes and antibodies. Eur J Immunol 30:1–7
Hong HS, Koch SD, Scheel B et al (2016) Distinct transcriptional changes in non-small cell lung cancer patients associated with multi-antigenic RNActive® CV9201 immunotherapy. Oncoimmunology 5:e1249560–e60
Hos BJ, Tondini E, van Kasteren SI et al (2018) Approaches to improve chemically defined synthetic peptide vaccines. Front Immunol 9:884–84
Iavarone C, O’Hagan DT, Yu D et al (2017) Mechanism of action of mRNA-based vaccines. Expert Rev Vaccines 16:871–881
Ibrahim MM (2010) Subcutaneous and visceral adipose tissue: structural and functional differences. Obes Rev 11:11–18
Ickenstein LM, Garidel P (2019) Lipid-based nanoparticle formulations for small molecules and RNA drugs. Expert Opin Drug Deliv 16:1205–1226
Inaba K, Metlay JP, Crowley MT et al (1990) Dendritic cells pulsed with protein antigens in vitro can prime antigen-specific, MHC-restricted T cells in situ. J Exp Med 172:631–640
Islam MA, Xu Y, Tao W et al (2018) Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat Biomed Eng 2:850–864
Jahn A, Reiner JE, Vreeland WN et al (2008) Preparation of nanoparticles by continuous-flow microfluidics. J Nanopart Res 10:925–934
Jayaraman M, Ansell SM, Mui BL et al (2012) Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew Chem Int Ed 51:8529–8533
Jeught KVd, Joe PT, Bialkowski L et al (2014) Intratumoral administration of mRNA encoding a fusokine consisting of IFN-β and the ectodomain of the TGF-β receptor II potentiates antitumor immunity. Oncotarget 5
Joe PT, Christopoulou I, van Hoecke L et al (2019) Intranodal administration of mRNA encoding nucleoprotein provides cross-strain immunity against influenza in mice. J Transl Med 17:242–42
Johansen P, Kundig TM (2014) Intralymphatic immunotherapy and vaccination in mice. J Vis Exp:e51031
Johansen P, Kündig TM (2015) Parenteral vaccine administration: tried and true In: Foged C, Rades T, Perrie Y, Hook S (eds) Subunit vaccine delivery. Springer New York, New York, pp 261–86
John S, Yuzhakov O, Woods A et al (2018) Multi-antigenic human cytomegalovirus mRNA vaccines that elicit potent humoral and cell-mediated immunity. Vaccine 36:1689–1699
Jones KL, Drane D, Gowans EJ (2007) Long-term storage of DNA-free RNA for use in vaccine studies. Biotechniques 43:675–681
Kaczmarek JC, Patel AK, Kauffman KJ et al. (2016). Polymer-lipid nanoparticles for systemic delivery of mRNA to the lungs. Angewandte Chemie (International ed. in English) 55:13808–13812
Kallen K-J, Heidenreich R, Schnee M et al (2013) A novel, disruptive vaccination technology: self-adjuvanted RNActive(®) vaccines. Human vaccines & immunotherapeutics 9:2263–2276
Kang Z, Meng Q, Liu K (2019) Peptide-based gene delivery vectors. Journal of Materials Chemistry B 7:1824–1841
Kariko K, Buckstein M, Ni H et al (2005) Suppression of RNA recognition by Toll-like receptors: the impact of nucleoside modification and the evolutionary origin of RNA. Immunity 23:165–175
Karikó K, Muramatsu H, Ludwig J et al (2011) Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res 39:e142–e42
Kariko K, Muramatsu H, Welsh FA et al (2008) Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther 16:1833–1840
Kashem SW, Haniffa M, Kaplan DH (2017) Antigen-presenting cells in the skin. Annu Rev Immunol 35:469–499
Kong N, Tao W, Ling X et al (2019) Synthetic mRNA nanoparticle-mediated restoration of p53 tumor suppressor sensitizes <em> p53 </em>-deficient cancers to mTOR inhibition. Sci Transl Med 11: eaaw1565
Kose N, Fox JM, Sapparapu G et al (2019) A lipid-encapsulated mRNA encoding a potently neutralizing human monoclonal antibody protects against chikungunya infection. Sci Immunol 4:eaaw6647
Kowalski PS, Rudra A, Miao L et al (2019) Delivering the messenger: advances in technologies for therapeutic mRNA delivery. Mol Ther
Kranz LM, Diken M, Haas H et al (2016) Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534:396
Kreiter S, Selmi A, Diken M et al (2010) Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res 70:9031–9040
Kubler H, Scheel B, Gnad-Vogt U et al (2015) Self-adjuvanted mRNA vaccination in advanced prostate cancer patients: a first-in-man phase I/IIa study. J Immunother Cancer 3:26
Leal L, Guardo AC, Morón-López S et al (2018) Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS 32:2533–2545
Lee JA (1981) Sydney Ringer (1834-1910) and Alexis Hartmann (1898-1964). Anaesthesia 36:1115–1121
Li B, Zhang X, Dong Y (2019) Nanoscale platforms for messenger RNA delivery. Wiley Interdisc Rev Nanomed Nanobiotechnol 11:e1530
Li W, Joshi MD, Singhania S et al (2014) Peptide vaccine: progress and challenges. Vaccines 2:515–536
Li M, Li Y, Peng K et al (2017a) Engineering intranasal mRNA vaccines to enhance lymph node trafficking and immune responses. Acta Biomater 64:237–248
Li W, Ma L, Guo L-P et al (2017b) West Nile virus infectious replicon particles generated using a packaging-restricted cell line is a safe reporter system. Sci Rep 7:3286
Liang F, Lindgren G, Lin A et al (2017) Efficient targeting and activation of antigen-presenting cells in vivo after modified mRNA vaccine administration in rhesus macaques. Mol Ther 25:2635–2647
Lim JP, Gleeson PA (2011) Macropinocytosis: an endocytic pathway for internalising large gulps. Immunol Cell Biol 89:836–843
Lindsay KE, Bhosle SM, Zurla C et al (2019) Visualization of early events in mRNA vaccine delivery in non-human primates via PET–CT and near-infrared imaging. Nat Biomed Eng 3:371–380
Lindsay KE, Vanover D, Thoresen M et al (2020) Aerosol delivery of synthetic mRNA to vaginal mucosa leads to durable expression of broadly neutralizing antibodies against HIV. Mol Ther 28:805–819
Liu C, Feng Q, Sun J (2018) Lipid nanovesicles by microfluidics: manipulation, synthesis, and drug delivery. Adv Mater 0:1804788
Lobaina Mato Y (2019) Nasal route for vaccine and drug delivery: features and current opportunities. Int J Pharm 572:118813
Lorenz C, Fotin-Mleczek M, Roth G et al (2011) Protein expression from exogenous mRNA: uptake by receptor-mediated endocytosis and trafficking via the lysosomal pathway. RNA Biol 8:627–636
Lorenzi JC, Trombone AP, Rocha CD et al (2010) Intranasal vaccination with messenger RNA as a new approach in gene therapy: use against tuberculosis. BMC Biotechnol 10:77
Lou B, De Koker S, Lau CYJ et al (2019) mRNA polyplexes with post-conjugated GALA peptides efficiently target, transfect, and activate antigen presenting cells. Bioconjug Chem 30:461–475
Lundstrom K (2016) Replicon RNA viral vectors as vaccines. Vaccines 4:39
Lurie N, Saville M, Hatchett R et al (2020) Developing Covid-19 vaccines at pandemic speed. New Engl J Med
Lv H, Zhang S, Wang B et al (2006) Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release Official J Control Release Soc 114:100–109
Macagno A, Bernasconi NL, Vanzetta F et al (2010) Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J Virol 84:1005
Magini D, Giovani C, Mangiavacchi S et al (2016) Self-amplifying mRNA vaccines expressing multiple conserved influenza antigens confer protection against homologous and heterosubtypic viral challenge. PLoS ONE 11:e0161193
Malone RW, Felgner PL, Verma IM (1989) Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci U S A 86:6077–6081
Manara C, Brazzoli M, Piccioli D et al (2019) Co-administration of GM-CSF expressing RNA is a powerful tool to enhance potency of SAM-based vaccines. Vaccine 37:4204–4213
Martinon F, Krishnan S, Lenzen G et al (1993) Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur J Immunol 23:1719–1722
Maruggi G, Chiarot E, Giovani C et al (2017) Immunogenicity and protective efficacy induced by self-amplifying mRNA vaccines encoding bacterial antigens. Vaccine 35:361–368
Maruggi G, Shaw CA, Otten GR et al (2013) Engineered alphavirus replicon vaccines based on known attenuated viral mutants show limited effects on immunogenicity. Virology 447:254–264
Maruggi G, Zhang C, Li J et al (2019) mRNA as a transformative technology for vaccine development to control infectious diseases. Mol Ther 27:757–772
Marzi A, Robertson SJ, Haddock E et al (2015) VSV-EBOV rapidly protects macaques against infection with the 2014/15 Ebola virus outbreak strain. Science (New York, N.Y.) 349:739
McCarthy HO, McCaffrey J, McCrudden CM et al (2014) Development and characterization of self-assembling nanoparticles using a bio-inspired amphipathic peptide for gene delivery. J Control Release: Official J Control Release Soc 189:141–149
McCullough KC, Bassi I, Milona P et al (2014) Self-replicating replicon-RNA delivery to dendritic cells by chitosan-nanoparticles for translation in vitro and in vivo. Mol Ther Nucleic Acids 3:e173
Melo M, Porter E, Zhang Y et al (2019) Immunogenicity of RNA replicons encoding HIV Env immunogens designed for self-assembly into nanoparticles. Mol Ther 27:2080–2090
Miao L, Li L, Huang Y et al (2019) Delivery of mRNA vaccines with heterocyclic lipids increases anti-tumor efficacy by STING-mediated immune cell activation. Nat Biotechnol 37:1174–1185
Midoux P, Pichon C (2015) Lipid-based mRNA vaccine delivery systems. Expert Rev Vaccines 14:221–234
Mitchell DA, Batich KA, Gunn MD et al (2015) Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature 519:366
Morrison J, Plotkin S (2016) Chapter 19—viral vaccines: fighting viruses with vaccines. In: Katze MG, Korth MJ, Law GL, Nathanson N (eds) Viral pathogenesis, 3rd edn. Academic Press, Boston, pp 253–269
Morse MA, Coleman RE, Akabani G et al (1999) Migration of human dendritic cells after injection in patients with metastatic malignancies. Can Res 59:56–58
Morse MA, Hobeika AC, Osada T et al (2010) An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J Clin Invest 120:3234–3241
Moyer TJ, Zmolek AC, Irvine DJ (2016) Beyond antigens and adjuvants: formulating future vaccines. J Clin Investig 126:799–808
Moyo N, Vogel AB, Buus S et al (2019) Efficient induction of T cells against conserved HIV-1 regions by mosaic vaccines delivered as self-amplifying mRNA. Mol Ther Methods Clin Dev 12:32–46
Nelson CS, Herold BC, Permar SR (2018) A new era in cytomegalovirus vaccinology: considerations for rational design of next-generation vaccines to prevent congenital cytomegalovirus infection. NPJ Vaccines 3:38
O’Hagan DT, Ott GS, De Gregorio E et al (2012) The mechanism of action of MF59—an innately attractive adjuvant formulation. Vaccine 30:4341–4348
Oberli MA, Reichmuth AM, Dorkin JR et al (2016) Lipid nanoparticle assisted mRNA delivery for potent cancer immunotherapy. Nano Lett 17:1326–1335
Ott G, Singh M, Kazzaz J et al (2002) A cationic sub-micron emulsion (MF59/DOTAP) is an effective delivery system for DNA vaccines. J Control Release 79:1–5
Papachristofilou A, Hipp MM, Klinkhardt U et al (2019) Phase Ib evaluation of a self-adjuvanted protamine formulated mRNA-based active cancer immunotherapy, BI1361849 (CV9202), combined with local radiation treatment in patients with stage IV non-small cell lung cancer. J Immunother Cancer 7:38
Pardi N, Hogan MJ, Pelc RS et al (2017a) Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543:248
Pardi N, Secreto AJ, Shan X et al (2017b) Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat Commun 8:14630
Pardi N, Hogan MJ, Porter FW et al (2018) mRNA vaccines—a new era in vaccinology. Nat Rev Drug Discov 17:261
Pardi N, Hogan MJ, Weissman D (2020) Recent advances in mRNA vaccine technology. Curr Opin Immunol 65:14–20
Pardi N, Tuyishime S, Muramatsu H et al (2015) Expression kinetics of nucleoside-modified mRNA delivered in lipid nanoparticles to mice by various routes. J Control Release 217:345–351
Patel AK, Kaczmarek JC, Bose S et al (2019a) Inhaled nanoformulated mRNA polyplexes for protein production in lung epithelium. Adv Mater 31:1805116
Patel S, Athirasala A, Menezes PP et al (2019b) Messenger RNA delivery for tissue engineering and regenerative medicine applications. Tissue Eng Part A 25:91–112
Patel S, Kim J, Herrera M et al (2019c) Brief update on endocytosis of nanomedicines. Adv Drug Deliv Rev 144:90–111
Pepini T, Pulichino A-M, Carsillo T et al (2017) Induction of an IFN-mediated antiviral response by a self-amplifying RNA vaccine: implications for vaccine design. J Immunol 198:4012
Perez CR, De Palma M (2019) Engineering dendritic cell vaccines to improve cancer immunotherapy. Nat Commun 10:5408
Persano S, Guevara ML, Li Z et al (2017) Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 125:81–89
Petsch B, Schnee M, Vogel AB et al (2012) Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol 30:1210
Podda A, Del Giudice G (2003) MF59-adjuvanted vaccines: increased immunogenicity with an optimal safety profile. Expert Rev Vaccines 2:197–203
Probst J, Weide B, Scheel B et al (2007) Spontaneous cellular uptake of exogenous messenger RNA in vivo is nucleic acid-specific, saturable and ion dependent. Gene Ther 14:1175
Pun SH, Hoffman AS (2013) B.8—nucleic acid delivery. In: Ratner BD, Hoffman AS, Schoen FJ, Lemons JE (eds) Biomaterial Science, 3rd edn. Academic Press, pp 1047–1054
Qiu Y, Man RCH, Liao Q et al (2019) Effective mRNA pulmonary delivery by dry powder formulation of PEGylated synthetic KL4 peptide. J Control Release Official J Control Release Soc 314:102–115
Rauch S, Jasny E, Schmidt KE et al (2018) New vaccine technologies to combat outbreak situations. Front Immunol 9:1963–63
Rausch S, Schwentner C, Stenzl A et al (2014) mRNA vaccine CV9103 and CV9104 for the treatment of prostate cancer. Human Vaccines Immunotherapeutics 10:3146–3152
Reche P, Flower DR, Fridkis-Hareli M et al (2018) Peptide-Based Immunotherapeutics and Vaccines 2017. J Immunol Res 2018:2
Redka DS, Gütschow M, Grinstein S et al (2018) Differential ability of proinflammatory and anti-inflammatory macrophages to perform macropinocytosis. Mol Biol Cell 29:53–65
Reichmuth AM, Oberli MA, Jaklenec A et al (2016) mRNA vaccine delivery using lipid nanoparticles. Ther Deliv 7:319–334
Richner JM, Himansu S, Dowd KA et al (2017a) Modified mRNA vaccines protect against Zika virus infection. Cell 168:1114–25.e10
Richner JM, Jagger BW, Shan C et al (2017b) Vaccine mediated protection against Zika Virus-induced congenital disease. Cell 170:273–83.e12
Riley RS, June CH, Langer R et al (2019) Delivery technologies for cancer immunotherapy. Nat Rev Drug Discovery 18:175–196
Ringer S (1882) Regarding the action of hydrate of soda, hydrate of ammonia, and hydrate of potash on the ventricle of the frog’s heart. J Physiol 3(195–202):6
Rini BI, Stenzl A, Zdrojowy R et al (2016) IMA901, a multipeptide cancer vaccine, plus sunitinib versus sunitinib alone, as first-line therapy for advanced or metastatic renal cell carcinoma (IMPRINT): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol 17:1599–1611
Roth C, Cantaert T, Colas C et al (2019) A modified mRNA vaccine targeting immunodominant NS epitopes protects against dengue virus infection in HLA class I transgenic mice. Front Immunol 10:1424–24
Sabari J, Ramirez KA, Schwarzenberger P et al (2019) Abstract B209: phase 1/2 study of mRNA vaccine therapy + durvalumab (durva) ± tremelimumab (treme) in patients with metastatic non-small cell lung cancer (NSCLC). Cancer Immunol Res 7:B209
Sabnis S, Kumarasinghe ES, Salerno T et al (2018) A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol Ther 26:1509–1519
Sahay G, Alakhova DY, Kabanov AV (2010) Endocytosis of nanomedicines. J Controlled Release 145:182–195
Sahay G, Querbes W, Alabi C et al (2013) Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat Biotechnol 31:653–658
Sahin U, Derhovanessian E, Miller M et al (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 547:222
Samsa MM, Dupuy LC, Beard CW et al (2019) Self-amplifying RNA vaccines for venezuelan equine encephalitis virus induce robust protective immunogenicity in mice. Mol Ther 27:850–865
Sato Y, Hashiba K, Sasaki K et al (2019) Understanding structure-activity relationships of pH-sensitive cationic lipids facilitates the rational identification of promising lipid nanoparticles for delivering siRNAs in vivo. J Controlled Release 295:140–152
Sayers EJ, Peel SE, Schantz A et al (2019) Endocytic profiling of cancer cell models reveals critical factors influencing LNP-mediated mRNA delivery and protein expression. Mol Ther 27:1950–1962
Scheel B, Aulwurm S, Probst J et al (2006) Therapeutic anti-tumor immunity triggered by injections of immunostimulating single-stranded RNA. Eur J Immunol 36:2807–2816
Scheel B, Braedel S, Probst J et al (2004) Immunostimulating capacities of stabilized RNA molecules. Eur J Immunol 34:537–547
Scheel B, Teufel R, Probst J et al (2005) Toll-like receptor-dependent activation of several human blood cell types by protamine-condensed mRNA. Eur J Immunol 35:1557–1566
Schlee M, Hartmann G (2016) Discriminating self from non-self in nucleic acid sensing. Nat Rev Immunol 16:566
Schnee M, Vogel AB, Voss D et al (2016) An mRNA vaccine encoding rabies virus glycoprotein induces protection against lethal infection in mice and correlates of protection in adult and newborn pigs. PLOS Negl Tropical Dis 10:e0004746
Sebastian M, Papachristofilou A, Weiss C et al (2014) Phase Ib study evaluating a self-adjuvanted mRNA cancer vaccine [RNActive(R)] combined with local radiation as consolidation and maintenance treatment for patients with stage IV non-small cell lung cancer. BMC Cancer 14:748
Sebastian M, Schroder A, Scheel B et al (2019) A phase I/IIa study of the mRNA-based cancer immunotherapy CV9201 in patients with stage IIIB/IV non-small cell lung cancer. Cancer Immunol Immunother 68:799–812
Sedic M, Senn JJ, Lynn A et al (2017) Safety evaluation of lipid nanoparticle-formulated modified mRNA in the Sprague-Dawley rat and cynomolgus monkey. Vet Pathol 55:341–354
Selmi A, Vascotto F, Kautz-Neu K et al (2016) Uptake of synthetic naked RNA by skin-resident dendritic cells via macropinocytosis allows antigen expression and induction of T-cell responses in mice. Cancer Immunol Immunother 65:1075–1083
Semple SC, Akinc A, Chen J et al (2010) Rational design of cationic lipids for siRNA delivery. Nat Biotechnol 28:172–176
Senti G, Kündig TM (2015) Intralymphatic Immunotherapy. World Allergy Organ J 8:9
Seubert A, Calabro S, Santini L et al (2011) Adjuvanticity of the oil-in-water emulsion MF59 is independent of Nlrp3 inflammasome but requires the adaptor protein MyD88. Proc Natl Acad Sci 108:11169–11174
Shay T, Jojic V, Zuk O et al (2013) Conservation and divergence in the transcriptional programs of the human and mouse immune systems. Proc Natl Acad Sci 110:2946
Sienkiewicz S, Palmunen JF (2017) Clinical nursing calculations, p xix, 591 p illustrations (some color); 28 cm. Jones & Bartlett Learning, Burlington, MA
Singer DF, Linderman JJ (1990) The relationship between antigen concentration, antigen internalization, and antigenic complexes: modeling insights into antigen processing and presentation. J Cell Biol 111:55–68
Sköld AE, van Beek JJP, Sittig SP et al (2015) Protamine-stabilized RNA as an ex vivo stimulant of primary human dendritic cell subsets. Cancer Immunol Immunother: CII 64:1461–1473
Son S, Nam J, Zenkov I et al (2020) Sugar-nanocapsules imprinted with microbial molecular patterns for mRNA vaccination. Nano Lett 20:1499–1509
Stadler CR, Bähr-Mahmud H, Celik L et al (2017) Elimination of large tumors in mice by mRNA-encoded bispecific antibodies. Nat Med 23:815
Stehle C, Hernández DC, Romagnani C (2018) Innate lymphoid cells in lung infection and immunity. Immunol Rev 286:102–119
Stewart MP, Langer R, Jensen KF (2018) Intracellular delivery by membrane disruption: mechanisms, strategies, and concepts. Chem Rev 118:7409–7531
Stewart MP, Sharei A, Ding X et al (2016) In vitro and ex vivo strategies for intracellular delivery. Nature 538:183
Stitz L, Vogel A, Schnee M et al (2017) A thermostable messenger RNA based vaccine against rabies. PLOS Negl Tropical Dis 11:e0006108
Taratula O, Garbuzenko O, Savla R et al (2011) Multifunctional nanomedicine platform for cancer specific delivery of siRNA by superparamagnetic iron oxide nanoparticles-dendrimer complexes. Curr Drug Deliv 8:59–69
Tateshita N, Miura N, Tanaka H et al (2019) Development of a lipoplex-type mRNA carrier composed of an ionizable lipid with a vitamin E scaffold and the KALA peptide for use as an ex vivo dendritic cell-based cancer vaccine. J Control Release: Official J Control Release Soc 310:36–46
Thran M, Mukherjee J, Pönisch M et al (2017) mRNA mediates passive vaccination against infectious agents, toxins, and tumors. EMBO Mol Med 9:1434–47
Tiwari PM, Vanover D, Lindsay KE et al (2018) Engineered mRNA-expressed antibodies prevent respiratory syncytial virus infection. Nat Commun 9:3999
Tuyaerts S, Noppe SM, Corthals J et al (2002) Generation of large numbers of dendritic cells in a closed system using Cell Factories™. J Immunol Methods 264:135–51
Udhayakumar VK, De Beuckelaer A, McCaffrey J et al (2017) Arginine-rich peptide-based mRNA nanocomplexes efficiently instigate cytotoxic T cell immunity dependent on the amphipathic organization of the peptide. Adv Healthc Mater 6
Usme-Ciro JA, Campillo-Pedroza N, Almazán F et al (2013) Cytoplasmic RNA viruses as potential vehicles for the delivery of therapeutic small RNAs. Virol J 10:185–85
Valencia PM, Farokhzad OC, Karnik R et al (2012) Microfluidic technologies for accelerating the clinical translation of nanoparticles. Nat Nanotechnol 7:623–29
Van Lint S, Goyvaerts C, Maenhout S et al (2012) Preclinical evaluation of TriMix and antigen mRNA-based antitumor therapy. Cancer Res 72:1661–71
Van Lint S, Renmans D, Broos K et al (2016) Intratumoral delivery of TriMix mRNA results in T-cell activation by cross-presenting dendritic cells. Cancer Immunol Res 4:146–56
Van Nuffel AMT, Benteyn D, Wilgenhof S et al (2012) Intravenous and intradermal TriMix-dendritic cell therapy results in a broad T-cell response and durable tumor response in a chemorefractory stage IV-M1c melanoma patient. Cancer Immunol Immunother 61:1033–43
Van Tendeloo VFI, Ponsaerts P, Lardon F et al (2001) Highly efficient gene delivery by mRNA electroporation in human hematopoietic cells: superiority to lipofection and passive pulsing of mRNA and to electroporation of plasmid cDNA for tumor antigen loading of dendritic cells. Blood 98:49–56
Vázquez-Calvo Á, Saiz J-C, McCullough KC et al (2012) Acid-dependent viral entry. Virus Res 167:125–37
Veiga N, Goldsmith M, Granot Y et al (2018) Cell specific delivery of modified mRNA expressing therapeutic proteins to leukocytes. Nat Commun 9:4493
Verbeke R, Lentacker I, Breckpot K et al (2019a) Broadening the message: a nanovaccine co-loaded with messenger RNA and α-GalCer induces antitumor immunity through conventional and natural killer T cells. ACS Nano 13:1655–1669
Verbeke R, Lentacker I, De Smedt SC et al (2019b) Three decades of messenger RNA vaccine development. Nano Today 100766
Versteeg L, Almutairi MM, Hotez PJ et al (2019) Enlisting the mRNA vaccine platform to combat parasitic infections. Vaccines (Basel) 7
Vesikari T, Forsten A, Herbinger KH et al (2012) Safety and immunogenicity of an MF59((R))-adjuvanted A/H5N1 pre-pandemic influenza vaccine in adults and the elderly. Vaccine 30:1388–96
Vogel AB, Lambert L, Kinnear E et al (2018) Self-amplifying RNA vaccines give equivalent protection against influenza to mRNA vaccines but at much lower doses. Mol Ther 26:446–55
von Herrath MG, Bot A (2003) Immune responsiveness, tolerance and dsRNA: implications for traditional paradigms. Trends Immunol 24:289–93
Warren L, Manos PD, Ahfeldt T et al (2010) Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell 7:618–30
Wculek SK, Cueto FJ, Mujal AM et al (2019) Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol
Wecker M, Gilbert P, Russell N et al (2012) Phase I safety and immunogenicity evaluations of an alphavirus replicon HIV-1 subtype C gag vaccine in healthy HIV-1-uninfected adults. Clin Vaccine Immunol 19:1651–60
Weide B, Pascolo S, Scheel B et al (2009) Direct injection of protamine-protected mRNA: results of a phase 1/2 vaccination trial in metastatic melanoma patients. J Immunother (Hagerstown, Md.: 1997) 32:498–507
Weissman D, Pardi N, Muramatsu H et al (2013) HPLC purification of in vitro transcribed long RNA. Methods Mol Biol 969:43–54
Weng Y, Li C, Yang T et al (2020) The challenge and prospect of mRNA therapeutics landscape. Biotechnol Adv 40:107534
White LJ, Sariol CA, Mattocks MD et al (2013) An alphavirus vector-based tetravalent dengue vaccine induces a rapid and protective immune response in macaques that differs qualitatively from immunity induced by live virus infection. J Virol 87:3409–24
Wilgenhof S, Van Nuffel AMT, Benteyn D et al (2013) A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol 24:2686–93
Wolff JA, Malone RW, Williams P et al (1990) Direct gene transfer into mouse muscle in vivo. Science (New York, N.Y.) 247:1465–1468
Xiong Q, Lee GY, Ding J et al (2018) Biomedical applications of mRNA nanomedicine. Nano Res 11:5281–309
Xue HY, Liu S, Wong HL (2014) Nanotoxicity: a key obstacle to clinical translation of siRNA-based nanomedicine. Nanomed (London, England) 9:295–312
Yanez Arteta M, Kjellman T, Bartesaghi S et al (2018) Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc Nat Acad Sci 115:E3351–E60
Yang X-Z, Dou S, Sun T-M et al (2011) Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy. J Control Release 156:203–11
Ying H, Zaks TZ, Wang RF et al (1999) Cancer therapy using a self-replicating RNA vaccine. Nat Med 5:823–7
Zeng C, Hou X, Yan J et al (2020) Leveraging mRNAs sequences to express SARS-CoV-2 antigens in vivo. bioRxiv: 2020.04.01.019877
Zhang C, Maruggi G, Shan H et al (2019) Advances in mRNA Vaccines for Infectious Diseases. Front Immuno 10
Zhang X, Li B, Luo X et al (2017) Biodegradable amino-ester nanomaterials for Cas9 mRNA delivery in vitro and in vivo. ACS Appl Mater Interfaces 9:25481–87
Zhou W-Z, Hoon D, Huang S et al (1999) RNA melanoma vaccine: induction of antitumor immunity by human glycoprotein 100 mRNA immunization. Human Gene Ther 10:2719–24
Zhuang X, Qi Y, Wang M et al (2020) mRNA vaccines encoding the HA protein of influenza A H1N1 virus delivered by cationic lipid nanoparticles induce protective immune responses in mice. Vaccines (Basel) 8
Zschaler J, Schlorke D, Arnhold J (2014) Differences in innate immune response between man and mouse. Crit Revi Immunol 34:433–54
Acknowledgments
Y.D. acknowledges the Maximizing Investigators’ Research Award R35GM119679 from the National Institute of General Medical Sciences. C.Zhang acknowledges the support from the Professor Sylvan G. Frank Graduate Fellowship.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2020 Springer Nature Switzerland AG
About this chapter

Cite this chapter
Zeng, C., Zhang, C., Walker, P.G., Dong, Y. (2020). Formulation and Delivery Technologies for mRNA Vaccines. In: Yu, D., Petsch, B. (eds) mRNA Vaccines. Current Topics in Microbiology and Immunology, vol 440. Springer, Cham. https://doi.org/10.1007/82_2020_217
Download citation
DOI: https://doi.org/10.1007/82_2020_217
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-031-18069-9
Online ISBN: 978-3-031-18070-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)