





ABSTRACT
This 21‐week, open‐label, phase 2a trial aimed to evaluate the pharmacodynamics and safety of multiple, escalating infusions of BPS804, a neutralizing, anti‐sclerostin antibody, in adults with moderate osteogenesis imperfecta (OI). Patients received BPS804 (three escalating doses each separated by 2 weeks [5, 10, and 20 mg/kg]) or no treatment (reference group). The primary efficacy endpoints were mean changes from baseline to day 43 in: procollagen type 1 N‐terminal propeptide (P1NP), procollagen type 1 C‐terminal propeptide (P1CP), bone‐specific alkaline phosphatase (BSAP), osteocalcin (OC), and type 1 collagen cross‐linked C‐telopeptide (CTX‐1). Mean change from baseline to day 141 in lumbar spine areal bone mineral density (aBMD) was also assessed. BPS804 safety and tolerability were assessed every 2 weeks. Overall, 14 adults were enrolled (BPS804 group: n = 9, mean age 30.7 years, mean aBMD Z‐score –2.6; reference group, n = 5, mean age 27.4 years, mean aBMD Z‐score –2.2). In the BPS804 group, P1NP, P1CP, BSAP, and OC were increased by 84% (p < 0.001), 53% (p = 0.003), 59% (p < 0.001), and 44% (p = 0.012), respectively, versus baseline (reference: P1NP, +6% [p = 0.651]; P1CP, +5% [p = 0.600]; BSAP, –13% [p = 0.582]; OC, –19% [p = 0.436]). BPS804 treatment downregulated CTX‐1 by 44% from baseline (reference: –7%; significance was not tested for this biomarker), and increased aBMD by 4% (p = 0.038; reference group: +1%; p = 0.138). BPS804 was generally well tolerated. There were 32 adverse events reported in nine patients; none was suspected to be treatment‐related. There were no treatment‐related fractures. BPS804 stimulates bone formation, reduces bone resorption, and increases lumbar spine aBMD in adults with moderate OI. This paves the way for a longer‐term, phase 3 trial into the efficacy, safety, and tolerability of BPS804 in patients with OI. © 2017 American Society for Bone and Mineral Research.
Introduction
Osteogenesis imperfecta (OI) is a rare genetic disorder of the connective tissues, characterized by reduced bone mass and bone fragility.(1) About 85% to 90% of cases are linked to mutations in either of the two genes encoding type I collagen (COL1A1 and COL1A2).(2, 3) The remaining cases may have mutations involving an enzyme complex responsible for posttranslational modifications of type 1 collagen or other, as yet unidentified, mutations.(3) OI has a prevalence of approximately 6 in 100,000 to 7 in 100,000.(2) As a result of various collagen mutations, bone quality is reduced in patients with OI, leading to frequent, spontaneous, and low‐impact fractures, which may cause bone deformities in the spine and limbs.(1, 3) Individuals may be affected by muscle weakness, hearing loss, fatigue, joint laxity, scoliosis, early‐onset arthritis, blue sclera, dentinogenesis imperfecta, and short stature.(3) Neurological complications may also occur.(4)
Historically, the clinical classification system, by Sillence, has divided OI into types I to IV, although further distinct types have recently been described.(1, 2, 5, 6, 7) Type I (classic non‐deforming) OI is typically a mild disease, without spine or limb deformities. Type II (perinatal lethal) OI is very severe; most individuals die early from respiratory failure. Type III is a progressively deforming disease and type IV is a common variable disease with normal sclera. The mildest form of OI (type I) accounts for approximately one‐half of the OI population.(2)
At present, there are no medical treatments for OI approved by the US Food and Drug Administration or European Medicines Agency. OI is managed by a combination of off‐label oral or intravenous bisphosphonates, calcium and vitamin D supplements, physiotherapy, physical aids and orthotics, and corrective surgery.(8, 9, 10, 11, 12, 13, 14, 15, 16) Thus, there is a clear need for new therapies for patients with OI.
Sclerostin is a key negative regulator of osteoblastic bone formation.(17) It acts on the Wnt signaling pathway, and inhibits osteoblast differentiation and bone formation.(18) Low levels of sclerostin have been implicated in several rare, genetic skeletal disorders in which there is high bone mineral density (BMD) and low fracture risk, such as sclerosteosis (OMIM #269500) and van Buchem disease (OMIM #239100).(17) Anti‐sclerostin antibody therapy might therefore be useful for patients with skeletal disorders that are characterized by bone fragility, such as OI.(17) Bone remodeling has been found to be significantly increased in patients with OI compared with healthy individuals,(19) which may further suggest a potential role for anti‐sclerostin antibody therapy in the management of the disease. Such therapy has been shown to stimulate osteoblast bone formation and improve trabecular and cortical bone mass in a knock‐in mouse model for moderately severe OI (Brtl/+),(20, 21, 22) as well as to reduce long‐bone fragility and increase femoral stiffness and strength.(20, 21) In a murine model of type III OI (oim/oim), anti‐sclerostin antibody therapy has been shown to reduce the number of fractures and improve tibia cortical thickness, ultimate load, and stiffness.(23, 24) Treatment was also associated with significant increases in areal BMD (aBMD).(24) In a severe disease model of OI, the response was less clear.(25)
BPS804 is a neutralizing, anti‐sclerostin, fully human IgG2λ monoclonal antibody.(17, 26) By binding to sclerostin, BPS804 alleviates the inhibitory effect of sclerostin on bone formation, leading to the production of new bone. BPS804 received Orphan Drug designation for OI in the United States in March 2016 and in the European Union in June 2016.(26, 27)
The primary objectives of the present randomized, open‐label, phase 2a trial were to evaluate the pharmacodynamics and safety of multiple, escalating infusions of BPS804 in adults with moderate OI (OI types I, III, or IV with a history of at least two fractures). The study included an untreated reference group to monitor the natural progression of disease with respect to bone biomarkers and aBMD.
Patients and Methods
Trial design
This was a 21‐week, open‐label, intrapatient, dose‐escalation, phase 2a study, with a planned population of 12 to 15 individuals. Patients were randomly assigned in a 2:1 ratio to receive BPS804 treatment or no treatment (reference group). Randomization was performed with a validated interactive voice response system. The study design is shown in Fig. 1. After assessments on day 1, patients in the treatment group were given three escalating doses of BPS804, each separated by a 2‐week period (day 1: 5 mg/kg; day 15: 10 mg/kg; day 29: 20 mg/kg). Every 2 weeks post‐dose, safety was assessed before proceeding to the next dose level. The risks of adverse events (AEs) were further minimized by staggering the dose escalation so that no more than one patient was treated per day. After the treatment period, patients were followed for 14 weeks and a study completion evaluation was carried out. Individuals who were randomized to the untreated reference group at screening subsequently visited the trial site only in week 7 and at the end of the trial; during these visits, pharmacodynamic (PD) and safety assessments were completed.
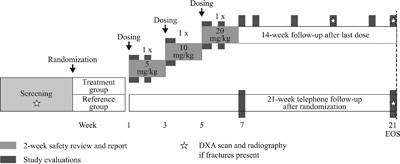
Study design. Screening took place for a maximum of 21 days prior to randomization. DXA = dual‐energy X‐ray absorptiometry; EOS = end of study.
Protocol modifications were made before patient recruitment and are listed in the Supporting Methods.
Trial objectives
The primary efficacy endpoints (PD assessments) were mean changes from baseline to day 43 in: the serum bone formation biomarkers procollagen type 1 N‐terminal propeptide (P1NP), procollagen type 1 C‐terminal propeptide (P1CP), bone‐specific alkaline phosphatase (BSAP), and osteocalcin (OC); and the serum bone resorption biomarker type 1 collagen cross‐linked C‐telopeptide (CTX‐1). Mean change from baseline to day 141 in lumbar spine aBMD was also assessed as a primary efficacy endpoint. The safety and tolerability of BPS804, administered as multiple, dose‐escalating, intravenous infusions, were also assessed as primary endpoints.
Ethics approval
The study protocol was approved by the appropriate independent ethics committee or institutional review board for each center, and complied with the ethical principles of the Declaration of Helsinki and good clinical practice. All patients provided written informed consent.
Participants
The trial was completed at six centers in Canada, Germany, Belgium, and the United States. The main study inclusion criteria were men and women aged 18 to 75 years with a diagnosis of moderate OI (defined as patients who have OI types I, III, or IV with a history of at least two fractures), confirmed by genetic testing; a lumbar spine aBMD Z‐score of –1.0 or lower and higher than –4.0, as determined by dual‐energy X‐ray absorptiometry (DXA) at screening; normal seasonal serum 25‐hydroxy vitamin D levels (≥25 nmol/L); and normal serum calcium levels (8.5–10.2 mg/dL [2.16–2.56 mmol/L]).
Key exclusion criteria were as follows: a history of skeletal malignancies or bone metastases, external beam radiation to the skeleton, or hypersensitivity to monoclonal antibodies; a history of or current diseases such as hypoparathyroidism/hyperparathyroidism, hypothyroidism/hyperthyroidism, Paget's disease, conditions that required previous neck surgery involving partial or complete thyroidectomy, or other endocrine disorders or conditions.
Additional exclusion criteria were as follows: bone fractures in the previous 2 weeks; suspected neural foraminal stenosis; treatment with any antiresorptive medication, bisphosphonates, and/or teriparatide in the 6 months before enrollment; exposure to blood products or monoclonal antibodies in the 12 months before initial dosing; any deformation of the spine that would preclude proper assessment of lumbar spine aBMD by DXA (assessed by clinical evaluation); presence of impaired renal function as indicated by clinically significant abnormal creatinine or blood urea nitrogen (BUN) and/or urea values, or abnormal urinary constituents (eg, albuminuria) at screening, and again at visit 2 in patients randomized to the treatment group; women who were pregnant or lactating; and women who were planning a pregnancy during the course of the study.
Decisions regarding discontinuation were discussed with the sponsor on a case‐by‐case basis. Concomitant medications required for treating AEs occurring between the start of screening and the end of the trial were permitted. Administration of acetaminophen was acceptable, but was required to be documented. For any patient who developed hypocalcemia (ie, a serum calcium level lower than 8.2 mg/dL [2.05 mmol/L] or an ionized calcium level lower than 4.4 mg/dL [1.1 mmol/L]), calcium supplementation was given until calcium levels had returned to within normal ranges.
Interventions
Patients in the treatment group received a total of three escalating doses of intravenous BPS804 solution, each separated by a 2‐week period (day 1: 5 mg/kg; day 15: 10 mg/kg; day 29: 20 mg/kg), at a rate of 2 mL/min (total administration time approximately 120 ± 10 min). Thirteen‐week toxicology studies in the mouse (up to a maximum dose of 100 mg/kg/week, which was the no‐observed‐adverse‐effect level) and in the cynomolgus monkey (up to a maximum dose of 200 mg/kg/week; the no‐observed‐adverse‐effect level was 20 mg/kg) provided toxicology coverage for the current study. The maximum intended dose in the current study was 20 mg/kg. This dose was equal to the maximum tested dose in a previous clinical study (CBPS804A2101), which was well tolerated and did not raise any overt safety concerns. Treatment was assigned to all eligible patients in accordance with entry into the study and was randomized using treatment allocation cards. The reference group received no intervention at all.
Outcomes
All blood samples were collected at 8:00 a.m. (±1 hour), when patients were in a fasted state. Efficacy assessments were carried out on blood samples taken at baseline, during drug administration (pre‐dose and 2 hours post‐dose), as well as on days 8, 43, 57, 85, and 113, and at the end of the study (day 141). All assessments were measured in a central laboratory. After collection, the blood samples were frozen and kept at –70°C until analysis.
P1NP was measured using the Elecsys total P1NP electrochemiluminescence sandwich‐based immunoassay with a lower limit of quantification (LLOQ) of 8.99 ng/mL; OC was measured using the Elecsys N‐mid OC electrochemiluminescence sandwich‐based immunoassay with an LLOQ of 6.54 ng/mL; and CTX‐1 was measured using the Elecsys β‐CrossLaps electroluminescence assay with an LLOQ of 0.082 ng/mL (all assays from Roche Diagnostics, Mannheim, Germany). P1CP was measured using the MicroVue P1CP sandwich enzyme immunoassay with a lower limit of detection of 1.0 ng/mL; and BSAP enzymatic activity was measured using the MicroVue BSAP enzyme immunoassay with a LLOQ of 4.95 U/L (both assays from Quidel Corporation, San Diego, CA, USA).
Standard pharmacokinetic (PK) parameters (the area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration [AUC0–last]; the area under the plasma concentration–time curve from time zero to infinity [AUC0–inf]; the observed maximum plasma concentration following drug administration [Cmax]; the time to reach Cmax after drug administration [Tmax]; and the terminal elimination half‐life [t½]) were measured.
Hematology, blood chemistry, and urine analysis were monitored for abnormalities at screening, pre‐dose on each day of treatment, 2 weeks after administration of the first dose, and at the end of the study. Levels of serum 25‐hydroxy vitamin D were assessed at screening only. Vital signs, physical condition, body weight, and electrocardiography results were assessed at screening, pre‐dose on each day of treatment, regularly during the follow‐up phase, and at the end of the study.
Electrocardiography results were also assessed 1, 2, and 5 weeks after administration of the first dose.
All AEs and serious AEs, and their severity and suspected relationship to the study drug, were recorded. A DXA scan was performed at screening, 12 weeks after administration of the first dose, and at the end of the study. The precision of this technique has been demonstrated.(28) A radiograph was obtained only in case of suspected fractures. If a fracture occurred in the 28 days before the first dose administration (treatment group) or randomization (reference group) and the last radiograph was obtained more than 2 weeks before the first administration/randomization, radiography was repeated.
Potential immunogenicity of BPS804 was assessed in patients who had been randomized to the treatment group. Serum anti‐BPS804 antibodies were measured using a validated homogenous AlphaLISA assay with an LLOQ of 250 ng/mL (PerkinElmer Inc., Waltham, MA, USA).
Sample size
A sample size of six patients in the BPS804 group completing the study was considered sufficient for the simultaneous rejection of the null hypothesis of no change from baseline (taken as the geometric mean of screening and day 1 pre‐dose values in the BPS804 group) in the three biomarker endpoints (P1NP, P1CP, and BSAP) or in lumbar spine aBMD.
This sample size provided >99% power to detect, at a two‐sided significance level of 0.05, a treatment difference of at least 0.7 ng/mL in P1NP and at least 0.48 mU/mL in BSAP between baseline and day 15 after dose administration (as based on a previous study, CBPS804A2101), assuming a variability of ∼0.07 ng/mL for P1NP and ∼0.04 mU/mL for BSAP. Even with only four evaluable patients, the power would be 93% for P1NP and 87% for BSAP. P1CP was assumed to follow a similar pattern to P1NP.
A sample size of six patients provided 93% power to detect, at a two‐sided significance level of 0.05, a treatment difference of at least 0.036 g/cm2 (with a variability of ∼0.0004 g/cm2) in lumbar spine aBMD between baseline and day 113 after dose administration (as observed when measured via DXA in a previous study, CBPS804A2101). Even with only five evaluable patients, the power would be 84%.
A sample size of three patients in the reference group was considered sufficient for the comparison of change from baseline between the BPS804 and reference groups. Using a one‐sided, two‐sample t test at the 10% level, the power to show a difference between groups with six patients in the BPS804 group and three patients in the reference group would be 98% for P1NP, 97% for BSAP, and 86% for aBMD. This assumed that the reference group would have a mean change of zero from baseline in P1NP, BSAP, and BMD levels, and that the variability in change from baseline would be the same as in the BPS804 group.
Study stopping rules are listed in the Supporting Methods.
Statistical analyses
The PD analysis set comprised all patients who had a baseline measurement and at least one post‐baseline measurement for at least one of the biomarkers analyzed (P1NP, P1CP, BSAP, CTX‐1, and OC); a baseline measurement and at least one post‐baseline measurement for aBMD; and no major protocol deviations that would impact on the PD data. The PK analysis set comprised all those who had at least one post‐baseline PK measurement (Cmax, Tmax) and no major protocol deviations that would impact on the PK data. The safety analysis set comprised all patients who received the study drug.
The comparisons versus study baseline were calculated as ratios on day 43 for P1NP, P1CP, BSAP, OC, and CTX‐1 and on day 141 for aBMD. The 90% confidence intervals (CI) for the change from study baseline were calculated (to describe the location of true treatment difference). It was considered a sign of efficacy if patients in the BPS804 group showed significant increases versus baseline in P1NP, P1CP, and BSAP on day 43 or a significant increase in lumbar spine aBMD on day 141 (in both cases statistical significance was defined at p < 0.05, based on two‐sided, one‐sample t tests).
Summary statistics were provided for the safety assessments.
Results
Patient demographics and baseline disease characteristics
In total, 14 adults with moderate OI were enrolled (BPS804 group, n = 9; reference group, n = 5). A patient flow diagram is shown in Fig. 2. All patients in the BPS804 group completed the study. One patient in the reference group provided samples for analysis of all efficacy and safety assessments up until day 43 but was subsequently lost to follow‐up and therefore did not receive a DXA scan at the end of the study. The first patient was enrolled on June 22, 2011; the last patient completed the study on December 5, 2012.
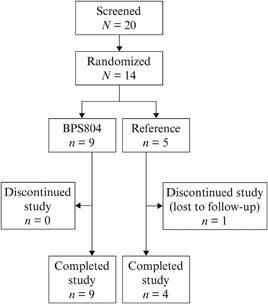
Patient flow diagram.
Patient baseline demographics and disease characteristics are shown in Table 1. The overall mean age was marginally higher in the BPS804 group (30.7 years) than in the reference group (27.4 years). The majority of patients were men (71.4%) and all were white. In the BPS804 group, four patients had type I OI and five had type III/IV OI. In the reference group, two patients had type I OI and three had type III/IV OI. The mean lumbar spine Z‐score in the BPS804 group was –2.6; in the reference group, the mean was –2.2. Two patients in the BPS804 group and one in the reference group had received bisphosphonates in the past; no patient received bisphosphonates in the 2 years prior to study entry. None of the patients in either study group was treated with hormonae replacement therapy before or during the study. One participant in the reference group was treated with melatonin 3 mg/day, 5 days a week (for insomnia) prior to study start; another participant in the reference group was fitted with a levonorgestrel‐releasing intrauterine device before the start of the study.
Patient Baseline Demographics and Disease Characteristics
| BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) | |
| Age (years) | |||
| Mean ± SD | 30.7 ± 13.5 | 27.4 ± 15.5 | 29.5 ± 13.7 |
| Median (range) | 25.0 (19.0 to 57.0) | 21.0 (19.0 to 55.0) | 21.5 (19.0 to 57.0) |
| Male, n (%) | 7 (77.8) | 3 (60.0) | 10 (71.4) |
| White, n (%) | 9 (100.0) | 5 (100.0) | 14 (100.0) |
| Type I OI, n | 4 | 2 | 6 |
| Type III/IV OI, n | 5 | 3 | 8 |
| Weight (kg) | |||
| Mean ± SD | 61.8 ± 14.4 | 58.2 ± 13.0 | 60.5 ± 13.5 |
| Median (range) | 63.9 (43.5 to 80.1) | 54.0 (44.0 to 75.0) | 59.5 (43.5 to 80.1) |
| Height (cm) | |||
| Mean ± SD | 161.6 ± 12.2 | 162.8 ± 13.9 | 162.0 ± 12.3 |
| Median (range) | 162.0 (142.0 to 178.0) | 161.0 (142.0 to 176.0) | 162.0 (142.0 to 178.0) |
| Lumbar spine Z‐score | |||
| Mean ± SD | −2.59 ± 1.19 | −2.18 ± 0.51 | −2.44 ± 1.00 |
| Median (range) | −2.30 (−4.9 to −1.1) | −2.07 (−2.9 to −1.5) | −2.19 (−4.9 to −1.1) |
| Years on bisphosphonates2 | |||
| Number of patients | 2 | 1 | 3 |
| Mean ± SD | 8.53 ± 4.88 | 15.46 | 10.84 ± 5.28 |
| Median (range) | 8.53 (5.1 to 12.0) | 15.46 (15.5 to 15.5) | 11.99 (5.1 to 15.5) |
| BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) | |
| Age (years) | |||
| Mean ± SD | 30.7 ± 13.5 | 27.4 ± 15.5 | 29.5 ± 13.7 |
| Median (range) | 25.0 (19.0 to 57.0) | 21.0 (19.0 to 55.0) | 21.5 (19.0 to 57.0) |
| Male, n (%) | 7 (77.8) | 3 (60.0) | 10 (71.4) |
| White, n (%) | 9 (100.0) | 5 (100.0) | 14 (100.0) |
| Type I OI, n | 4 | 2 | 6 |
| Type III/IV OI, n | 5 | 3 | 8 |
| Weight (kg) | |||
| Mean ± SD | 61.8 ± 14.4 | 58.2 ± 13.0 | 60.5 ± 13.5 |
| Median (range) | 63.9 (43.5 to 80.1) | 54.0 (44.0 to 75.0) | 59.5 (43.5 to 80.1) |
| Height (cm) | |||
| Mean ± SD | 161.6 ± 12.2 | 162.8 ± 13.9 | 162.0 ± 12.3 |
| Median (range) | 162.0 (142.0 to 178.0) | 161.0 (142.0 to 176.0) | 162.0 (142.0 to 178.0) |
| Lumbar spine Z‐score | |||
| Mean ± SD | −2.59 ± 1.19 | −2.18 ± 0.51 | −2.44 ± 1.00 |
| Median (range) | −2.30 (−4.9 to −1.1) | −2.07 (−2.9 to −1.5) | −2.19 (−4.9 to −1.1) |
| Years on bisphosphonates2 | |||
| Number of patients | 2 | 1 | 3 |
| Mean ± SD | 8.53 ± 4.88 | 15.46 | 10.84 ± 5.28 |
| Median (range) | 8.53 (5.1 to 12.0) | 15.46 (15.5 to 15.5) | 11.99 (5.1 to 15.5) |
SD = standard deviation; OI = osteogenesis imperfecta.
No patient had received bisphosphonates in the 2 years prior to study entry.
Patient Baseline Demographics and Disease Characteristics
| BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) | |
| Age (years) | |||
| Mean ± SD | 30.7 ± 13.5 | 27.4 ± 15.5 | 29.5 ± 13.7 |
| Median (range) | 25.0 (19.0 to 57.0) | 21.0 (19.0 to 55.0) | 21.5 (19.0 to 57.0) |
| Male, n (%) | 7 (77.8) | 3 (60.0) | 10 (71.4) |
| White, n (%) | 9 (100.0) | 5 (100.0) | 14 (100.0) |
| Type I OI, n | 4 | 2 | 6 |
| Type III/IV OI, n | 5 | 3 | 8 |
| Weight (kg) | |||
| Mean ± SD | 61.8 ± 14.4 | 58.2 ± 13.0 | 60.5 ± 13.5 |
| Median (range) | 63.9 (43.5 to 80.1) | 54.0 (44.0 to 75.0) | 59.5 (43.5 to 80.1) |
| Height (cm) | |||
| Mean ± SD | 161.6 ± 12.2 | 162.8 ± 13.9 | 162.0 ± 12.3 |
| Median (range) | 162.0 (142.0 to 178.0) | 161.0 (142.0 to 176.0) | 162.0 (142.0 to 178.0) |
| Lumbar spine Z‐score | |||
| Mean ± SD | −2.59 ± 1.19 | −2.18 ± 0.51 | −2.44 ± 1.00 |
| Median (range) | −2.30 (−4.9 to −1.1) | −2.07 (−2.9 to −1.5) | −2.19 (−4.9 to −1.1) |
| Years on bisphosphonates2 | |||
| Number of patients | 2 | 1 | 3 |
| Mean ± SD | 8.53 ± 4.88 | 15.46 | 10.84 ± 5.28 |
| Median (range) | 8.53 (5.1 to 12.0) | 15.46 (15.5 to 15.5) | 11.99 (5.1 to 15.5) |
| BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) | |
| Age (years) | |||
| Mean ± SD | 30.7 ± 13.5 | 27.4 ± 15.5 | 29.5 ± 13.7 |
| Median (range) | 25.0 (19.0 to 57.0) | 21.0 (19.0 to 55.0) | 21.5 (19.0 to 57.0) |
| Male, n (%) | 7 (77.8) | 3 (60.0) | 10 (71.4) |
| White, n (%) | 9 (100.0) | 5 (100.0) | 14 (100.0) |
| Type I OI, n | 4 | 2 | 6 |
| Type III/IV OI, n | 5 | 3 | 8 |
| Weight (kg) | |||
| Mean ± SD | 61.8 ± 14.4 | 58.2 ± 13.0 | 60.5 ± 13.5 |
| Median (range) | 63.9 (43.5 to 80.1) | 54.0 (44.0 to 75.0) | 59.5 (43.5 to 80.1) |
| Height (cm) | |||
| Mean ± SD | 161.6 ± 12.2 | 162.8 ± 13.9 | 162.0 ± 12.3 |
| Median (range) | 162.0 (142.0 to 178.0) | 161.0 (142.0 to 176.0) | 162.0 (142.0 to 178.0) |
| Lumbar spine Z‐score | |||
| Mean ± SD | −2.59 ± 1.19 | −2.18 ± 0.51 | −2.44 ± 1.00 |
| Median (range) | −2.30 (−4.9 to −1.1) | −2.07 (−2.9 to −1.5) | −2.19 (−4.9 to −1.1) |
| Years on bisphosphonates2 | |||
| Number of patients | 2 | 1 | 3 |
| Mean ± SD | 8.53 ± 4.88 | 15.46 | 10.84 ± 5.28 |
| Median (range) | 8.53 (5.1 to 12.0) | 15.46 (15.5 to 15.5) | 11.99 (5.1 to 15.5) |
SD = standard deviation; OI = osteogenesis imperfecta.
No patient had received bisphosphonates in the 2 years prior to study entry.
Efficacy
Biomarkers of bone formation and bone resorption
BPS804 treatment provided significant increases from baseline to day 43 after first treatment in the activity of serum biomarkers of bone formation. P1NP was increased by 84% compared with baseline in the BPS804 group (p < 0.001) and by 6% compared with baseline in the reference group (p = 0.651; Table 2, Fig. 3A). P1CP was increased by 53% compared with baseline in the BPS804 group (p = 0.003) and by 5% compared with baseline in the reference group (p = 0.600; Table 2, Fig. 3B). BSAP was increased by 59% compared with baseline in the BPS804 group (p < 0.001) and decreased by 13% compared with baseline in the reference group (p = 0.582; Table 2, Fig. 3C). OC was increased by 44% compared with baseline in the BPS804 group (p = 0.012) and decreased by 19% compared with baseline in the reference group (p = 0.436; Table 2, Fig. 3D).
Activity of Serum Bone Formation and Bone Resorption Biomarkers on Day 43 After First Treatment with BPS804
| BPS804 (n = 9) | Reference (n = 5) | |||||
| Parameter | Ratio of geometric mean (90% CI) | Change from baseline (%) | p | Ratio of geometric mean (90% CI) | Change from baseline (%) | p |
| P1NP | 1.84 (1.65–2.06) | 84 | <0.001 | 1.06 (0.83–1.34) | 6 | 0.651 |
| P1CP | 1.53 (1.27–1.84) | 53 | 0.003 | 1.05 (0.87–1.26) | 5 | 0.600 |
| BSAP | 1.59 (1.36–1.86) | 59 | <0.001 | 0.87 (0.53–1.42) | −13 | 0.582 |
| OC | 1.44 (1.17–1.78) | 44 | 0.012 | 0.81 (0.48–1.36) | −19 | 0.436 |
| CTX‐14 | 0.554 (−) | −44 | NT | 0.932 (−) | −7 | NT |
| BPS804 (n = 9) | Reference (n = 5) | |||||
| Parameter | Ratio of geometric mean (90% CI) | Change from baseline (%) | p | Ratio of geometric mean (90% CI) | Change from baseline (%) | p |
| P1NP | 1.84 (1.65–2.06) | 84 | <0.001 | 1.06 (0.83–1.34) | 6 | 0.651 |
| P1CP | 1.53 (1.27–1.84) | 53 | 0.003 | 1.05 (0.87–1.26) | 5 | 0.600 |
| BSAP | 1.59 (1.36–1.86) | 59 | <0.001 | 0.87 (0.53–1.42) | −13 | 0.582 |
| OC | 1.44 (1.17–1.78) | 44 | 0.012 | 0.81 (0.48–1.36) | −19 | 0.436 |
| CTX‐14 | 0.554 (−) | −44 | NT | 0.932 (−) | −7 | NT |
CI = confidence interval; P1NP = procollagen type 1 N‐terminal propeptide; P1CP = procollagen type 1 C‐terminal propeptide; BSAP = bone‐specific alkaline phosphatase; OC = osteocalcin; CTX‐1 = type 1 collagen cross‐linked C‐telopeptide; NT = not tested.
For CTX‐1, n = 8; 90% CI values and p values were not determined for CTX‐1.
Activity of Serum Bone Formation and Bone Resorption Biomarkers on Day 43 After First Treatment with BPS804
| BPS804 (n = 9) | Reference (n = 5) | |||||
| Parameter | Ratio of geometric mean (90% CI) | Change from baseline (%) | p | Ratio of geometric mean (90% CI) | Change from baseline (%) | p |
| P1NP | 1.84 (1.65–2.06) | 84 | <0.001 | 1.06 (0.83–1.34) | 6 | 0.651 |
| P1CP | 1.53 (1.27–1.84) | 53 | 0.003 | 1.05 (0.87–1.26) | 5 | 0.600 |
| BSAP | 1.59 (1.36–1.86) | 59 | <0.001 | 0.87 (0.53–1.42) | −13 | 0.582 |
| OC | 1.44 (1.17–1.78) | 44 | 0.012 | 0.81 (0.48–1.36) | −19 | 0.436 |
| CTX‐14 | 0.554 (−) | −44 | NT | 0.932 (−) | −7 | NT |
| BPS804 (n = 9) | Reference (n = 5) | |||||
| Parameter | Ratio of geometric mean (90% CI) | Change from baseline (%) | p | Ratio of geometric mean (90% CI) | Change from baseline (%) | p |
| P1NP | 1.84 (1.65–2.06) | 84 | <0.001 | 1.06 (0.83–1.34) | 6 | 0.651 |
| P1CP | 1.53 (1.27–1.84) | 53 | 0.003 | 1.05 (0.87–1.26) | 5 | 0.600 |
| BSAP | 1.59 (1.36–1.86) | 59 | <0.001 | 0.87 (0.53–1.42) | −13 | 0.582 |
| OC | 1.44 (1.17–1.78) | 44 | 0.012 | 0.81 (0.48–1.36) | −19 | 0.436 |
| CTX‐14 | 0.554 (−) | −44 | NT | 0.932 (−) | −7 | NT |
CI = confidence interval; P1NP = procollagen type 1 N‐terminal propeptide; P1CP = procollagen type 1 C‐terminal propeptide; BSAP = bone‐specific alkaline phosphatase; OC = osteocalcin; CTX‐1 = type 1 collagen cross‐linked C‐telopeptide; NT = not tested.
For CTX‐1, n = 8; 90% CI values and p values were not determined for CTX‐1.
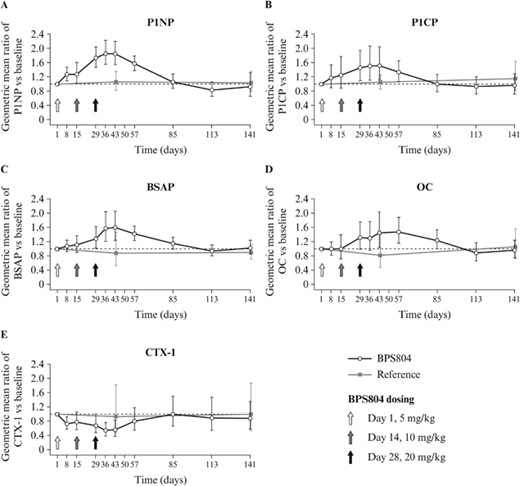
Changes from baseline to week 20 in (A–D) serum levels of bone formation and (E) bone resorption biomarkers in the BPS804 group and the reference group. P1NP, P1CP, OC, and CTX‐1 were measured in ng/mL; BSAP was measured in mU/mL. BSAP = bone‐specific alkaline phosphatase; CTX‐1 = type 1 collagen cross‐linked C‐telopeptide; OC = osteocalcin; P1CP = procollagen type 1 C‐terminal propeptide; P1NP = procollagen type 1 N‐terminal propeptide.
BPS804 treatment downregulated serum levels of the bone resorption biomarker CTX‐1 by 44% from baseline to day 43 after first treatment (statistical significance was not tested). A downregulation of 7% was observed in the reference group (Table 2, Fig. 3E).
Lumbar spine aBMD
Treatment with BPS804 significantly increased lumbar spine aBMD by 4% on day 141 compared with baseline (p = 0.038; ratio of geometric mean [90% CI] 1.04 [1.01 to 1.07]). In the reference group, lumbar spine aBMD was increased by 1% compared with baseline (p = 0.138; 1.01 [1.00 to 1.01]).
Pharmacokinetics
BPS804 exhibited a favorable PK profile, consistent with that for a fully human IgG2λ monoclonal antibody. All nine patients included in the PK analysis dataset had quantifiable serum BPS804 concentrations throughout most of the three‐period treatment phases. Peak serum concentrations were 175 µg/mL, 365 µg/mL, and 746 µg/mL for the 5, 10, and 20 mg/kg doses, respectively, and were reached shortly after the end of each infusion (median Tmax of 2.12, 2.07, and 2.20 hours, respectively, for the three doses). Summary statistics of PK parameters are presented in Table 3.
Pharmacokinetic Parameters for BPS804
| Parameter | BPS804 5 mg/kg (n = 9) | BPS804 10 mg/kg (n = 9) | BPS804 20 mg/kg (n = 9) |
| AUC0–last (day·μg/mL) | 604.2 ± 160.0 | ND | 8458.3 ± 2512.5 |
| AUC0–inf (day·μg/mL) | ND | ND | 8541.7 ± 2533.3 |
| Cmax (μg/mL) | 175 ± 34 | 365 ± 86 | 746 ± 169 |
| Tmax (hours), median (min–max) | 2.12 (1.95–4.18) | 2.07 (1.97–2.38) | 2.20 (2.00–11.1) |
| t½ (days) | ND | ND | 9.58 ± 1.39 |
| Parameter | BPS804 5 mg/kg (n = 9) | BPS804 10 mg/kg (n = 9) | BPS804 20 mg/kg (n = 9) |
| AUC0–last (day·μg/mL) | 604.2 ± 160.0 | ND | 8458.3 ± 2512.5 |
| AUC0–inf (day·μg/mL) | ND | ND | 8541.7 ± 2533.3 |
| Cmax (μg/mL) | 175 ± 34 | 365 ± 86 | 746 ± 169 |
| Tmax (hours), median (min–max) | 2.12 (1.95–4.18) | 2.07 (1.97–2.38) | 2.20 (2.00–11.1) |
| t½ (days) | ND | ND | 9.58 ± 1.39 |
Data shown are mean ± SD unless otherwise indicated.
AUC0–last = area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration; ND = not done; AUC0–inf = area under the plasma concentration–time curve from time zero to infinity; Cmax = observed maximum plasma concentration following drug administration; Tmax = time to reach Cmax after drug administration; t½ = terminal elimination half‐life; SD = standard deviation.
Pharmacokinetic Parameters for BPS804
| Parameter | BPS804 5 mg/kg (n = 9) | BPS804 10 mg/kg (n = 9) | BPS804 20 mg/kg (n = 9) |
| AUC0–last (day·μg/mL) | 604.2 ± 160.0 | ND | 8458.3 ± 2512.5 |
| AUC0–inf (day·μg/mL) | ND | ND | 8541.7 ± 2533.3 |
| Cmax (μg/mL) | 175 ± 34 | 365 ± 86 | 746 ± 169 |
| Tmax (hours), median (min–max) | 2.12 (1.95–4.18) | 2.07 (1.97–2.38) | 2.20 (2.00–11.1) |
| t½ (days) | ND | ND | 9.58 ± 1.39 |
| Parameter | BPS804 5 mg/kg (n = 9) | BPS804 10 mg/kg (n = 9) | BPS804 20 mg/kg (n = 9) |
| AUC0–last (day·μg/mL) | 604.2 ± 160.0 | ND | 8458.3 ± 2512.5 |
| AUC0–inf (day·μg/mL) | ND | ND | 8541.7 ± 2533.3 |
| Cmax (μg/mL) | 175 ± 34 | 365 ± 86 | 746 ± 169 |
| Tmax (hours), median (min–max) | 2.12 (1.95–4.18) | 2.07 (1.97–2.38) | 2.20 (2.00–11.1) |
| t½ (days) | ND | ND | 9.58 ± 1.39 |
Data shown are mean ± SD unless otherwise indicated.
AUC0–last = area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration; ND = not done; AUC0–inf = area under the plasma concentration–time curve from time zero to infinity; Cmax = observed maximum plasma concentration following drug administration; Tmax = time to reach Cmax after drug administration; t½ = terminal elimination half‐life; SD = standard deviation.
Safety
BPS804 was generally well tolerated. Table 4 shows the number of patients affected for each category of AE. There were 32 different AEs reported overall (Supporting Table 1; 22 different AEs were reported in the BPS804 group and 13 were reported in the reference group); none was suspected by the investigators to be related to BPS804. Three AEs (occurring in two patients receiving BPS804) were moderate in intensity; the remaining AEs were mild in intensity. Headache, influenza, arthralgia, and fatigue were the most common AEs in both groups (Supporting Table 1). There were no deaths reported in the study. There were no serious AEs reported in the BPS804 treatment group. One serious AE (goiter) was reported in the reference group, which was mild in intensity and was resolved before the end of the study. The goiter was euthyroid and the patient was hospitalized on the same day as the event occurred and treated with radioiodine therapy. The event was not suspected to be related to the study medication by the study investigator. No other clinically relevant AEs were reported. No abnormal serum calcium level changes, or clinically significant changes in chemistry parameters (including creatinine), were observed. Fractures were reported in three patients receiving BPS804 (one fracture per patient, in the ankle [minor avulsion of the right malleolus sustained when the patient jumped down a flight of stairs], foot, and scapula, respectively; Supporting Table 1); all were deemed unrelated to BPS804 and not clinically significant. Of those fractures, two occurred 2 days after the first BPS804 dose and the third one occurred on day 48, before aBMD levels had reached their peak amplitude. No fractures were reported in the reference group; although based on medical histories, all patients had had previous fractures. Given the short duration of the study and the small sample size, the clinical significance of this observation is unclear, and may be a chance finding.
Incidence of Adverse Events in the BPS804 Treatment Group and the Reference Group by Primary System Organ Class
| Adverse event | BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) |
| Total | 9 (100) | 4 (80.0) | 13 (92.9) |
| Infections and infestations | 3 (33.3) | 2 (40.0) | 5 (35.7) |
| Injury, poisoning, and procedural complications | 5 (55.6) | 0 | 5 (35.7) |
| General disorders and administration site conditions | 3 (33.3) | 1 (20.0) | 4 (28.6) |
| Nervous system disorders | 2 (22.2) | 2 (40.0) | 4 (28.6) |
| Musculoskeletal and connective tissue disorders | 2 (22.2) | 1 (20.0) | 3 (21.4) |
| Gastrointestinal disorders | 1 (11.1) | 1 (20.0) | 2 (14.3) |
| Respiratory, thoracic, and mediastinal disorders | 2 (22.2) | 0 | 2 (14.3) |
| Endocrine disorders | 0 | 1 (20.0) | 1 (7.1) |
| Psychiatric disorders | 0 | 1 (20.0) | 1 (7.1) |
| Reproductive system and breast disorders | 0 | 1 (20.0) | 1 (7.1) |
| Adverse event | BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) |
| Total | 9 (100) | 4 (80.0) | 13 (92.9) |
| Infections and infestations | 3 (33.3) | 2 (40.0) | 5 (35.7) |
| Injury, poisoning, and procedural complications | 5 (55.6) | 0 | 5 (35.7) |
| General disorders and administration site conditions | 3 (33.3) | 1 (20.0) | 4 (28.6) |
| Nervous system disorders | 2 (22.2) | 2 (40.0) | 4 (28.6) |
| Musculoskeletal and connective tissue disorders | 2 (22.2) | 1 (20.0) | 3 (21.4) |
| Gastrointestinal disorders | 1 (11.1) | 1 (20.0) | 2 (14.3) |
| Respiratory, thoracic, and mediastinal disorders | 2 (22.2) | 0 | 2 (14.3) |
| Endocrine disorders | 0 | 1 (20.0) | 1 (7.1) |
| Psychiatric disorders | 0 | 1 (20.0) | 1 (7.1) |
| Reproductive system and breast disorders | 0 | 1 (20.0) | 1 (7.1) |
Values are n (%).
Incidence of Adverse Events in the BPS804 Treatment Group and the Reference Group by Primary System Organ Class
| Adverse event | BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) |
| Total | 9 (100) | 4 (80.0) | 13 (92.9) |
| Infections and infestations | 3 (33.3) | 2 (40.0) | 5 (35.7) |
| Injury, poisoning, and procedural complications | 5 (55.6) | 0 | 5 (35.7) |
| General disorders and administration site conditions | 3 (33.3) | 1 (20.0) | 4 (28.6) |
| Nervous system disorders | 2 (22.2) | 2 (40.0) | 4 (28.6) |
| Musculoskeletal and connective tissue disorders | 2 (22.2) | 1 (20.0) | 3 (21.4) |
| Gastrointestinal disorders | 1 (11.1) | 1 (20.0) | 2 (14.3) |
| Respiratory, thoracic, and mediastinal disorders | 2 (22.2) | 0 | 2 (14.3) |
| Endocrine disorders | 0 | 1 (20.0) | 1 (7.1) |
| Psychiatric disorders | 0 | 1 (20.0) | 1 (7.1) |
| Reproductive system and breast disorders | 0 | 1 (20.0) | 1 (7.1) |
| Adverse event | BPS804 (n = 9) | Reference (n = 5) | Total (n = 14) |
| Total | 9 (100) | 4 (80.0) | 13 (92.9) |
| Infections and infestations | 3 (33.3) | 2 (40.0) | 5 (35.7) |
| Injury, poisoning, and procedural complications | 5 (55.6) | 0 | 5 (35.7) |
| General disorders and administration site conditions | 3 (33.3) | 1 (20.0) | 4 (28.6) |
| Nervous system disorders | 2 (22.2) | 2 (40.0) | 4 (28.6) |
| Musculoskeletal and connective tissue disorders | 2 (22.2) | 1 (20.0) | 3 (21.4) |
| Gastrointestinal disorders | 1 (11.1) | 1 (20.0) | 2 (14.3) |
| Respiratory, thoracic, and mediastinal disorders | 2 (22.2) | 0 | 2 (14.3) |
| Endocrine disorders | 0 | 1 (20.0) | 1 (7.1) |
| Psychiatric disorders | 0 | 1 (20.0) | 1 (7.1) |
| Reproductive system and breast disorders | 0 | 1 (20.0) | 1 (7.1) |
Values are n (%).
Antibodies to BPS804 were detected in the serum of five patients receiving BPS804; of these, four patients tested positive for antibodies to BPS804 at baseline, before dosing.
Discussion
BPS804, administered as intravenous infusions at doses of 5, 10, and 20 mg/kg, significantly increased biomarkers of bone formation; the results were consistent with a gain in bone mass, as demonstrated by the significant increase in lumbar spine aBMD.
The primary objectives of this phase 2a trial were to determine the pharmacodynamics (as shown by changes from baseline to day 43 in serum biomarkers of bone formation and resorption, and changes from baseline to day 141 in lumbar spine aBMD), and to evaluate the PK profile, safety, and tolerability of three intrapatient escalating doses of BPS804 in adults with moderate OI. An untreated reference group was included for monitoring and observation of the natural progression of OI.
P1NP, P1CP, BSAP, and OC are widely used markers of bone formation.(29) Antiresorptive treatment with bisphosphonates is associated with an increase in aBMD and a reduction in bone formation and resorption biomarkers.(10, 30, 31, 32) In contrast, treatment with BPS804 increased bone formation biomarkers (P1NP, P1CP, BSAP, and OC) and decreased the bone resorption biomarker CTX‐1. These results are consistent with published data on osteoanabolic responses using anti‐sclerostin antibodies for the treatment of postmenopausal osteoporosis.(33, 34)
BPS804 exhibited a predictable PK profile, as would be expected for a fully human IgG2 monoclonal antibody binding to a soluble ligand. Four patients tested positive for antibodies to BPS804 at baseline, before dosing. This finding is not readily explained, but likely indicates an underperforming assay.
In this study, BPS804 was generally well tolerated, with only mild or moderate AEs reported. Three fractures were reported in the BPS804 group, all of which were deemed unrelated to treatment. No fractures were reported in the reference group. Whether the increase in aBMD observed will result in a reduction in the fracture incidence needs to be investigated in larger and longer‐term trials.
This trial demonstrates that BPS804 treatment stimulates bone formation and reduces bone resorption in adults with moderate OI and is associated with an increase in lumbar spine aBMD. BPS804 interferes with Wnt signaling by binding to the inhibitory component sclerostin, thereby producing an osteoanabolic response. A limitation of the study was the small number of participants; further studies enrolling a larger cohort of patients with OI are warranted to confirm these data.
Current treatment concepts in OI rely primarily on bisphosphonates, which enhance mechanical properties and bone architecture on a macroscale by reducing cortical porosity and improving cortical thickness and bone width. This leads to reduced bone resorption and bone turnover.(35) However, it is still unclear whether the effects of bisphosphonates on bone metabolism are favorable for bone regeneration and bone material properties on a molecular level, particularly when considering the need for long‐term treatment regimens. Furthermore, the apparent significant decrease in the incidence of fractures associated with bisphosphonates remains a matter of debate.(36, 37, 38, 39, 40) Hence, the bone‐remodeling‐based concept of anti‐sclerostin treatment may provide a promising approach to cover relevant treatment periods as part of a long‐term medication strategy that enables phases of bone regeneration and formation. The findings of this study pave the way for a longer‐term, phase 3 trial to assess the efficacy, safety, and tolerability of BPS804 in patients with OI.
Disclosures
FHG is a consultant to Novartis, Amgen, and Mereo BioPharma. J‐PD received materials, but not financial support, from Amgen to study oim/oim mice in preclinical studies; he also received travel grants for oral presentations at meetings from Amgen. SH is an employee of Novartis and owns shares in Novartis. UJ was an employee of Novartis at the time of the study. FJ and LS have conducted clinical trials on sclerostin antibodies sponsored by Novartis and Amgen. MD, SG, JR, and PJW have no disclosures to report.
Acknowledgments
This research was funded by Novartis Institutes for BioMedical Research, Basel, Switzerland, and the Shriners of North America. Medical writing support was provided by Noëlle L O'Regan, PhD, of PharmaGenesis London, London, UK, and was funded by Mereo BioPharma.
Authors’ roles: Study concept and design, data acquisition, data analysis and interpretation, drafting manuscript, revising manuscript content, approving final version of manuscript: FHG, J‐PD, MD, SG, SH, FJ, UJ, JR, LS, and PJW. All authors are accountable for all aspects of the work and for ensuring that any questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved. FHG, as corresponding author, accepts responsibility for the integrity of the data analysis.
References
Mereo BioPharma Group. Mereo's BPS‐804 granted EU Orphan Drug status for Osteogenesis Imperfecta [Internet]. 2016 Jun 30 [cited 2017 Apr 7]. Available from: http://mereobiopharma.com/news‐and‐media/press‐releases/2016/mereo‐s‐bps‐804‐granted‐eu‐orphan‐drug‐status‐for‐osteogenesis‐imperfecta/mereo‐s‐bps‐804‐granted‐eu‐orphan‐drug‐status‐for‐osteogenesis‐imperfecta/
Author notes
Public clinical trial registration: http://clinicaltrials.gov/show/NCT01417091. A Randomized, Open Label Intra‐patient Dose Escalation Study With an Untreated Reference Group to Evaluate Safety and Tolerability, Pharmacokinetics, and Pharmacodynamics of Multiple Infusions of BPS804 in Adults With Moderate Osteogenesis Imperfecta

{kind=link}
{kind=link}
{kind=link}